
Abstract
Over the past few decades, research on Alzheimer’s disease (AD) has focused on pathomechanisms linked to two of the major pathological hallmarks of extracellular deposition of beta-amyloid peptides and intra-neuronal formation of neurofibrils. Recently, a third disease component, the neuroinflammatory reaction mediated by cerebral innate immune cells, has entered the spotlight, prompted by findings from genetic, pre-clinical, and clinical studies. Various proteins that arise during neurodegeneration, including beta-amyloid, tau, heat shock proteins, and chromogranin, among others, act as danger-associated molecular patterns, that—upon engagement of pattern recognition receptors—induce inflammatory signaling pathways and ultimately lead to the production and release of immune mediators. These may have beneficial effects but ultimately compromise neuronal function and cause cell death. The current review, assembled by participants of the Chiclana Summer School on Neuroinflammation 2016, provides an overview of our current understanding of AD-related immune processes. We describe the principal cellular and molecular players in inflammation as they pertain to AD, examine modifying factors, and discuss potential future therapeutic targets.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Neuroinflammation plays an important part in the pathogenesis of Alzheimer’s disease (AD), with both positive and negative consequences. |
Induction of inflammatory signaling pathways leads to the production and release of immune mediators, which ultimately compromises neuronal function and causes cell death. |
Anti-inflammatory therapeutic approaches to modify AD progression are the basis for ongoing and future therapeutic trials in this area. |
1 Introduction
Dementias and related diseases of cognitive decline pose an enormous and growing disease burden on our societies and health economies. According to the most recent World Health Organization (WHO) global disease burden report [1], deaths from neurological diseases have risen by 114% over the past 20 years to 1.2 million in 2010. The increase is largely driven by neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease and by an aging population in general. Not surprisingly, the development of strategies to curb this frightening surge is a high priority for life science research. The responsible allocation of these resources requires the identification of valid therapeutic targets. The immune system is particularly alluring in this regard.
How the immune system and peripheral infections contribute to cognitive decline remains incompletely understood, but the past 15 years have established a key role for inflammation in the progression of age-related neurodegeneration. The immune privilege of the brain is clearly not absolute, and cells of the central nervous system (CNS) are sensitive to inflammatory events occurring both within and outside of the brain.
We summarize the current state of neuroinflammation research from cellular to molecular mechanisms, as they pertain to the pathogenesis of AD. Further, we outline leverage points for preventive strategies and therapeutic approaches to stem the daunting surge in dementia diseases facing our society.
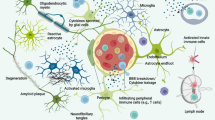
2 Cellular Players
2.1 Microglia
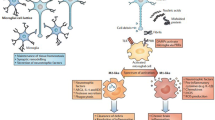
Microglia are the principal innate immune cells in the brain, and they are often considered the macrophages of the CNS. Recent studies have shed light on their origin from erythromyeloid progenitors from the yolk sac [2, 3], which migrate into the brain at embryonic day 7.5 where they further differentiate into microglial cells [2]. Microglia exhibit the capacity of self-renewal within the brain [4, 5], likely arising from a newly identified progenitor [6]. Microglia continuously survey their microenvironment and monitor ongoing synaptic activity, including synapse remodeling, debris clearance, and trophic support for neurons. In addition, they drive a major part of the innate immune response. Microglia react to pathological triggers via pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) [7,8,9].
Microglia are also phagocytic cells and can ingest amyloid β (Aβ) through a range of cell surface receptors, including cluster of differentiation (CD)-14, toll-like receptor (TLR)-2, TLR4, α6β1 integrin, CD47, and scavenger receptors, such as CD36 [10,11,12,13]. It has been suggested that, in AD, a key factor in the accumulation of Aβ throughout the brain is the failure of microglia to remove extracellular amyloid [14,15,16]. Indeed, in cortical tissue specimens from patients with AD, the microglia surrounding plaques are impaired at Aβ uptake [15, 17, 18]. Newly developed positron emission tomography (PET) techniques employ radio ligands to detect activated microglia in vivo [19,20,21]. Many tracers target the 18 kDa translocator protein (TSPO) [19], an outer mitochondrial membrane protein present in microglia, which is upregulated during activation [22,23,24]. The 11C-PK11195 ligand was the first prototypical TSPO ligand, although second-generation tracers have been developed more recently with improved signal-to-noise ratios [25]. However, a common polymorphism significantly influences the binding affinity of these new compounds [26], thus making genetic screening a necessary step for accurate quantification [27]. TSPO upregulation has been described in prodromal AD and in manifest AD dementia, using both 11C-PK11195 [28,29,30,31] and second-generation tracers [32,33,34,35,36] in regions known to be affected by AD pathology and beyond. Mixed evidence has emerged regarding the relationship between in vivo microglial activation and Aβ plaque burden [29, 31, 32, 37, 38].
2.2 Astrocytes
Under pathological conditions, astrocytes exhibit morphological changes, including hypertrophy and upregulation of glial fibrillary acidic protein (GFAP). Astrocytes can detect aggregated proteins such as Aβ or respond to inflammatory molecules (e.g., cytokines, chemokines, see below). Indeed, significant astrocyte reactivity has been reported in sporadic [39,40,41] and familial AD [42]. Similar to microglia, reactive astrocytes can polarize their processes around amyloid plaques and are capable of amyloid plaque degradation [43, 44]. Altered calcium signaling [45], impaired glutamate homeostasis [46, 47], and increased production of inflammatory mediators by astrocytes are also observed in AD.
2.3 Oligodendrocytes
The involvement of oligodendrocytes in AD remains poorly understood, although there is emerging evidence that these cells contribute to the pathogenesis and progression of neurodegenerative disorders, including AD [48]. Bartzokis et al. [49,50,51,52] demonstrated that the loss of myelin integrity that normally occurs during aging is strongly aggravated in human presenilin-1 familial, preclinical, and sporadic AD cases, particularly near Aβ plaques. In addition, focal loss of oligodendrocytes has been observed in sporadic cases of AD. This demyelination was also found in transgenic mouse models of AD, specifically at the core of Aβ plaques [52]. Focal oligodendrocyte loss has also been detected in Tg2576 and APP/PS1 transgenic mice [52], a phenomenon that may negatively influence cortical processing and neurite formation. Several cellular processes such as neuroinflammation, oxidative stress, and/or apoptosis may contribute to oligodendrocyte dysfunction and death [52]. In addition, Aβ can impair the survival and maturation of oligodendrocyte progenitor cells and the formation of the myelin sheath [53].
2.4 Myeloid Cells Other than Microglia
In addition to microglia, a variety of other monocytic cells have been found in the brain, including perivascular cells, meningeal macrophages, choroid plexus macrophages, and peripheral blood-derived monocytes [54]. These cells may, under certain circumstances, also phagocytize and degrade amyloid plaques in a transgenic model of AD [55]. Migration of peripheral monocytes is dependent on C-C chemokine receptor type 2 (CCR2), as its ablation in Tg2576 mice results in decreased recruitment of these cells and a corresponding increase in amyloid pathology [56]. In contrast, blocking transforming growth factor (TGF)-β signaling increased peripheral myeloid cell infiltration into the CNS and significantly reduced the amyloid burden [57]. Glatiramer acetate has also been shown to increase recruitment of peripheral monocytes to the CNS, and this reduces amyloid deposition. Ablation of bone marrow-derived myeloid cells in this model exacerbated amyloid pathology [58]. In contrast, when resident microglia were ablated from the APP/PS1 and APP23 mouse models, recruitment of peripheral myeloid cells was not sufficient to clear amyloid load [59, 60]. Furthermore, a recent parabiosis experiment found no evidence of monocyte infiltration around amyloid plaques [61]. Thus, the extent of myeloid infiltration into the brain and its contribution to damage or clearance of pathological proteins is still not fully understood. A particularly critical aspect of this body of work is the complexity and toxicity of experimental approaches used.
3 PAMPs and DAMPs: Inducers and Modulators of Neuroinflammation in Alzheimer’s Disease
During periods of pathogen invasion or tissue damage, DAMPs and PAMPs alert the immune system of the host and trigger an appropriate response to the insult.
DAMPs encompass a diverse class of molecules. A well-characterized group of DAMPs consists of intracellular proteins that are expressed at a basal level within a cell and are released after injury. These include high-mobility group protein B1 (HMGB1), S100 proteins, heat shock proteins (HSPs), chromogranin A, and Aβ. A second class of DAMPs comprises nucleic acids and nucleotide derivatives, such as mitochondrial DNA (mt-DNA), DNA, and adenosine triphosphate (ATP) [62]. In contrast, PAMPs mainly include microbial molecules that are normally not present in human cells, such as lipid A, flagellin, lipoproteins from Gram-positive and Gram-negative bacteria, bacterial DNA containing particular CpG motifs, and fragments of bacterial peptidoglycan [63].
Both PAMPs and DAMPs contribute to neuroinflammation in AD. Aβ can induce inflammatory responses [64] via activation of pattern recognition receptors (PRRs) of the innate immune system, including TLR2 [65], TLR4, and TLR6, as well as their co-receptors, CD36, CD14, and CD47. Neutralization by CD14 antibodies can reduce the Aβ-induced microglial activation [66]. Furthermore, the NLRP1 and NLRP3 inflammasome can sense a range of aggregated proteins, including Aβ [67]. Indeed, lack of NLRP3 and caspase-1 has been shown to protect mice from AD pathology [67, 68].
HMGB1 levels are increased in AD brains and are associated with senile plaques, promoting their stabilization [69]. It has been shown that microglia stimulation by HMGB1 can reduce Aβ phagocytosis [69]. HMGB1 promotes the migration and proliferation of immune cells through binding to advanced glycation end-product receptors (RAGE) and TLRs [70]. HMGB1 can also act in concert with other factors such as chemokines, growth factors, and PAMPs, together promoting immune system activation [71, 72].
Chromogranin A is associated with microglial activation in neurodegeneration [73, 74] and induces the release of interleukin (IL)-1β, indicating that TLRs and the NLRP3 inflammasome are involved in this pathway [75]. In AD, increased levels of chromogranin A have been observed in senile plaque dystrophic neurites [76]. Interestingly, the immune stimulatory potential appears almost identical to bacterial lipopolysaccharide (LPS), at least in vitro [77].
Many S100 proteins are involved in AD, including S100A9, S100A8, and S100B. S100A8 and S100A9 form a complex that is increased in the brain and cerebrospinal fluid (CSF) of patients with AD [78, 79] and can activate microglia through TLR4. Furthermore, S100A8-mediated inflammatory stimuli are connected with the upregulation of the β-site β-amyloid precursor protein (APP)-cleaving enzyme BACE1, which is involved in APP processing [80, 81]. S100B has been observed in both Aβ plaques and in the CSF [82, 83], and overexpression of human S100B exacerbates amyloidosis and gliosis in the Tg2576 AD mouse model [84].
Likewise, mt-DNA and DNA can be released from the cells and act as DAMPs upon entering the blood circulation, causing inflammation [85]. mt-DNA can bind to TLR-9 and mediate the release of tumor necrosis factor (TNF)-α and type I interferons (IFNs) [86]. Moreover, cell free DNA can bind to TLR and non-TLR receptors. Upon TLR binding, DNA activates the nuclear factor (NF)-κB pathway, thereby promoting pro-inflammatory cytokine production [87]. DNA can also bind to the absent in melanoma 2 (AIM2) inflammasome, releasing IL-1β, through the caspase-1 activation pathway.
HSPs bind to several receptors, such as TLR2 and TLR4, resulting in the production of inflammatory cytokines, such as TNFα and IL-1β [88,89,90]. Furthermore, HSPs may also exert beneficial effects in AD, thus, HSP70 can bind to APP and reduce the secretion of Aβ1-40 and Aβ1-42 through interference with the APP processing pathway [91]. HSP70, together with HSP90, can also interact with tau and Aβ oligomers and degrade them by employing proteasomal degradation [92].
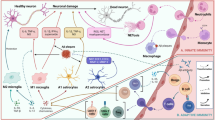
4 Endogenous Modulators
Neurotransmitters such as ATP, glutamate, dopamine, and various neurotrophic factors, e.g., brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF), can act as endogenous modulators. Microglial cells are equipped with a plethora of neurotransmitter receptors, which makes them a primary target, particularly as sites of non-synaptic release [93]. In AD, ATP production from neurons declines. Mitochondrial dysfunction, as evidenced by reduced ATP, is related to oxidative stress in AD pathology [94]. Oxidative stress can also initiate inflammatory responses and contributes to the etiopathology of AD [95].
In AD, glutamatergic neurotransmission is disturbed because of an increased amount of soluble Aβ oligomers [96]. The possible inflammatory process occurs subsequently through activation of microglia with TNFα release, synergizing with N-methyl-d-aspartate (NMDA)-mediated neurodegeneration [97]. However, the modulation of glutamate can be either pro- or anti-inflammatory depending on the expression of different groups of glutamate receptors (GluRs) on microglia and, most likely, on astroglial uptake capabilities [98, 99].
Dopamine possibly mediates the activation of microglia by triggering the mitogen-activated protein kinase (MAPK)–NFκB cascade and inducing toxicity versus dopaminergic neurons [100, 101]. In general, acetylcholine prevents the inflammatory response in microglia via α7-nicotinic acetylcholine receptors, mediated by the PLC/IP3/Ca2+ signaling pathway [102]. In patients with AD, significant loss of cholinergic neurons is tightly related to the progression of the disease. Failure in cholinergic neurotransmission decreases the cholinergic input to microglia, which in turn results in microglial activation [103]. In addition, stimulation of microglia with norepinephrine suppressed inflammation through cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) signaling cascades [104, 105]. Of note, the locus ceruleus, the chief source of noradrenaline (NA) in the human brain, degenerates very early in the disease course. Thus, its projection regions, most prominently the limbic system and neocortex, experience decreased levels of NA. Modeling this in rodents increased Aβ-induced inflammation [106, 107] and substantially increased neuronal death and memory deficits [108]. Using 2-photon laser microscopy, it was demonstrated that depletion of NA in APP/PS1 transgenic mice caused complete inhibition of microglial Aβ clearance, and, subsequently, an increase in the number and volume of Aβ deposits [104]. The replenishment of NA levels in the cortex and hippocampal after treatment with l-threo-DOPS, an NA precursor, partly rescued this phenotype.
The levels of BDNF and NGF are severely altered in the brains of patients with AD. BDNF is released by activated microglia and inhibits the release of TNF-α and IFN-γ, whereas it promotes the expression of anti-inflammatory cytokines IL-4, IL-10, and IL-11 [109]. However, an in vitro study has also shown prolonged microglial activation through a positive feedback loop by autocrine BDNF [110], demonstrating that BDNF may modulate inflammation at various levels. In addition, evidence from a human microglial cell line suggested that NGF synthesis is potentially stimulated by inflammatory signals (cytokines and complement factors), as well as by exposure to Aβ25-35, through NFκB-dependent and -independent mechanisms.
5 Inflammatory Mediators
5.1 Cytokines
Cytokines are released by glial cells, such as astrocytes and microglia, upon every inflammatory challenge [111,112,113]. Many cytokines, such as IL-1β and IL-12, have been related to the progression of AD pathology [114, 115]. Increased IL-1β serum levels have been linked to AD and patients with mild cognitive impairment, a putative prodromal phase of dementia [114, 116]. Several studies have reported associations between IL-1β polymorphisms and the onset of AD pathology [115, 117, 118], linking both IL-1β polymorphisms and apolipoprotein E (APOE)-ε4 to higher levels of IL-1β in the blood and sleep disturbance in patients [119]. IL-12 is related to the regulation of the adaptive and the innate immune system [120], and an IL-12 polymorphism has been linked to AD in a Han Chinese population [121]. Vom Berg et al. [122] suggested that inhibition of the IL-12/IL-23 pathway may attenuate AD pathology and cognitive deficits due to a decrease in the IL-12p40 subunit and its receptor activity [122]. In this study, the concentration of IL-12p40 was increased in the CSF of patients with AD. Regarding anti-inflammatory cytokines, IL-10 deletion attenuated AD-related deficits, such as altered synaptic integrity and behavioral deficits in APP/PS1 mice [123]. Chakrabarty et al. [124] showed that the overexpression of IL-10 using adeno-associated viruses (AAVs) increased amyloid deposition, behavioral deficits, and synaptic alterations and impaired microglial phagocytosis of Aβ in the APP transgenic mouse model [124].
Another major regulator of inflammation is TGF-β. Increased TGF-β has been observed in amyloid plaques [125] and in the CSF of patients with AD [126, 127]. However, this cytokine plays a dual role in AD. Overexpression of TGF-β in vivo induces Aβ deposition in cerebral blood vessels, but it may also decrease microgliosis while increasing Aβ phagocytosis [128]. A link between TGF-β and neuro-fibrillary tangles (NFTs) has also been reported [129].
5.2 Chemokines
Chemokines participate in the chemoattraction of immune cells from the periphery to the brain and in the recruitment and activation of resident glial cells. In AD, chemokines are implicated in both the resolution and the propagation of pathology [130]. The most intensively studied chemokines in AD are CX3C chemokine ligand 1 (CX3CL1) and chemokine ligand 2 (CCL2).
CX3CL1, also termed fractalkine, is expressed by neurons, whereas its receptor, CX3CR1, is predominantly expressed by microglia [131, 132]. The participation of this chemokine in the pathophysiology of AD is complex since CX3CL1/CX3CR1 signaling can have a beneficial role in the context of tau pathology [132,133,135] or a detrimental role in an amyloid context [136]. In fact, in an amyloid model, deficiency of CX3CR1 decreased Aβ deposition [136] but worsened tau pathology and lowered cognitive performance [133, 134]. Moreover, the expression of CX3CL1 has been shown to be increased in tau-injured neurons but decreased in the brains of APP transgenic mice [137]. However, in human patients, the level of CX3CL1 is inversely correlated with AD severity [138]. Together, this may point to the possibility that the same inflammatory mediator may adopt various, if not opposing, effects and properties during disease progression.
CCL2 has also been associated with a dominant role in chronic inflammation [139]. A recent study has demonstrated that the CCL2/CCR2 pathway of astrocyte-induced microglial activation is associated with “M1-polarised” and enhanced microglial activity [140]. In AD, CCL2 levels were increased in mild but not in severe AD, suggesting that elevated CCL2 may play a pathogenic role during early AD stages [141]. In agreement with this, Westin et al. [142] showed that CCL2 is associated with a faster cognitive decline in early disease stages. Kiyota et al. [143] found accelerated neurodegeneration in APP/CCL2 transgenic mice, indirectly suggesting that direct inhibition of CCL2 signaling may modify microglial activation, resulting in lower Aβ deposition and improving behavioral outcomes. CCL2 overexpression accelerated oligomeric and diffuse Aβ deposition and led to spatial and working memory deficits by affecting Aβ seeding in Tg2576 mice [143].
5.3 Other Mediators
Nitric oxide (NO) is synthesized by three different isoforms of NO synthase (NOS). Each isoform plays a role in either AD progression or prevention, suggesting that NO can be neuroprotective or neurotoxic. High doses of LPS induced robust CNS inflammation and microglia-induced release of NO. NO in the CNS can influence many signaling pathways, including protein nitrosylation, impairment of long-term potentiation, or inhibition of mitochondrial respiration. The impact of NO signaling depends on the local cellular environment. In the AD brain, NO mainly derives from the inducible isoform of NOS, NOS2, which is expressed by neurons [144], microglia, and astrocytes [145]. Nitrosative stress has been shown to affect all types of cellular proteins, including, but not restricted to, synaptic proteins. Post-translational protein modification can take place either by s-nitrosylation of cysteine residues or by nitration of tyrosine residues. Importantly, Aβ itself represents a nitration target at tyrosine 10 of its amino acid sequence. Nitration at this position strongly increases the peptide’s propensity to aggregate, and nitrated Aβ predominantly resides in the core of the deposits, suggesting that this mechanism contributes to the initiation of deposition [146].
6 Effect of Neuroinflammation on Neuronal Function
6.1 Cytokines and Synaptic Scaling
Synaptic plasticity is strongly influenced by basal levels of cytokines [147]. Emblematic is the case of “synaptic scaling,” a well-defined form of homeostatic plasticity that regulates the density of GluRs at presynaptic and postsynaptic sites [148]. A homeostatic reduction of neuronal excitability by withdrawal of GluRs is termed down-scaling, whereas the increase of neuronal excitability (by accumulation of GluRs) is known as up-scaling. TNFα has been shown to support synaptic up-scaling by increasing AMPA receptor-dependent miniature excitatory postsynaptic currents (mEPSC). Importantly, TNFα required for up-scaling synapses is derived from glial cells [149] and not from neurons themselves. Such evidence implies that glial cells are able to release cytokines in response to changes in neuronal activity. By contrast, enhanced release of inflammatory cytokines, for instance during chronic peripheral inflammation, can disrupt the physiological mechanisms of synaptic plasticity, promoting neuronal hyper-excitability and increased susceptibility to seizure generation [150].
A growing body of evidence demonstrates that microglia can actively respond to increased neuronal excitability, and microglial processes make physical contact with excitatory synapses [151,152,153,154]. This type of microglia–synapse interaction has been shown to reduce neuronal excitability [110, 155], potentially as a form of a regulatory mechanism for preventing glutamatergic excitotoxicity [152].
6.2 Microglia and Synaptic Pruning
Microglia can actively participate in remodeling synaptic connections (“synaptic pruning”). A pathological form of synaptic pruning may represent a commonly shared mechanism among several neurological conditions of different nature: a recent study in a murine model of chronic stress, showed electron-dense (dark) microglia co-localized with synaptic terminals. This microglial phenotype associated with synaptic pruning appeared clearly reactive, possibly accounting for an increased loss of synapses during chronic inflammation [156]. Microglia may also remove synapses in a complement-dependent manner in a mouse model of West Nile virus-induced neuroinflammation [157]. Mice with either a deficit in the number of microglia (IL-34−/−) or a deficiency of complement components (such as C3 protein or complement receptor 3 knock-out) were protected from inflammation-induced synaptic loss [157].
An alternative hypothesis suggests that pathological pruning of synapses during inflammation may also represent a form of “tissue remodeling” for auto-protective purposes [158]. A study suggested that upon LPS injection, microglia pruned preferentially GABAergic terminals, thereby increasing excitatory synaptic activity and induction of neurotrophic pathways in downstream neurons. This mechanism has been interpreted as an attempt to promote neuronal viability in a pathological context, although the price was a temporary imbalance of synaptic connectivity [159].
In mouse models of Aβ deposition, complement protein C1q was elevated as early as 1 month of age in both DG and frontal cortex. At this timepoint, neither plaques nor synaptic loss are detectable. At a later age (mice aged 3–4 months), the number of synapses decreased significantly; however, synaptic loss was rescued almost completely in the absence of either C1q, C3, or CR3. Additionally, intracerebroventricular (ICV) injection of oligomeric Aβ in wild-type mice induced synapse loss and activated a phagocytic phenotype in microglia. Moreover, synapse loss in response to oligomeric Aβ was not observed in C1q or CR3 knock-out mice [160] (Table 1).
Similar findings have been obtained in a mouse model deficient for the progranulin gene, typically associated with frontotemporal dementia (FTD) in humans [161, 162]. Lack of progranulin has been shown to trigger an exaggerated inflammatory reaction in microglia and macrophages [161, 163]. Interestingly, the brains of progranulin-deficient mice showed increased levels of complement proteins, a prominent pro-phagocytic activation of microglia, and enhanced pruning of synapses [164].
6.3 Inflammation and Neurogenesis
Under homeostasis, immunological signals can actively shape adult neurogenesis. Microglia were shown to rapidly engulf and remove apoptotic neuronal progenitors, remarkably, without any trace of inflammatory reaction [165, 166]. Other evidence has pinpointed a close interplay between different immune proteins and the neurogenic process [167, 168]. IL-1β has often received particular attention because of its anti-neurogenic activity [169,170,171,172,173,174,175]. One may assume that microglia are primarily responsible for this reduction of neurogenesis during inflammatory challenges. An interesting molecular player is the CX3C axis between neurons and microglia, which is known to preserve the microglial “resting” phenotype under physiological conditions [176, 177]. Several consistent findings showed reduced neurogenesis in CX3CR1-deficient mice, along with increased NF-κB activation and IL-1β expression in microglia [178,179,180,181]. Consistently, when an inflammatory challenge is applied under CX3CR1-deficient conditions, microglia release an increased and uncontrolled amount of inflammatory mediators and free radicals, causing neurotoxicity and cognitive/behavioral deficits [132, 182]. Interestingly, during ageing, neurons decrease expression of CX3CL1 [183], which likely would result in a general downregulation of the CX3C axis and pro-inflammatory skewing of microglia [184, 185]. Pro-inflammatory microglial priming has been suggested as a possible mechanism leading to the dysregulated microglial function and the ensuing stepwise decline of neurogenesis (and potentially neurodegeneration) during senility [186,187,188,189,190,191,192]. In contrast, several lines of evidence point towards the pro-neurogenic function of microglia, especially during the period of early brain development. These indications suggest that microglia play an important role during brain development, axonal guidance, and formation of neuronal networks.
6.4 Astrocytes and Glutamate Reuptake
In the CNS, extracellular levels of glutamate are tightly regulated by astrocytes in order to modulate GluR activity and prevent potential excitotoxicity [193]. Once in the synaptic cleft, excess glutamate is promptly scavenged by the excitatory amino acid transporters (EAATs) expressed on both neurons and astrocytes [194]. The astrocytic EAAT2 is thought to be responsible for about 90% of all glutamate uptake in the brain [195, 196]. There are no synaptic enzymes that otherwise would degrade glutamate. Therefore, astrocyte-mediated glutamate uptake represents the primary mechanism for the homeostatic regulation of glutamate bioavailability [197]. Impairment of glutamate uptake causes excitotoxicity characterized by overload of cellular calcium, generation of free radicals, and protein/lipid oxidation. Notably, astrocyte glutamate transporters (EAAT1 and EAAT2) were shown to be reduced in the cortex and hippocampus of patients with AD [46, 198]. Moreover, Aβ-induced neurotoxicity in vivo has been associated with NMDA receptor-dependent excitotoxicity [199]. In conclusion, pharmaceutical compounds aiming to modulate glutamate excitotoxicity have revealed a certain therapeutic potential for neurodegenerative diseases (Table 2).
6.5 Function and Dysfunction of the Blood–Brain Barrier
Dysfunction of the blood–brain barrier (BBB) is a relatively new frontier in AD research [200,201,202]. The fully functional BBB is a highly specialized monolayer of endothelial cells lining the cerebrovasculature and separating the circulating blood from the brain parenchyma. The integrity of the BBB depends critically on the functional state of the associated pericytes, astrocytes, and microglia and is compromised during neuroinflammation [203]. Aβ binds to low-density lipoprotein receptor-related protein-1 (LPR1) on the endothelial cells of the brain capillaries and is then released into the bloodstream [204, 205]. Vice versa, in the BBB, RAGE are upregulated with aging and facilitate the influx of Aβ from the blood into the brain [206]. Deficient Aβ clearance from the brain parenchyma is thus proposed to be, at least in part, the result of its faulty transport across the BBB [207,208,209].
7 Modifiable Risk Factors for AD
A plethora of exogenous factors exert both beneficial and detrimental modulating effects on the inflammatory state of an organism. This, in turn, has direct and important consequences for the risk of developing AD. As these factors are amenable to non-pharmacological interventions and can be mitigated (or promoted) by preventive measures or lifestyle choices, they deserve special attention.
7.1 Infections
The evidence pointing to infections as risk factors for AD stems from epidemiological and neuropathological studies. Prospective cohort studies show that infection represents an important risk factor in the progression of dementia and AD [210, 211]. A case–control study suggested that multiple infections double the risk of developing dementia [212]. In studies of a large AD patient cohort, peripheral infection was associated with accelerated cognitive decline [213, 214]. Conversely, the frequency of various infections, including urinary and respiratory tract infections is higher among individuals with AD than among healthy, age-matched controls [215]. Indeed, pneumonia is one of the most common causes of death in AD [204, 205, 216–218]. In contrast, vaccination against influenza and other infectious conditions is associated with a significantly lower risk of developing AD [219, 220].
A number of specific viral, bacterial, and fungal infections has been detected by polymerase chain reaction (PCR) in human AD brain tissue and have been implicated in AD development. One example is herpes simplex virus type 1 (HSV-1) [221,222,223], which is an AD risk factor in people carrying the APOE4 allele [224, 225]. Chlamydia pneumonia, a Gram-negative bacteria, has been detected via PCR in the brain tissue of patients with AD [226, 227], where it was found to have infected microglia, astrocytes, and neurons [227]. Interestingly, fungal proteins and DNA have been identified in the brain tissue and CSF of patients with AD [228, 229]. In postmortem AD brains, co-infection with many fungi has also been reported, with fungal material identified inside neuronal cells and in many regions [230, 231]. The significance of these findings remains uncertain because several factors, including postmortem time and handling, must be considered before conclusions are drawn. In addition, causality is notoriously difficult to ascertain in these scenarios. Systemic inflammation certainly has an impact on the brain, which is not at all “immune privileged,” as textbooks still suggest. Thus, higher rates of cognitive decline have been observed in patients with AD with acute systemic inflammation [214]. Leaky gut may be one of the drivers of systemic inflammation and is directly related to an imbalance of gut microbiota [232].
7.2 Traumatic Brain Injury
Traumatic brain injury (TBI) leads to damaged blood vessels, axons, nerve cells, and glia of the brain in a focal, multifocal, or diffuse pattern, resulting in impaired brain function [233, 234]. A single moderate or severe TBI may increase the risk of developing late-onset AD, whereas repetitive mild TBI (e.g., through contact sport) is associated with an elevated risk of chronic traumatic encephalopathy [235, 236]. Two key meta-analyses of case–control studies found a significant association between moderate-severe TBI and AD [237, 238]. Furthermore, human pathological studies evince abnormal accumulation of AD-related pathological proteins, including soluble and insoluble Aβ and hyperphosphorylated tau aggregates, following TBI. This, in turn, is supported by studies in large animals [239, 240]. Aggregation and deposition of Aβ is accelerated after an acute TBI event, with changes within a mere 24 h up to 2 months after injury in animal studies [240,241,242,243,244]. Further, aggregation and deposition of Aβ have been associated with memory impairments in 3xTg-AD mice [244]. Aberrant tau phosphorylation has also been described in several models after TBI [245,246,247,248]. The formation of misfolded Aβ and tau oligomeric seeds triggered by TBI may lead to spreading of the pathology in a prion-like manner, causing a faster and more severe onset of the disease [249].
7.3 Smoking
The role of smoking as a modifiable risk factor in AD is controversial. Some early case–control studies reported smoking had a beneficial effect on AD [250, 251]. In contrast, more recent cohort studies without affiliation to the tobacco industry clearly point towards a deleterious impact [252, 253]. Meta-analyses of these studies showed that smoking during a lifetime is associated with at least a 1.7 times higher risk of AD [254]. Although this increase obviously correlates with smoking intensity and duration [255], the findings regarding former smoking status are more heterogeneous. Reitz et al. [252] observed no association between past smoking and AD, whereas Aggarwal et al. [253] reported a lower risk for former smokers carrying the ApoEε4 allele than for those who never smoked.
It is estimated that, today, smoking accounts for 4.7 million AD cases worldwide [256]. Evidence from various in vitro and in vivo studies suggests that sustained cigarette smoke exposure facilitates the emergence of regional Aβ and tau pathology [257, 258]. Several possible pathways are likely to contribute to the development of pathological AD features in smokers. These include cerebrovascular dysfunction [259], neuroinflammation [258], and protein misfolding and aggregation [260, 261], which all may be triggered by an increase in oxidative stress [262,263,264]. However, human postmortem studies yielded contradictory results concerning the link between smoking and AD neuropathology, as Aβ levels were reduced in the brains of active smoker AD cases compared with never-smoking patients [265]. Remarkably, nicotine and some related compounds exert neuroprotective effects in a variety of model systems [266,267,268], for example via activation of nicotinic acetylcholine receptors [269] or direct binding to Aβ fibrils [270].
7.4 Physical Activity
A case–control study showed patients with AD were less active in midlife [271]. Physical inactivity is accompanied by several secondary effects, including obesity, metabolic syndrome, type 2 diabetes mellitus (T2DM), and cardiovascular disease [272]. In contrast, regular physical exercise positively influences neurogenesis, brain plasticity, and metabolic function, reduces levels of pro-inflammatory cytokines and oxidative stress [273,274,275], and can alter disease-related biomarkers in patients with dementia [276]. Thus, it is not surprising that cognitive function and mental processing speed in elderly people could be significantly improved with leisure time activities and exercise programs [256, 277,278,279]. However, Küster et al. [280] showed that a (self-reported) active lifestyle rather than the exercise itself is associated with a decreased risk of AD. Whether physical exercise benefits all patient populations equally remains controversial [276, 281]. Some studies report a stronger effect of physical activity among APOE4 carriers compared with non-carriers [282,283,284], whereas others could not replicate these results [285,286,287]. Analysis of different animal models suggests positive effects of physical exercise on BDNF levels, oxidative stress and even Aβ and tau pathology, resulting in delayed disease onset and progression [288–291].
7.5 Diet and Obesity
Many specific dietary components have been studied in relation to AD. In clinical studies, a higher intake of unsaturated fatty acids, antioxidants, and vitamins B12 and folate have been associated with a lower risk for AD and cognitive decline [292,293,294]. However, the opposite or even no effect has also been found for these factors [295,296,297]. Instead of focusing on individual dietary components, the effect of overall dietary patterns (which incorporates nutrient interaction) has been examined, including the Mediterranean Diet (MeDi) [298], Dietary Approaches to Stop Hypertension (DASH) [299], and the Mediterranean-DASH Intervention for Neurodegenerative Delay (MIND) [300]. These studies point towards diet as having a protective effect against cognitive decline and development of AD [299, 301,302,303,304,305,306,307].
In contrast, overnutrition can lead to obesity, which in turn has been associated with AD development. Obesity is characterized by leptin and insulin resistance, leading to impaired energy metabolism and chronic inflammation [308]. This chronic inflammatory status can cause cellular stress and neurodegeneration and is thought to be the link between obesity and its adverse effects on cognitive performance and AD development [309,310,311,312,313]. Most importantly, some of these effects may occur as early as midlife. Thus, increased body mass index and sagittal abdominal diameter in men aged 40–45 years has been associated with an increased risk of AD in later life [314].
7.6 Lifetime Distress
Lifetime frequency of stress exposure is consistently associated with the incidence of mild cognitive impairment (MCI) and may increase the risk of late-onset AD [315,316,317]. In particular, higher levels of the stress hormone cortisol are associated with an accelerated age-related decline in cognition [318, 319]. The hypothalamic–pituitary–adrenal (HPA) axis regulates the release of cortisol in humans or the corresponding corticosterone in rodents. This system is dysregulated in patients with AD, with higher cortisol levels found in the blood plasma and CSF of subjects with AD than in age-matched controls [318, 320].
Interestingly, exposing rodents to stressful experiences increases corticosterone levels and glucocorticoid receptor activation, resulting in aggravation of AD-related neuropathology in various transgenic models [321,322,323,324,325,326]. Microglia are highly responsive to glucocorticoids, with abundant glucocorticoid receptor expression levels [327]. Furthermore, glucocorticoids can induce a pro-inflammatory microglial phenotype upon stress, especially following a secondary inflammatory challenge [328,329,330].
7.7 Diabetes Mellitus
T2DM affects approximately 370 million people worldwide, accounting for 90–95% of all patients with diabetes [331]. The disease is characterized by hyperglycemia, insulin resistance, and relative lack of insulin [332]. People with T2DM have a 73% greater risk of developing dementia [333] and decreased white and grey matter volume of the temporal and frontal lobes [334, 335]. Cortical and hippocampal atrophies have also been observed in diabetic mice (db/db) [336].
Importantly, insulin resistance leads to the generation of NFTs: decreased activation of protein kinase B in T2DM results in ineffective inhibition of glycogen synthase kinase 3, thus mediating tau phosphorylation and formation of NFTs [337, 338]. In addition, insulin levels in patients with diabetes mean the insulin-degrading enzyme is sequestered away from Aβ, which fosters the accumulation of Aβ in the brain [339, 340]. Patients with T2DM have impaired immunological defense mechanisms, resulting in frequent infections, which may contribute to the development of AD [341]. The concentration of pro-inflammatory cytokines in the CSF is increased in patients with T2DM [342]; indeed, chronic sub-acute inflammation can also induce insulin resistance and cause T2DM [343].
8 Protection by Anti-inflammatory Strategies
8.1 Past and Present Strategies
Various anti-inflammatory therapeutic approaches have been taken to modify AD progression over the past 2 decades, ranging from non-steroidal anti-inflammatory drugs (NSAIDs) to TNFα inhibition.
8.1.1 Non-steroidal Anti-inflammatory Drugs (NSAIDs)
One of those approaches was ADAPT (Alzheimer’s Disease Anti-inflammatory Prevention Trial). This trial was constructed to examine whether NSAIDs could prevent or delay the onset of AD and whether such treatment could impact cognitive decline associated with aging [344].
Early epidemiological studies had suggested that long-term treatment with NSAIDs decreased the risk of AD development [345,346,347,348]. Additionally, strong experimental evidence has emerged supporting the positive effect of NSAIDs in AD animal models [80, 349]. NSAIDs have been shown to reduce Aβ secretion and accumulation, both in vitro and in vivo, to modulate γ-secretase activity, to exert an anti-inflammatory effect, and to improve cognitive function in AD mouse models [350,351,352,353,354,355].
However, most NSAIDs have not convincingly shown any beneficial effects during clinical trials in patients with AD [356]. Only a small, early study using indomethacin in patients with AD [357], which has not been replicated, and a follow-up analysis from the ADAPT research group using naproxen [358] have shown positive effects. Aspirin also did not prove effective against AD but increased the risk of serious bleeds (AD2000 trial).
8.1.2 Non-NSAIDs
Peroxisome proliferator-activated receptor (PPAR)-γ agonism has consistently been shown to reduce the production of inflammatory cytokines and amyloid accumulation in AD mouse models [351, 359,360,361,362]. Rosiglitazone induces activation of the ERK pathway, leading to cognitive enhancement in AD models [363,364,365,366]. Pioglitazone has been found to improve cognition and cerebral blood flow in mild AD [367]. Additionally, pioglitazone treatment reduced dementia risk in patients with initially non-insulin-dependent diabetes mellitus in a case–control study [368]. However, a pilot randomized clinical trial for the safety of this drug in patients with AD found no significant effect [369]. Rosiglitazone has been found to delay cognitive decline in patients with early AD and MCI [370]. Another study showed improvement in cognitive function using pioglitazone, which was restricted to APOEε4 non-carriers [371]. The TOMORRW study is ongoing and will evaluate the efficacy of pioglitazone versus placebo in delaying the onset of MCI-AD in cognitively normal participants who are at high risk for developing MCI within the next 5 years (NCT01931566).
8.1.3 Tumor Necrosis Factor-α Inhibitors
Inhibiting TNFα signaling has also become an interesting and promising approach to the treatment of AD. A clinical case report found that intrathecal administration of infliximab (an antibody against TNFα already approved for other indications) reduced Aβ plaques and tau pathology in APP/PS1 mice and enhanced cognitive function. Additionally, two small pilot clinical studies using a different TNFα inhibitor, etanercept, showed cognitive improvement in patients with AD [372, 373]. However, these studies used small sample sizes and an open-label design and lacked a placebo group. Thus, a larger well-designed placebo-controlled study would be necessary to assess the possible utility of TNFα in AD [349].
8.1.4 Other Anti-inflammatory Drugs
Trials have examined other anti-inflammatory drugs, such as prednisone, hydroxychloroquine, simvastatin, and atorvastatin, but have shown no significant positive cognitive effects in patients with AD [368, 374,375,376,377,378].
8.2 Future Strategies
Recently, fenamate NSAIDs including mefenamic acid were found to selectively inhibit NLRP3 through the inhibition of volume-regulated ion channels (VRACs), thereby preventing cognitive impairments in rodent models of AD. Mefenamic acid is already on the market and is used for abdominal pain in premenstrual syndrome. MCC950, a new potent NLRP3-selective inhibitor has been developed but is not yet available for clinical use [379]. Anakinra, an IL-1 receptor antagonist, and a neutralizing antibody, canakinumab, have been proposed to work by inhibiting this NLRP3 axis, but the cost benefit and bioavailability in the brain remains a concern [380].
CSP-1103 (also known as CHF 5074 or Itanapraced) is now in phase III clinical trials as a microglia modulator. It may inhibit Aβ plaque deposition, reduce tau pathology, restore normal microglial function by increasing phagocytosis, and decrease production of pro-inflammatory cytokines [381].
Some other new therapeutic targets have been proposed. MAPKα inhibitors (e.g., Neflamapimod [VX-745]) could reduce IL-1β levels [382] (NCT02423200). Administration of low-dose IL-2 could increase plaque-associated microglia and improve cognitive performance [383]. C3aR antagonist SB290157 could decrease amyloid load and microgliosis [384]. PD-1 inhibitors could reduce plaque load and improve cognition [385]. Blocking the p40 common subunit of IL-12 and IL-23 could decrease amyloid burden [122, 359, 386]. A CD33 inhibitor might promote microglial phagocytosis of Aβ [387].
9 Summary and Conclusions
Neuroinflammation in AD is likely to arise from the recognition of Aβ by PRRs on the surface of innate immune cells in the brain. Once initiated, sustained inflammation and neurodegeneration may unleash further factors, which, in turn, act as DAMPs and thereby contribute to the persisting and chronic sterile immune reaction in the brain. Several mechanisms of interaction by which inflammatory processes contribute to disease progression have been identified. Given that deposition of Aβ occurs decades prior to the first amnestic and cognitive deficits, such mechanisms may represent promising therapeutic targets. Identification of suitable mode and site of intervention models, which better target the human cerebral innate immune system, and associated biomarkers, is urgently required.
References
World Health Organization. The global burden of disease: 2004 update. 2008.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–5.
Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16(3):273–80.
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10(12):1538–43.
Tay TL, Hagemeyer N, Prinz M. The force awakens: insights into the origin and formation of microglia. Curr Opin Neurobiol. 2016;39:30–7.
Elmore MRP, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–97.
Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15(8):738–48.
Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci Off J Soc Neurosci. 2005;25(36):8240–9.
Sondag CM, Dhawan G, Combs CK. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflamm. 2009;6:1.
Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci Off J Soc Neurosci. 2003;23(7):2665–74.
Ries M, Sastre M. Mechanisms of Aβ clearance and degradation by glial cells. Front Aging Neurosci (Internet). 2016;8:160. https://doi.org/10.3389/fnagi.2016.00160.
Tahara K, Kim H-D, Jin J-J, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain J Neurol. 2006;129(Pt 11):3006–19.
Wilkinson K, El Khoury J. Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer’s disease. Int J Alzheimers Dis. 2012;2012:489456.
Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci Off J Soc Neurosci. 2008;28(33):8354–60.
Thériault P, ElAli A, Rivest S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res Ther. 2015;7(1):41.
Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer’s disease. Nat Rev Immunol. 2006;6(5):404–16.
Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol (Berl). 1992;84(3):225–33.
Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, et al. Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PLOS One. 2013;8(4):e60921.
Jacobs AH, Tavitian B, INMiND consortium. Noninvasive molecular imaging of neuroinflammation. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2012;32(7):1393–415.
Stefaniak J, O’Brien J. Imaging of neuroinflammation in dementia: a review. J Neurol Neurosurg Psychiatry. 2016;87(1):21–8.
Varley J, Brooks DJ, Edison P. Imaging neuroinflammation in Alzheimer’s disease and other dementias: recent advances and future directions. Alzheimers Dement J Alzheimers Assoc. 2015;11(9):1110–20.
Lavisse S, Guillermier M, Hérard A-S, Petit F, Delahaye M, Van Camp N, et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci Off J Soc Neurosci. 2012;32(32):10809–18.
Liu G-J, Middleton RJ, Hatty CR, Kam WW-Y, Chan R, Pham T, et al. The 18 kDa translocator protein, microglia and neuroinflammation. Brain Pathol Zurich Switz. 2014;24(6):631–53.
Venneti S, Lopresti BJ, Wiley CA. The peripheral benzodiazepine receptor (translocator protein 18 kDa) in microglia: from pathology to imaging. Prog Neurobiol. 2006;80(6):308–22.
Chauveau F, Boutin H, Van Camp N, Dollé F, Tavitian B. Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur J Nucl Med Mol Imaging. 2008;35(12):2304–19.
Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2012;32(1):1–5.
Turkheimer FE, Rizzo G, Bloomfield PS, Howes O, Zanotti-Fregonara P, Bertoldo A, et al. The methodology of TSPO imaging with positron emission tomography. Biochem Soc Trans. 2015;43(4):586–92.
Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, et al. In-vivo measurement of activated microglia in dementia. Lancet Lond Engl. 2001;358(9280):461–7.
Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, et al. Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32(3):412–9.
Fan Z, Aman Y, Ahmed I, Chetelat G, Landeau B, Ray Chaudhuri K, et al. Influence of microglial activation on neuronal function in Alzheimer’s and Parkinson’s disease dementia. Alzheimers Dement J Alzheimers Assoc. 2015;11(6):608–21.e7.
Okello A, Edison P, Archer HA, Turkheimer FE, Kennedy J, Bullock R, et al. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009;72(1):56–62.
Hamelin L, Lagarde J, Dorothée G, Leroy C, Labit M, Comley RA, et al. Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain J Neurol. 2016;139(Pt 4):1252–64.
Kreisl WC, Lyoo CH, McGwier M, Snow J, Jenko KJ, Kimura N, et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain J Neurol. 2013;136(Pt 7):2228–38.
Suridjan I, Pollock BG, Verhoeff NPLG, Voineskos AN, Chow T, Rusjan PM, et al. In-vivo imaging of grey and white matter neuroinflammation in Alzheimer’s disease: a positron emission tomography study with a novel radioligand, [18F]-FEPPA. Mol Psychiatry. 2015;20(12):1579–87.
Varrone A, Oikonen V, Forsberg A, Joutsa J, Takano A, Solin O, et al. Positron emission tomography imaging of the 18-kDa translocator protein (TSPO) with [18F]FEMPA in Alzheimer’s disease patients and control subjects. Eur J Nucl Med Mol Imaging. 2015;42(3):438–46.
Yasuno F, Kosaka J, Ota M, Higuchi M, Ito H, Fujimura Y, et al. Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with [11C]DAA1106. Psychiatry Res. 2012;203(1):67–74.
Fan Z, Okello AA, Brooks DJ, Edison P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain J Neurol. 2015;138(Pt 12):3685–98.
Yokokura M, Mori N, Yagi S, Yoshikawa E, Kikuchi M, Yoshihara Y, et al. In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2011;38(2):343–51.
Carter SF, Schöll M, Almkvist O, Wall A, Engler H, Långström B, et al. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med Off Publ Soc Nucl Med. 2012;53(1):37–46.
Choo ILH, Carter SF, Schöll ML, Nordberg A. Astrocytosis measured by 11C-deprenyl PET correlates with decrease in gray matter density in the parahippocampus of prodromal Alzheimer’s patients. Eur J Nucl Med Mol Imaging. 2014;41(11):2120–6.
Santillo AF, Gambini JP, Lannfelt L, Långström B, Ulla-Marja L, Kilander L, et al. In vivo imaging of astrocytosis in Alzheimer’s disease: an 11C-L-deuteriodeprenyl and PIB PET study. Eur J Nucl Med Mol Imaging. 2011;38(12):2202–8.
Rodriguez-Vieitez E, Saint-Aubert L, Carter SF, Almkvist O, Farid K, Schöll M, et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain J Neurol. 2016;139(Pt 3):922–36.
Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang H-Y. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003;971(2):197–209.
Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9(4):453–7.
Vincent AJ, Gasperini R, Foa L, Small DH. Astrocytes in Alzheimer’s disease: emerging roles in calcium dysregulation and synaptic plasticity. J Alzheimers Dis. 2010;22(3):699–714.
Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann Neurol. 1996;40(5):759–66.
Scimemi A, Meabon JS, Woltjer RL, Sullivan JM, Diamond JS, Cook DG. Amyloid-β1-42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J Neurosci Off J Soc Neurosci. 2013;33(12):5312–8.
Ettle B, Schlachetzki JCM, Winkler J. Oligodendroglia and myelin in neurodegenerative diseases: more than just bystanders? Mol Neurobiol. 2016;53(5):3046–62.
Bartzokis G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol Aging. 2004;25(1):49–62.
Bartzokis G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol Aging. 2011;32(8):1341–71.
Bartzokis G, Lu PH, Mintz J. Quantifying age-related myelin breakdown with MRI: novel therapeutic targets for preventing cognitive decline and Alzheimer’s disease. J Alzheimers Dis. 2004;6(6 Suppl):S53–9.
Mitew S, Kirkcaldie MTK, Halliday GM, Shepherd CE, Vickers JC, Dickson TC. Focal demyelination in Alzheimer’s disease and transgenic mouse models. Acta Neuropathol (Berl). 2010;119(5):567–77.
Desai MK, Mastrangelo MA, Ryan DA, Sudol KL, Narrow WC, Bowers WJ. Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am J Pathol. 2010;177(3):1422–35.
Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15(5):300–12.
Simard AR, Soulet D, Gowing G, Julien J-P, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49(4):489–502.
El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13(4):432–8.
Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, et al. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14(6):681–7.
Koronyo Y, Salumbides BC, Sheyn J, Pelissier L, Li S, Ljubimov V, et al. Therapeutic effects of glatiramer acetate and grafted CD115+ monocytes in a mouse model of Alzheimer’s disease. Brain J Neurol. 2015;138(Pt 8):2399–422.
Prokop S, Miller KR, Drost N, Handrick S, Mathur V, Luo J, et al. Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer’s disease-like mice. J Exp Med. 2015;212(11):1811–8.
Varvel NH, Grathwohl SA, Degenhardt K, Resch C, Bosch A, Jucker M, et al. Replacement of brain-resident myeloid cells does not alter cerebral amyloid-deposition in mouse models of Alzheimer’s disease. J Exp Med. 2015;212(11):1803–9.
Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016;213(5):667–75.
Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81(1):1–5.
Vance RE, Isberg RR, Portnoy DA. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe. 2009;6(1):10–21.
Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Inflammation in Alzheimer’s disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol. 2009;87(3):181–94.
Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, et al. TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J Immunol Baltim Md 1950. 2012;188(3):1098–107.
Fassbender K, Walter S, Kühl S, Landmann R, Ishii K, Bertsch T, et al. The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J Off Publ Fed Am Soc Exp Biol. 2004;18(1):203–5.
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9(8):857–65.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–8.
Takata K, Kitamura Y, Kakimura J, Shibagaki K, Tsuchiya D, Taniguchi T, et al. Role of high mobility group protein-1 (HMG1) in amyloid-beta homeostasis. Biochem Biophys Res Commun. 2003;301(3):699–703.
Degryse B, Bonaldi T, Scaffidi P, Müller S, Resnati M, Sanvito F, et al. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152(6):1197–206.
Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009;86(3):573–6.
Castellani P, Balza E, Rubartelli A. Inflammation, DAMPs, tumor development, and progression: a vicious circle orchestrated by redox signaling. Antioxid Redox Signal. 2014;20(7):1086–97.
Ciesielski-Treska J, Ulrich G, Chasserot-Golaz S, Zwiller J, Revel MO, Aunis D, et al. Mechanisms underlying neuronal death induced by chromogranin A-activated microglia. J Biol Chem. 2001;276(16):13113–20.
Kingham PJ, Cuzner ML, Pocock JM. Apoptotic pathways mobilized in microglia and neurones as a consequence of chromogranin A-induced microglial activation. J Neurochem. 1999;73(2):538–47.
Terada K, Yamada J, Hayashi Y, Wu Z, Uchiyama Y, Peters C, et al. Involvement of cathepsin B in the processing and secretion of interleukin-1beta in chromogranin A-stimulated microglia. Glia. 2010;58(1):114–24.
Weiler R, Lassmann H, Fischer P, Jellinger K, Winkler H. A high ratio of chromogranin A to synaptin/synaptophysin is a common feature of brains in Alzheimer and Pick disease. FEBS Lett. 1990;263(2):337–9.
Taupenot L, Remacle JE, Helle KB, Aunis D, Bader MF. Recombinant human chromogranin A: expression, purification and characterization of the N-terminal derived peptides. Regul Pept. 1995;56(1):71–88.
Maezawa I, Zimin PI, Wulff H, Jin L-W. Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem. 2011;286(5):3693–706.
Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci Off J Soc Neurosci. 2009;29(38):11982–92.
Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer’s disease. Nat Immunol. 2015;16(3):229–36.
Kummer MP, Hammerschmidt T, Martinez A, Terwel D, Eichele G, Witten A, et al. Ear2 deletion causes early memory and learning deficits in APP/PS1 mice. J Neurosci Off J Soc Neurosci. 2014;34(26):8845–54.
Moran O, Conti F, Tammaro P. Sodium channel heterologous expression in mammalian cells and the role of the endogenous beta1-subunits. Neurosci Lett. 2003;336(3):175–9.
Van Eldik LJ, Griffin WS. S100 beta expression in Alzheimer’s disease: relation to neuropathology in brain regions. Biochim Biophys Acta. 1994;1223(3):398–403.
Mori T, Koyama N, Arendash GW, Horikoshi-Sakuraba Y, Tan J, Town T. Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer’s disease. Glia. 2010;58(3):300–14.
Thundyil J, Lim K-L. DAMPs and neurodegeneration. Ageing Res Rev. 2015;24(Pt A):17–28.
Hauser CJ, Sursal T, Rodriguez EK, Appleton PT, Zhang Q, Itagaki K. Mitochondrial damage associated molecular patterns from femoral reamings activate neutrophils through formyl peptide receptors and P44/42 MAP kinase. J Orthop Trauma. 2010;24(9):534–8.
Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29(1):185–214.
Luong M, Zhang Y, Chamberlain T, Zhou T, Wright JF, Dower K, et al. Stimulation of TLR4 by recombinant HSP70 requires structural integrity of the HSP70 protein itself. J Inflamm Lond Engl. 2012;9(1):11.
Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277(17):15028–34.
Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. 2000;12(11):1539–46.
Yang Y, Turner RS, Gaut JR. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J Biol Chem. 1998;273(40):25552–5.
Dickey C, Kraft C, Jinwal U, Koren J, Johnson A, Anderson L, et al. Aging analysis reveals slowed tau turnover and enhanced stress response in a mouse model of tauopathy. Am J Pathol. 2009;174(1):228–38.
Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30(10):527–35.
Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta. 2010;1802(1):2–10.
Mao P, Manczak M, Calkins MJ, Truong Q, Reddy TP, Reddy AP, et al. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid β production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum Mol Genet. 2012;21(13):2973–90.
De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29(9):1334–47.
Wenk GL, Parsons CG, Danysz W. Potential role of N-methyl-d-aspartate receptors as executors of neurodegeneration resulting from diverse insults: focus on memantine. Behav Pharmacol. 2006;17(5–6):411–24.
Farso MC, O’Shea RD, Beart PM. Evidence group I mGluR drugs modulate the activation profile of lipopolysaccharide-exposed microglia in culture. Neurochem Res. 2009;34(10):1721–8.
Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci Off J Soc Neurosci. 2008;28(9):2221–30.
Beaulieu J-M, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63(1):182–217.
Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lança AJ, et al. Dopamine D1 and D2 receptor Co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem. 2004;279(34):35671–8.
Suzuki T, Hide I, Matsubara A, Hama C, Harada K, Miyano K, et al. Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J Neurosci Res. 2006;83(8):1461–70.
Carnevale D, De Simone R, Minghetti L. Microglia-neuron interaction in inflammatory and degenerative diseases: role of cholinergic and noradrenergic systems. CNS Neurol Disord Drug Targets. 2007;6(6):388–97.
Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, et al. Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci USA. 2010;107(13):6058–63.
Tanaka KF, Kashima H, Suzuki H, Ono K, Sawada M. Existence of functional beta1- and beta2-adrenergic receptors on microglia. J Neurosci Res. 2002;70(2):232–7.
Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O’Banion MK, et al. Noradrenergic depletion potentiates beta -amyloid-induced cortical inflammation: implications for Alzheimer’s disease. J Neurosci Off J Soc Neurosci. 2002;22(7):2434–42.
Heneka MT, Gavrilyuk V, Landreth GE, O’Banion MK, Weinberg G, Feinstein DL. Noradrenergic depletion increases inflammatory responses in brain: effects on IkappaB and HSP70 expression. J Neurochem. 2003;85(2):387–98.
Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, et al. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci Off J Soc Neurosci. 2006;26(5):1343–54.
Makar TK, Bever CT, Singh IS, Royal W, Sahu SN, Sura TP, et al. Brain-derived neurotrophic factor gene delivery in an animal model of multiple sclerosis using bone marrow stem cells as a vehicle. J Neuroimmunol. 2009;210(1–2):40–51.
Zhang J, Malik A, Choi HB, Ko RWY, Dissing-Olesen L, MacVicar BA. Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron. 2014;82(1):195–207.
Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K, et al. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol Aging. 2014;35(12):2746–60.
Su F, Bai F, Zhang Z. Inflammatory cytokines and Alzheimer’s disease: a review from the perspective of genetic polymorphisms. Neurosci Bull. 2016;32(5):469–80.
Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2(1):a006346.
Dursun E, Gezen-Ak D, Hanağası H, Bilgiç B, Lohmann E, Ertan S, et al. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J Neuroimmunol. 2015;15(283):50–7.
Sciacca FL, Ferri C, Licastro F, Veglia F, Biunno I, Gavazzi A, et al. Interleukin-1B polymorphism is associated with age at onset of Alzheimer’s disease. Neurobiol Aging. 2003;24(7):927–31.
Forlenza OV, Diniz BS, Talib LL, Mendonça VA, Ojopi EB, Gattaz WF, et al. Increased serum IL-1beta level in Alzheimer’s disease and mild cognitive impairment. Dement Geriatr Cogn Disord. 2009;28(6):507–12.
Payão SLM, Gonçalves GM, de Labio RW, Horiguchi L, Mizumoto I, Rasmussen LT, et al. Association of interleukin 1β polymorphisms and haplotypes with Alzheimer’s disease. J Neuroimmunol. 2012;247(1–2):59–62.
Yuan H, Xia Q, Ge P, Wu S. Genetic polymorphism of interleukin 1β-511C/T and susceptibility to sporadic Alzheimer’s disease: a meta-analysis. Mol Biol Rep. 2013;40(2):1827–34.
Yin Y, Liu Y, Pan X, Chen R, Li P, Wu H-J, et al. Interleukin-1β promoter polymorphism enhances the risk of sleep disturbance in Alzheimer’s disease. PloS One. 2016;11(3):e0149945.
Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity. 2003;19(5):641–4.
Zhu X-C, Tan L, Jiang T, Tan M-S, Zhang W, Yu J-T. Association of IL-12A and IL-12B polymorphisms with Alzheimer’s disease susceptibility in a Han Chinese population. J Neuroimmunol. 2014;274(1–2):180–4.
Vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat Med. 2012;18(12):1812–9.
Guillot-Sestier M-V, Doty KR, Gate D, Rodriguez J, Leung BP, Rezai-Zadeh K, et al. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron. 2015;85(3):534–48.
Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B, et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. 2015;85(3):519–33.
van der Wal EA, Gómez-Pinilla F, Cotman CW. Transforming growth factor-beta 1 is in plaques in Alzheimer and Down pathologies. Neuroreport. 1993;4(1):69–72.
Chao CC, Ala TA, Hu S, Crossley KB, Sherman RE, Peterson PK, et al. Serum cytokine levels in patients with Alzheimer’s disease. Clin Diagn Lab Immunol. 1994;1(4):433–6.
Zetterberg H, Andreasen N, Blennow K. Increased cerebrospinal fluid levels of transforming growth factor-beta1 in Alzheimer’s disease. Neurosci Lett. 2004;367(2):194–6.
Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, et al. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7(5):612–8.
Chalmers KA, Love S. Neurofibrillary tangles may interfere with Smad 2/3 signaling in neurons. J Neuropathol Exp Neurol. 2007;66(2):158–67.
Le Thuc O, Blondeau N, Nahon JL, Rovère C. The complex contribution of chemokines to neuroinflammation: switching from beneficial to detrimental effects. Ann N Y Acad Sci. 2015;1351(1):127–40.
Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci USA. 1998;95(18):10896–901.
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9(7):917–24.
Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19–31.
Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P, et al. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem. 2011;286(37):32713–22.
Nash KR, Lee DC, Hunt JB, Morganti JM, Selenica ML, Moran P, et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol Aging. 2013;34(6):1540–8.
Lee DC, Rizer J, Selenica M-LB, Reid P, Kraft C, Johnson A, et al. LPS-induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflamm. 2010;7:56.
Duan RS, Yang X, Chen ZG, Lu MO, Morris C, Winblad B, et al. Decreased fractalkine and increased IP-10 expression in aged brain of APPswe transgenic mice. Neurochem Res. 2008;33(6):1085–9.
Kim T-S, Lim H-K, Lee JY, Kim D-J, Park S, Lee C, et al. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci Lett. 2008;436(2):196–200.
Sokolova A, Hill MD, Rahimi F, Warden LA, Halliday GM, Shepherd CE. Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer’s disease. Brain Pathol. 2009;19(3):392–8.
He M, Dong H, Huang Y, Lu S, Zhang S, Qian Y, et al. Astrocyte-derived CCL2 is associated with m1 activation and recruitment of cultured microglial cells. Cell Physiol Biochem. 2016;38(3):859–70.
Galimberti D, Fenoglio C, Lovati C, Venturelli E, Guidi I, Corrà B, et al. Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer’s disease. Neurobiol Aging. 2006;27(12):1763–8.
Westin K, Buchhave P, Nielsen H, Minthon L, Janciauskiene S, Hansson O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS One. 2012;7(1):1–6.
Kiyota T, Yamamoto M, Xiong H, Lambert MP, Klein WL, Gendelman HE, et al. CCL2 accelerates microglia-mediated A? Oligomer formation and progression of neurocognitive dysfunction. PLoS One. 2009;4(7):e6197.
Vodovotz Y, Lucia MS, Flanders KC, Chesler L, Xie QW, Smith TW, et al. Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer’s disease. J Exp Med. 1996;184(4):1425–33.
Heneka MT, Wiesinger H, Dumitrescu-Ozimek L, Riederer P, Feinstein DL, Klockgether T. Neuronal and glial coexpression of argininosuccinate synthetase and inducible nitric oxide synthase in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60(9):906–16.
Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, et al. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. 2011;71(5):833–44.
Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25(2):181–213.
Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135(3):422–35.
Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440(7087):1054–9.
Riazi K, Galic MA, Kuzmiski JB, Ho W, Sharkey KA, Pittman QJ. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci USA. 2008;105(44):17151–6.
Dissing-Olesen L, LeDue JM, Rungta RL, Hefendehl JK, Choi HB, MacVicar BA. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J Neurosci Off J Soc Neurosci. 2014;34(32):10511–27.
Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu L-J. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J Neurosci Off J Soc Neurosci. 2014;34(32):10528–40.
Fontainhas AM, Wang M, Liang KJ, Chen S, Mettu P, Damani M, et al. Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PloS One. 2011;6(1):e15973.
Li Y, Du X-F, Liu C-S, Wen Z-L, Du J-L. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell. 2012;23(6):1189–202.
Ji K, Akgul G, Wollmuth LP, Tsirka SE. Microglia actively regulate the number of functional synapses. PloS One. 2013;8(2):e56293.
Bisht K, Sharma KP, Lecours C, Sánchez MG, El Hajj H, Milior G, et al. Dark microglia: a new phenotype predominantly associated with pathological states. Glia. 2016;64(5):826–39.
Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534(7608):538–43.
Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, et al. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci Off J Soc Neurosci. 2012;32(34):11706–15.
Chen Z, Jalabi W, Hu W, Park H-J, Gale JT, Kidd GJ, et al. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat Commun. 2014;22(5):4486.
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–6.
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–9.
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–4.
Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207(1):117–28.
Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang H-Y, et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell. 2016;165(4):921–35.
Abiega O, Beccari S, Diaz-Aparicio I, Nadjar A, Layé S, Leyrolle Q, et al. Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol. 2016;14(5):e1002466.
Sierra A, Encinas JM, Deudero JJP, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7(4):483–95.
Boulanger LM. Immune proteins in brain development and synaptic plasticity. Neuron. 2009;64(1):93–109.
Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64(1):61–78.
Gemma C, Bachstetter AD, Cole MJ, Fister M, Hudson C, Bickford PC. Blockade of caspase-1 increases neurogenesis in the aged hippocampus. Eur J Neurosci. 2007;26(10):2795–803.
Green HF, Nolan YM. Unlocking mechanisms in interleukin-1β-induced changes in hippocampal neurogenesis–a role for GSK-3β and TLX. Transl Psychiatry. 2012;20(2):e194.
Guadagno J, Swan P, Shaikh R, Cregan SP. Microglia-derived IL-1β triggers p53-mediated cell cycle arrest and apoptosis in neural precursor cells. Cell Death Dis. 2015;4(6):e1779.
Koo JW, Duman RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci USA. 2008;105(2):751–6.
Wu MD, Montgomery SL, Rivera-Escalera F, Olschowka JA, O’Banion MK. Sustained IL-1β expression impairs adult hippocampal neurogenesis independent of IL-1 signaling in nestin + neural precursor cells. Brain Behav Immun. 2013;32:9–18.
Zhang K, Xu H, Cao L, Li K, Huang Q. Interleukin-1β inhibits the differentiation of hippocampal neural precursor cells into serotonergic neurons. Brain Res. 2013;15(1490):193–201.
Zunszain PA, Anacker C, Cattaneo A, Choudhury S, Musaelyan K, Myint AM, et al. Interleukin-1β: a new regulator of the kynurenine pathway affecting human hippocampal neurogenesis. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2012;37(4):939–49.
Biber K, Neumann H, Inoue K, Boddeke HWGM. Neuronal “On” and “Off” signals control microglia. Trends Neurosci. 2007;30(11):596–602.
Hellwig S, Heinrich A, Biber K. The brain’s best friend: microglial neurotoxicity revisited. Front Cell Neurosci. 2013;7:71.
Maggi L, Scianni M, Branchi I, D’Andrea I, Lauro C, Limatola C. CX(3)CR1 deficiency alters hippocampal-dependent plasticity phenomena blunting the effects of enriched environment. Front Cell Neurosci. 2011;5:22.
Reshef R, Kreisel T, Beroukhim Kay D, Yirmiya R. Microglia and their CX3CR1 signaling are involved in hippocampal-but not olfactory bulb-related memory and neurogenesis. Brain Behav Immun. 2014;41:239–50.
Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci Off J Soc Neurosci. 2011;31(45):16241–50.
Sellner S, Paricio-Montesinos R, Spieß A, Masuch A, Erny D, Harsan LA, et al. Microglial CX3CR1 promotes adult neurogenesis by inhibiting Sirt 1/p65 signaling independent of CX3CL1. Acta Neuropathol Commun. 2016;4(1):102.
Corona AW, Norden DM, Skendelas JP, Huang Y, O’Connor JC, Lawson M, et al. Indoleamine 2,3-dioxygenase inhibition attenuates lipopolysaccharide induced persistent microglial activation and depressive-like complications in fractalkine receptor (CX(3)CR1)-deficient mice. Brain Behav Immun. 2013;31:134–42.
Bachstetter AD, Morganti JM, Jernberg J, Schlunk A, Mitchell SH, Brewster KW, et al. Fractalkine and CX 3 CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol Aging. 2011;32(11):2030–44.
Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT. Age-related alterations in the dynamic behavior of microglia. Aging Cell. 2011;10(2):263–76.
Hefendehl JK, Neher JJ, Sühs RB, Kohsaka S, Skodras A, Jucker M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell. 2014;13(1):60–9.
Eggen BJL, Raj D, Hanisch U-K, Boddeke HWGM. Microglial phenotype and adaptation. J Neuroimmune Pharmacol Off J Soc NeuroImmune Pharmacol. 2013;8(4):807–23.
Gemma C, Bachstetter AD. The role of microglia in adult hippocampal neurogenesis. Front Cell Neurosci. 2013;22(7):229.
Harry GJ. Microglia during development and aging. Pharmacol Ther. 2013;139(3):313–26.
Niraula A, Sheridan JF, Godbout JP. Microglia priming with aging and stress. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2017;42(1):318–33.
Perry VH. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol (Berl). 2010;120(3):277–86.
Raj DDA, Jaarsma D, Holtman IR, Olah M, Ferreira FM, Schaafsma W, et al. Priming of microglia in a DNA-repair deficient model of accelerated aging. Neurobiol Aging. 2014;35(9):2147–60.
Wong WT. Microglial aging in the healthy CNS: phenotypes, drivers, and rejuvenation. Front Cell Neurosci. 2013;7:22.
Dong X, Wang Y, Qin Z. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30(4):379–87.
Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258(5087):1498–501.
Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105.
Holmseth S, Scott HA, Real K, Lehre KP, Leergaard TB, Bjaalie JG, et al. The concentrations and distributions of three C-terminal variants of the GLT1 (EAAT2; slc1a2) glutamate transporter protein in rat brain tissue suggest differential regulation. Neuroscience. 2009;162(4):1055–71.
Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2011;13(1):22–37.
Scott HA, Gebhardt FM, Mitrovic AD, Vandenberg RJ, Dodd PR. Glutamate transporter variants reduce glutamate uptake in Alzheimer’s disease. Neurobiol Aging. 2011;32(3):553.e1–11.
Miguel-Hidalgo JJ, Alvarez XA, Cacabelos R, Quack G. Neuroprotection by memantine against neurodegeneration induced by beta-amyloid(1-40). Brain Res. 2002;958(1):210–21.
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12(12):723–38.
Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta. 2016;1862(5):887–900.
Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: a matter of blood–brain barrier dysfunction? J Exp Med. 2017;214(11):3151–69.
Rustenhoven J, Jansson D, Smyth LC, Dragunow M. Brain pericytes as mediators of neuroinflammation. Trends Pharmacol Sci. 2017;38(3):291–304.
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood–brain barrier. J Clin Investig. 2000;106(12):1489–99.
Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, et al. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007;13(9):1029–31.
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nat Med. 2003;9(7):907–13.
Zlokovic BV, Yamada S, Holtzman D, Ghiso J, Frangione B. Clearance of amyloid beta-peptide from brain: transport or metabolism? Nat Med. 2000;6(7):718–9.
Selkoe DJ. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis. 2001;3(1):75–80.
Holtzman DM, Zlokovic B. Role of Aβ transport and clearance in the pathogenesis and treatment of Alzheimer’s disease. In: Sisodia SS, Tanzi RE editors. Alzheimer’s disease (Internet). Boston: Springer; 2007. pp 179–98. https://doi.org/10.1007/978-0-387-35135-3_11.
Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304(16):1787–94.
McManus RM, Heneka MT. Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res Ther. 2017;9(1):14.
Dunn N, Mullee M, Perry VH, Holmes C. Association between dementia and infectious disease: evidence from a case–control study. Alzheimer Dis Assoc Disord. 2005;19(2):91–4.
Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74(6):788–9.
Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73(10):768–74.
Natalwala A, Potluri R, Uppal H, Heun R. Reasons for hospital admissions in dementia patients in Birmingham, UK, during 2002-2007. Dement Geriatr Cogn Disord. 2008;26(6):499–505.
Fitzpatrick AL, Kuller LH, Lopez OL, Kawas CH, Jagust W. Survival following dementia onset: Alzheimer’s disease and vascular dementia. J Neurol Sci. 2005;15(229–230):43–9.
Foley NC, Affoo RH, Martin RE. A systematic review and meta-analysis examining pneumonia-associated mortality in dementia. Dement Geriatr Cogn Disord. 2015;39(1–2):52–67.
Magaki S, Yong WH, Khanlou N, Tung S, Vinters HV. Comorbidity in dementia: update of an ongoing autopsy study. J Am Geriatr Soc. 2014;62(9):1722–8.
Tyas SL, Manfreda J, Strain LA, Montgomery PR. Risk factors for Alzheimer’s disease: a population-based, longitudinal study in Manitoba, Canada. Int J Epidemiol. 2001;30(3):590–7.
Verreault R, Laurin D, Lindsay J, Serres GD. Past exposure to vaccines and subsequent risk of Alzheimer’s disease. CMAJ Can Med Assoc J. 2001;165(11):1495–8.
Jamieson GA, Maitland NJ, Wilcock GK, Craske J, Itzhaki RF. Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J Med Virol. 1991;33(4):224–7.
Jamieson GA, Maitland NJ, Wilcock GK, Yates CM, Itzhaki RF. Herpes simplex virus type 1 DNA is present in specific regions of brain from aged people with and without senile dementia of the Alzheimer type. J Pathol. 1992;167(4):365–8.
Wozniak MA, Shipley SJ, Combrinck M, Wilcock GK, Itzhaki RF. Productive herpes simplex virus in brain of elderly normal subjects and Alzheimer’s disease patients. J Med Virol. 2005;75(2):300–6.
Honjo K, van Reekum R, Verhoeff NPLG. Alzheimer’s disease and infection: do infectious agents contribute to progression of Alzheimer’s disease? Alzheimers Dement J Alzheimers Assoc. 2009;5(4):348–60.
Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet Lond Engl. 1997;349(9047):241–4.
Gérard HC, Wildt KL, Whittum-Hudson JA, Lai Z, Ager J, Hudson AP. The load of Chlamydia pneumoniae in the Alzheimer’s brain varies with APOE genotype. Microb Pathog. 2005;39(1–2):19–26.
Gérard HC, Dreses-Werringloer U, Wildt KS, Deka S, Oszust C, Balin BJ, et al. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol Med Microbiol. 2006;48(3):355–66.
Alonso R, Pisa D, Marina AI, Morato E, Rábano A, Carrasco L. Fungal infection in patients with Alzheimer’s disease. J Alzheimers Dis. 2014;41(1):301–11.
Alonso R, Pisa D, Rábano A, Rodal I, Carrasco L. Cerebrospinal fluid from Alzheimer’s disease patients contains fungal proteins and DNA. J Alzheimers Dis. 2015;47(4):873–6.
Pisa D, Alonso R, Rábano A, Rodal I, Carrasco L. Different brain regions are infected with fungi in Alzheimer’s disease. Sci Rep. 2015;15(5):15015.
Pisa D, Alonso R, Juarranz A, Rábano A, Carrasco L. Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. J Alzheimers Dis. 2015;43(2):613–24.
Jakobsson HE, Rodríguez-Piñeiro AM, Schütte A, Ermund A, Boysen P, Bemark M, et al. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015;16(2):164–77.
Faden AI, Loane DJ. Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurother J Am Soc Exp Neurother. 2015;12(1):143–50.
Faden AI, Wu J, Stoica BA, Loane DJ. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br J Pharmacol. 2016;173(4):681–91.
Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol. 2013;9(4):211–21.
Washington PM, Villapol S, Burns MP. Polypathology and dementia after brain trauma: does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp Neurol. 2016;275(Pt 3):381–8.
Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 2003;74(7):857–62.
Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case–control studies. EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20(Suppl 2):S28–35.
Johnson VE, Stewart W, Smith DH. Widespread τ and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol Zurich Switz. 2012;22(2):142–9.
Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, et al. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58(9):982–92.
Bennett RE, Esparza TJ, Lewis HA, Kim E, Mac Donald CL, Sullivan PM, et al. Human apolipoprotein E4 worsens acute axonal pathology but not amyloid-β immunoreactivity after traumatic brain injury in 3xTG-AD mice. J Neuropathol Exp Neurol. 2013;72(5):396–403.
Chen X-H, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 2004;165(2):357–71.
Iwata A, Chen X-H, McIntosh TK, Browne KD, Smith DH. Long-term accumulation of amyloid-beta in axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J Neuropathol Exp Neurol. 2002;61(12):1056–68.
Shishido H, Kishimoto Y, Kawai N, Toyota Y, Ueno M, Kubota T, et al. Traumatic brain injury accelerates amyloid-β deposition and impairs spatial learning in the triple-transgenic mouse model of Alzheimer’s disease. Neurosci Lett. 2016;26(629):62–7.
Kane MJ, Angoa-Pérez M, Briggs DI, Viano DC, Kreipke CW, Kuhn DM. A mouse model of human repetitive mild traumatic brain injury. J Neurosci Methods. 2012;203(1):41–9.
Luo J, Nguyen A, Villeda S, Zhang H, Ding Z, Lindsey D, et al. Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Front Neurol (Internet). 2014;5:12. https://doi.org/10.3389/fneur.2014.00012.
McAteer KM, Corrigan F, Thornton E, Turner RJ, Vink R. Short and long term behavioral and pathological changes in a novel rodent model of repetitive mild traumatic brain injury. PLOS One. 2016;11(8):e0160220.
Petraglia AL, Plog BA, Dayawansa S, Dashnaw ML, Czerniecka K, Walker CT, et al. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg Neurol Int. 2014;5:184.
Gerson J, Castillo-Carranza DL, Sengupta U, Bodani R, Prough DS, DeWitt DS, et al. Tau oligomers derived from traumatic brain injury cause cognitive impairment and accelerate onset of pathology in htau mice. J Neurotrauma. 2016;33(22):2034–43.
Hillier V, Salib E. A case–control study of smoking and Alzheimer’s disease. Int J Geriatr Psychiatry. 1997;12(3):295–300.
Brenner DE, Kukull WA, van Belle G, Bowen JD, McCormick WC, Teri L, et al. Relationship between cigarette smoking and Alzheimer’s disease in a population-based case–control study. Neurology. 1993;43(2):293–300.
Reitz C, den Heijer T, van Duijn C, Hofman A, Breteler MMB. Relation between smoking and risk of dementia and Alzheimer disease: The Rotterdam Study. Neurology. 2007;69(10):998–1005.
Aggarwal NT, Bienias JL, Bennett DA, Wilson RS, Morris MC, Schneider JA, et al. The relation of cigarette smoking to incident Alzheimer’s disease in a biracial urban community population. Neuroepidemiology. 2006;26(3):140–6.
Cataldo JK, Prochaska JJ, Glantz SA. Cigarette smoking is a risk factor for Alzheimer’s disease: an analysis controlling for tobacco industry affiliation. J Alzheimers Dis. 2010;19(2):465–80.
Tyas SL, White LR, Petrovitch H, Webster Ross G, Foley DJ, Heimovitz HK, et al. Mid-life smoking and late-life dementia: the Honolulu-Asia Aging Study. Neurobiol Aging. 2003;24(4):589–96.
Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819–28.
Giunta B, Deng J, Jin J, Sadic E, Rum S, Zhou H, et al. Evaluation of how cigarette smoke is a direct risk factor for Alzheimer’s disease. Technol Innov. 2012;14(1):39–48.
Moreno-Gonzalez I, Estrada LD, Sanchez-Mejias E, Soto C. Smoking exacerbates amyloid pathology in a mouse model of Alzheimer’s disease. Nat Commun. 2013;4:1495.
Gons RAR, van Norden AGW, de Laat KF, van Oudheusden LJB, van Uden IWM, Zwiers MP, et al. Cigarette smoking is associated with reduced microstructural integrity of cerebral white matter. Brain J Neurol. 2011;134(Pt 7):2116–24.
Kenche H, Baty CJ, Vedagiri K, Shapiro SD, Blumental-Perry A. Cigarette smoking affects oxidative protein folding in endoplasmic reticulum by modifying protein disulfide isomerase. FASEB J Off Publ Fed Am Soc Exp Biol. 2013;27(3):965–77.
Shen C, Chen Y, Liu H, Zhang K, Zhang T, Lin A, et al. Hydrogen peroxide promotes Aβ production through JNK-dependent activation of γ-Secretase. J Biol Chem. 2008;283(25):17721–30.
Ho Y-S, Yang X, Yeung S-C, Chiu K, Lau C-F, Tsang AW-T, et al. Cigarette smoking accelerated brain aging and induced pre-Alzheimer-like neuropathology in rats. PLOS One. 2012;7(5):e36752.
Seet RCS, Lee C-YJ, Loke WM, Huang SH, Huang H, Looi WF, et al. Biomarkers of oxidative damage in cigarette smokers: which biomarkers might reflect acute versus chronic oxidative stress? Free Radic Biol Med. 2011;50(12):1787–93.
Baldeiras I, Santana I, Proença MT, Garrucho MH, Pascoal R, Rodrigues A, et al. Oxidative damage and progression to Alzheimer’s disease in patients with mild cognitive impairment. J Alzheimers Dis. 2010;21(4):1165–77.
Hellström-Lindahl E, Mousavi M, Ravid R, Nordberg A. Reduced levels of Abeta 40 and Abeta 42 in brains of smoking controls and Alzheimer’s patients. Neurobiol Dis. 2004;15(2):351–60.
Gao J, Adam B-L, Terry AV. Evaluation of nicotine and cotinine analogs as potential neuroprotective agents for Alzheimer’s disease. Bioorg Med Chem Lett. 2014;24(6):1472–8.
Rosa AO, Egea J, Gandía L, López MG, García AG. Neuroprotection by nicotine in hippocampal slices subjected to oxygen-glucose deprivation: involvement of the alpha7 nAChR subtype. J Mol Neurosci MN. 2006;30(1–2):61–2.
Nordberg A, Hellström-Lindahl E, Lee M, Johnson M, Mousavi M, Hall R, et al. Chronic nicotine treatment reduces beta-amyloidosis in the brain of a mouse model of Alzheimer’s disease (APPsw). J Neurochem. 2002;81(3):655–8.
Kihara T, Shimohama S, Sawada H, Kimura J, Kume T, Kochiyama H, et al. Nicotinic receptor stimulation protects neurons against beta-amyloid toxicity. Ann Neurol. 1997;42(2):159–63.
Ono K, Hasegawa K, Yamada M, Naiki H. Nicotine breaks down preformed Alzheimer’s beta-amyloid fibrils in vitro. Biol Psychiatry. 2002;52(9):880–6.
Friedland RP, Fritsch T, Smyth KA, Koss E, Lerner AJ, Chen CH, et al. Patients with Alzheimer’s disease have reduced activities in midlife compared with healthy control-group members. Proc Natl Acad Sci USA. 2001;98(6):3440–5.
Booth FW, Laye MJ, Lees SJ, Rector RS, Thyfault JP. Reduced physical activity and risk of chronic disease: the biology behind the consequences. Eur J Appl Physiol. 2008;102(4):381–90.
Baker LD, Frank LL, Foster-Schubert K, Green PS, Wilkinson CW, McTiernan A, et al. Effects of aerobic exercise on mild cognitive impairment: a controlled trial. Arch Neurol. 2010;67(1):71–9.
Nascimento CMC, Pereira JR, de Andrade LP, Garuffi M, Talib LL, Forlenza OV, et al. Physical exercise in MCI elderly promotes reduction of pro-inflammatory cytokines and improvements on cognition and BDNF peripheral levels. Curr Alzheimer Res. 2014;11(8):799–805.
Radak Z, Hart N, Sarga L, Koltai E, Atalay M, Ohno H, et al. Exercise plays a preventive role against Alzheimer’s disease. J Alzheimers Dis. 2010;20(3):777–83.
Jensen CS, Hasselbalch SG, Waldemar G, Simonsen AH. Biochemical markers of physical exercise on mild cognitive impairment and dementia: systematic review and perspectives. Front Neurol. 2015;6:187.
Cheng S-T, Chow PK, Song Y-Q, Yu ECS, Chan ACM, Lee TMC, et al. Mental and physical activities delay cognitive decline in older persons with dementia. Am J Geriatr Psychiatry Off J Am Assoc Geriatr Psychiatry. 2014;22(1):63–74.
Sofi F, Valecchi D, Bacci D, Abbate R, Gensini GF, Casini A, et al. Physical activity and risk of cognitive decline: a meta-analysis of prospective studies. J Intern Med. 2011;269(1):107–17.
Sampaio A, Marques EA, Mota J, Carvalho J. Effects of a multicomponent exercise program in institutionalized elders with Alzheimer’s disease. Dement Lond Engl. 2016. https://doi.org/10.1177/1471301216674558.
Küster OC, Fissler P, Laptinskaya D, Thurm F, Scharpf A, Woll A, et al. Cognitive change is more positively associated with an active lifestyle than with training interventions in older adults at risk of dementia: a controlled interventional clinical trial. BMC Psychiatry. 2016;16(1):315.
Smith JC, Nielson KA, Woodard JL, Seidenberg M, Rao SM. Physical activity and brain function in older adults at increased risk for Alzheimer’s disease. Brain Sci. 2013;3(1):54–83.
Rovio S, Kåreholt I, Helkala E-L, Viitanen M, Winblad B, Tuomilehto J, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4(11):705–11.
Schuit AJ, Feskens EJ, Launer LJ, Kromhout D. Physical activity and cognitive decline, the role of the apolipoprotein e4 allele. Med Sci Sports Exerc. 2001;33(5):772–7.
Etnier JL, Caselli RJ, Reiman EM, Alexander GE, Sibley BA, Tessier D, et al. Cognitive performance in older women relative to ApoE-epsilon4 genotype and aerobic fitness. Med Sci Sports Exerc. 2007;39(1):199–207.
Cheng S-T, Chow PK, Song Y-Q, Yu ECS, Chan ACM, Lee TMC, et al. Mental and physical activities delay cognitive decline in older persons with dementia. Am J Geriatr Psychiatry Off J Am Assoc Geriatr Psychiatry. 2014;22(1):63–74.
Podewils LJ, Guallar E, Kuller LH, Fried LP, Lopez OL, Carlson M, et al. Physical activity, APOE genotype, and dementia risk: findings from the Cardiovascular Health Cognition Study. Am J Epidemiol. 2005;161(7):639–51.
Eggermont LHP, Swaab DF, Hol EM, Scherder EJA. Walking the line: a randomised trial on the effects of a short term walking programme on cognition in dementia. J Neurol Neurosurg Psychiatry. 2009;80(7):802–4.
Kim B-K, Shin M-S, Kim C-J, Baek S-B, Ko Y-C, Kim Y-P. Treadmill exercise improves short-term memory by enhancing neurogenesis in amyloid beta-induced Alzheimer disease rats. J Exerc Rehabil. 2014;10(1):2–8.
Leem Y-H, Lim H-J, Shim S-B, Cho J-Y, Kim B-S, Han P-L. Repression of tau hyperphosphorylation by chronic endurance exercise in aged transgenic mouse model of tauopathies. J Neurosci Res. 2009;87(11):2561–70.
Marosi K, Bori Z, Hart N, Sárga L, Koltai E, Radák Z, et al. Long-term exercise treatment reduces oxidative stress in the hippocampus of aging rats. Neuroscience. 2012;13(226):21–8.
Um HS, Kang EB, Leem YH, Cho IH, Yang CH, Chae KR, et al. Exercise training acts as a therapeutic strategy for reduction of the pathogenic phenotypes for Alzheimer’s disease in an NSE/APPsw-transgenic model. Int J Mol Med. 2008;22(4):529–39.
Commenges D, Scotet V, Renaud S, Jacqmin-Gadda H, Barberger-Gateau P, Dartigues JF. Intake of flavonoids and risk of dementia. Eur J Epidemiol. 2000;16(4):357–63.
Morris MC. Nutritional determinants of cognitive aging and dementia. Proc Nutr Soc. 2012;71(1):1–13.
Wang HX, Wahlin A, Basun H, Fastbom J, Winblad B, Fratiglioni L. Vitamin B(12) and folate in relation to the development of Alzheimer’s disease. Neurology. 2001;56(9):1188–94.
Crystal HA, Ortof E, Frishman WH, Gruber A, Hershman D, Aronson M. Serum vitamin B12 levels and incidence of dementia in a healthy elderly population: a report from the Bronx Longitudinal Aging Study. J Am Geriatr Soc. 1994;42(9):933–6.
Engelhart MJ, Geerlings MI, Ruitenberg A, Van Swieten JC, Hofman A, Witteman JCM, et al. Diet and risk of dementia: does fat matter? Neurology. 2002;59(12):1915–21.
Luchsinger JA, Tang M-X, Shea S, Mayeux R, et al. Antioxidant vitamin intake and risk of Alzheimer disease. Arch Neurol. 2003;60(2):203.
Willett WC, Sacks F, Trichopoulou A, Drescher G, Ferro-Luzzi A, Helsing E, et al. Mediterranean diet pyramid: a cultural model for healthy eating. Am J Clin Nutr. 1995;61(6 Suppl):1402S–6S.
Tangney CC, Li H, Wang Y, Barnes L, Schneider JA, Bennett DA, et al. Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology. 2014;83(16):1410–6.
Morris MC, Tangney CC, Wang Y, Sacks FM, Barnes LL, Bennett DA, et al. MIND diet slows cognitive decline with aging. Alzheimers Dement J Alzheimers Assoc. 2015;11(9):1015–22.
Morris MC, Tangney CC, Wang Y, Sacks FM, Bennett DA, Aggarwal NT. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc. 2015;11(9):1007–14.
Morris MC, Tangney CC, Wang Y, Sacks FM, Barnes LL, Bennett DA, et al. MIND diet slows cognitive decline with aging. Alzheimers Dement J Alzheimers Assoc. 2015;11(9):1015–22.
Scarmeas N, Stern Y, Mayeux R, Luchsinger JA. Mediterranean diet, Alzheimer disease, and vascular mediation. Arch Neurol. 2006;63(12):1709–17.
Scarmeas N, Stern Y, Tang M-X, Mayeux R, Luchsinger JA. Mediterranean diet and risk for Alzheimer’s disease. Ann Neurol. 2006;59(6):912–21.
Scarmeas N, Luchsinger JA, Schupf N, Brickman AM, Cosentino S, Tang MX, et al. Physical activity, diet, and risk of Alzheimer disease. JAMA. 2009;302(6):627–37.
Tangney CC, Kwasny MJ, Li H, Wilson RS, Evans DA, Morris MC. Adherence to a Mediterranean-type dietary pattern and cognitive decline in a community population. Am J Clin Nutr. 2011;93(3):601–7.
Tsivgoulis G, Judd S, Letter AJ, Alexandrov AV, Howard G, Nahab F, et al. Adherence to a Mediterranean diet and risk of incident cognitive impairment. Neurology. 2013;80(18):1684–92.
Schmidt FM, Weschenfelder J, Sander C, Minkwitz J, Thormann J, Chittka T, et al. Inflammatory cytokines in general and central obesity and modulating effects of physical activity. PloS One. 2015;10(3):e0121971.
Cai D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol Metab. 2013;24(1):40–7.
Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB. Obesity, diabetes and cognitive deficit: The Framingham Heart Study. Neurobiol Aging. 2005;26(Suppl 1):11–6.
Engelhart MJ, Geerlings MI, Meijer J, Kiliaan A, Ruitenberg A, van Swieten JC, et al. Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol. 2004;61(5):668–72.
Tan ZS, Beiser AS, Vasan RS, Roubenoff R, Dinarello CA, Harris TB, et al. Inflammatory markers and the risk of Alzheimer disease The Framingham Study. Neurology. 2007;68(22):1902–8.
Waldstein SR, Katzel LI. Interactive relations of central versus total obesity and blood pressure to cognitive function. Int J Obes 2005. 2006;30(1):201–7.
Whitmer RA, Gustafson DR, Barrett-Connor E, Haan MN, Gunderson EP, Yaffe K. Central obesity and increased risk of dementia more than three decades later. Neurology. 2008;71(14):1057–64.
Johansson L, Guo X, Duberstein PR, Hällström T, Waern M, Ostling S, et al. Midlife personality and risk of Alzheimer disease and distress: a 38-year follow-up. Neurology. 2014;83(17):1538–44.
Wilson RS, Evans DA, Bienias JL, Mendes de Leon CF, Schneider JA, Bennett DA. Proneness to psychological distress is associated with risk of Alzheimer’s disease. Neurology. 2003;61(11):1479–85.
Wilson RS, Schneider JA, Boyle PA, Arnold SE, Tang Y, Bennett DA. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68(24):2085–92.
Arsenault-Lapierre G, Chertkow H, Lupien S. Seasonal effects on cortisol secretion in normal aging, mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging. 2010;31(6):1051–4.
Comijs HC, Gerritsen L, Penninx BWJH, Bremmer MA, Deeg DJH, Geerlings MI. The association between serum cortisol and cognitive decline in older persons. Am J Geriatr Psychiatry Off J Am Assoc Geriatr Psychiatry. 2010;18(1):42–50.
Csernansky JG, Dong H, Fagan AM, Wang L, Xiong C, Holtzman DM, et al. Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am J Psychiatry. 2006;163(12):2164–9.
Baglietto-Vargas D, Chen Y, Suh D, Ager RR, Rodriguez-Ortiz CJ, Medeiros R, et al. Short-term modern life-like stress exacerbates Aβ-pathology and synapse loss in 3xTg-AD mice. J Neurochem. 2015;134(5):915–26.
Dong H, Yuede CM, Yoo H-S, Martin MV, Deal C, Mace AG, et al. Corticosterone and related receptor expression are associated with increased beta-amyloid plaques in isolated Tg2576 mice. Neuroscience. 2008;155(1):154–63.
Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci Off J Soc Neurosci. 2006;26(35):9047–56.
Huang H, Wang L, Cao M, Marshall C, Gao J, Xiao N, et al. Isolation housing exacerbates Alzheimer’s disease-like pathophysiology in aged APP/PS1 mice. Int J Neuropsychopharmacol (Internet). 2015;18(7):pyu116. https://doi.org/10.1093/ijnp/pyu116.
Jeong YH, Park CH, Yoo J, Shin KY, Ahn S-M, Kim H-S, et al. Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV717I-CT100 transgenic mice, an Alzheimer’s disease model. FASEB J Off Publ Fed Am Soc Exp Biol. 2006;20(6):729–31.
Rothman SM, Herdener N, Camandola S, Texel SJ, Mughal MR, Cong W-N, et al. 3xTgAD mice exhibit altered behavior and elevated Aβ after chronic mild social stress. Neurobiol Aging. 2012;33(4):830.e1–12.
Sierra A, Gottfried-Blackmore A, Milner TA, McEwen BS, Bulloch K. Steroid hormone receptor expression and function in microglia. Glia. 2008;56(6):659–74.
Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends Endocrinol Metab. 2013;24(3):109–19.
Perez Nievas BG, Hammerschmidt T, Kummer MP, Terwel D, Leza JC, Heneka MT. Restraint stress increases neuroinflammation independently of amyloid β levels in amyloid precursor protein/PS1 transgenic mice. J Neurochem. 2011;116(1):43–52.
Walker FR, Nilsson M, Jones K. Acute and chronic stress-induced disturbances of microglial plasticity, phenotype and function. Curr Drug Targets. 2013;14(11):1262–76.
Verdile G, Fuller SJ, Martins RN. The role of type 2 diabetes in neurodegeneration. Neurobiol Dis. 2015;84:22–38.
Kandimalla R, Thirumala V, Reddy PH. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim Biophys Acta. 2017;1863(5):1078–89.
Biessels GJ, Strachan MWJ, Visseren FLJ, Kappelle LJ, Whitmer RA. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. Lancet Diabetes Endocrinol. 2014;2(3):246–55.
Brundel M, Kappelle LJ, Biessels GJ. Brain imaging in type 2 diabetes. Eur Neuropsychopharmacol J Eur Coll Neuropsychopharmacol. 2014;24(12):1967–81.
Moran C, Phan TG, Chen J, Blizzard L, Beare R, Venn A, et al. Brain atrophy in type 2 diabetes: regional distribution and influence on cognition. Diabetes Care. 2013;36(12):4036–42.
Ramos-Rodriguez JJ, Molina-Gil S, Ortiz-Barajas O, Jimenez-Palomares M, Perdomo G, Cozar-Castellano I, et al. Central Proliferation and Neurogenesis Is Impaired in Type 2 Diabetes and Prediabetes Animal Models. PLOS One. 2014;9(2):e89229.
Kroner Z. The relationship between Alzheimer’s disease and diabetes: type 3 diabetes? Altern Med Rev J Clin Ther. 2009;14(4):373–9.
Lizcano JM, Alessi DR. The insulin signalling pathway. Curr Biol CB. 2002;12(7):R236–8.
Haque R, Nazir A. Insulin-degrading enzyme: a link between Alzheimer’s and type 2 diabetes mellitus. CNS Neurol Disord Drug Targets. 2014;13(2):259–64.
Qiu WQ, Folstein MF. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: review and hypothesis. Neurobiol Aging. 2006;27(2):190–8.
Wu C, Xu G, Tsai SA, Freed WJ, Lee CT. Transcriptional profiles of type 2 diabetes in human skeletal muscle reveal insulin resistance, metabolic defects, apoptosis, and molecular signatures of immune activation in response to infections. Biochem Biophys Res Commun. 2017;482(2):282–8.
Fishel MA, Watson GS, Montine TJ, Wang Q, Green PS, Kulstad JJ, et al. Hyperinsulinemia provokes synchronous increases in central inflammation and beta-amyloid in normal adults. Arch Neurol. 2005;62(10):1539–44.
Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Investig. 2006;116(7):1793–801.
Meinert CL, Breitner JCS. Chronic disease long-term drug prevention trials: lessons from the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT). Alzheimers Dement J Alzheimers Assoc. 2008;4(1 Suppl 1):S7–14.
Andersen K, Launer LJ, Ott A, Hoes AW, Breteler MM, Hofman A. Do nonsteroidal anti-inflammatory drugs decrease the risk for Alzheimer’s disease? The Rotterdam Study. Neurology. 1995;45(8):1441–5.
Breitner JC, Gau BA, Welsh KA, Plassman BL, McDonald WM, Helms MJ, et al. Inverse association of anti-inflammatory treatments and Alzheimer’s disease: initial results of a co-twin control study. Neurology. 1994;44(2):227–32.
Gasparini L, Ongini E, Wenk G. Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease: old and new mechanisms of action. J Neurochem. 2004;91(3):521–36.
Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology. 1997;48(3):626–32.
Shadfar S, Hwang CJ, Lim M-S, Choi D-Y, Hong JT. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch Pharm Res. 2015;38(12):2106–19.
Carreras I, McKee AC, Choi J-K, Aytan N, Kowall NW, Jenkins BG, et al. R-flurbiprofen improves tau, but not Aβ pathology in a triple transgenic model of Alzheimer’s disease. Brain Res. 2013;6(1541):115–27.
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, et al. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain J Neurol. 2005;128(Pt 6):1442–53.
Kukar T, Prescott S, Eriksen JL, Holloway V, Murphy MP, Koo EH, et al. Chronic administration of R-flurbiprofen attenuates learning impairments in transgenic amyloid precursor protein mice. BMC Neurosci. 2007;24(8):54.
Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, et al. Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging. 2001;22(6):983–91.
McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S, et al. Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 2008;1(1207):225–36.
Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414(6860):212–6.
Jaturapatporn D, Isaac MGEKN, McCleery J, Tabet N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst Rev. 2012;2:CD006378.
Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, et al. Clinical trial of indomethacin in Alzheimer’s disease. Neurology. 1993;43(8):1609–11.
Breitner JC, Baker LD, Montine TJ, Meinert CL, Lyketsos CG, Ashe KH, et al. Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement J Alzheimers Assoc. 2011;7(4):402–11.
Hu WT, Holtzman DM, Fagan AM, Shaw LM, Perrin R, Arnold SE, et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology. 2012;79(9):897–905.
Heneka MT, Landreth GE, Feinstein DL. Role for peroxisome proliferator-activated receptor-gamma in Alzheimer’s disease. Ann Neurol. 2001;49(2):276.
Landreth GE, Heneka MT. Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer’s disease. Neurobiol Aging. 2001;22(6):937–44.
Mandrekar-Colucci S, Landreth GE. Nuclear receptors as therapeutic targets for Alzheimer’s disease. Expert Opin Ther Targets. 2011;15(9):1085–97.
Alaynick WA. Nuclear receptors, mitochondria and lipid metabolism. Mitochondrion. 2008;8(4):329–37.
Denner LA, Rodriguez-Rivera J, Haidacher SJ, Jahrling JB, Carmical JR, Hernandez CM, et al. Cognitive enhancement with rosiglitazone links the hippocampal PPARγ and ERK MAPK signaling pathways. J Neurosci Off J Soc Neurosci. 2012;32(47):16725–35a.
Inestrosa NC, Toledo EM. The role of Wnt signaling in neuronal dysfunction in Alzheimer’s disease. Mol Neurodegener. 2008;24(3):9.
Jahrling JB, Hernandez CM, Denner L, Dineley KT. PPARγ recruitment to active ERK during memory consolidation is required for Alzheimer’s disease-related cognitive enhancement. J Neurosci Off J Soc Neurosci. 2014;34(11):4054–63.
Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging. 2011;32(9):1626–33.
Heneka MT, Fink A, Doblhammer G. Effect of pioglitazone medication on the incidence of dementia. Ann Neurol. 2015;78(2):284–94.
Geldmacher DS, Fritsch T, McClendon MJ, Landreth G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch Neurol. 2011;68(1):45–50.
Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry Off J Am Assoc Geriatr Psychiatry. 2005;13(11):950–8.
Risner ME, Saunders AM, Altman JFB, Ormandy GC, Craft S, Foley IM, et al. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenom J. 2006;6(4):246–54.
Tobinick EL, Gross H. Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration. J Neuroinflamm. 2008;9(5):2.
Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. MedGenMed Medscape Gen Med. 2006;8(2):25.
Aisen PS, Davis KL, Berg JD, Schafer K, Campbell K, Thomas RG, et al. A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology. 2000;54(3):588–93.
Feldman HH, Doody RS, Kivipelto M, Sparks DL, Waters DD, Jones RW, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010;74(12):956–64.
Simons M, Schwärzler F, Lütjohann D, von Bergmann K, Beyreuther K, Dichgans J, et al. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: a 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol. 2002;52(3):346–50.
Sparks DL, Sabbagh MN, Connor DJ, Lopez J, Launer LJ, Browne P, et al. Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Arch Neurol. 2005;62(5):753–7.
Van Gool WA, Weinstein HC, Scheltens P, Walstra GJ, Scheltens PK. Effect of hydroxychloroquine on progression of dementia in early Alzheimer’s disease: an 18-month randomised, double-blind, placebo-controlled study. Lancet Lond Engl. 2001;358(9280):455–60.
Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–55.
Dinarello CA, Simon A, van der Meer JWM. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11(8):633–52.
Porrini V, Lanzillotta A, Branca C, Benarese M, Parrella E, Lorenzini L, et al. CHF5074 (CSP-1103) induces microglia alternative activation in plaque-free Tg2576 mice and primary glial cultures exposed to beta-amyloid. Neuroscience. 2015;27(302):112–20.
Alam JJ. Selective brain-targeted antagonism of p38 MAPKα reduces hippocampal IL-1β levels and improves morris water maze performance in aged rats. J Alzheimers Dis. 2015;48(1):219–27.
Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain J Neurol. 2016;139(Pt 4):1237–51.
Mathieu M-C, Sawyer N, Greig GM, Hamel M, Kargman S, Ducharme Y, et al. The C3a receptor antagonist SB 290157 has agonist activity. Immunol Lett. 2005;100(2):139–45.
Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat Med. 2016;22(2):135–7.
Teng MWL, Bowman EP, McElwee JJ, Smyth MJ, Casanova J-L, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. 2015;21(7):719–29.
Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78(4):631–43.
AD2000 Collaborative Group, Bentham P, Gray R, Sellwood E, Hills R, Crome P, et al. Aspirin in Alzheimer’s disease (AD2000): a randomised open-label trial. Lancet Neurol. 2008;7(1):41–9.
de Jong D, Jansen R, Hoefnagels W, Jellesma-Eggenkamp M, Verbeek M, Borm G, et al. No effect of one-year treatment with indomethacin on Alzheimer’s disease progression: a randomized controlled trial. PloS One. 2008;3(1):e1475.
Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289(21):2819–26.
Aisen PS, Schmeidler J, Pasinetti GM. Randomized pilot study of nimesulide treatment in Alzheimer’s disease. Neurology. 2002;58(7):1050–4.
Thal LJ, Ferris SH, Kirby L, Block GA, Lines CR, Yuen E, et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2005;30(6):1204–15.
Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, et al. Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62(1):66–71.
ADAPT Research Group, Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JCS, et al. Cognitive function over time in the Alzheimer’s disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008;65(7):896–905.
Alzheimer’s Disease Anti-inflammatory Prevention Trial Research Group. Results of a follow-up study to the randomized Alzheimer’s disease Anti-Inflammatory Prevention Trial (ADAPT). Alzheimers Dement J Alzheimers Assoc. 2013;9(6):714–23.
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405.
Gold M, Alderton C, Zvartau-Hind M, Egginton S, Saunders AM, Irizarry M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord. 2010;30(2):131–46.
Harrington C, Sawchak S, Chiang C, Davies J, Donovan C, Saunders AM, et al. Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimer’s disease: two phase 3 studies. Curr Alzheimer Res. 2011;8(5):592–606.
Butchart J, Brook L, Hopkins V, Teeling J, Püntener U, Culliford D, et al. Etanercept in Alzheimer disease: a randomized, placebo-controlled, double-blind, phase 2 trial. Neurology. 2015;84(21):2161–8.
Ross J, Sharma S, Winston J, Nunez M, Bottini G, Franceschi M, et al. CHF5074 reduces biomarkers of neuroinflammation in patients with mild cognitive impairment: a 12-week, double-blind, placebo-controlled study. Curr Alzheimer Res. 2013;10(7):742–53.
Relkin NR, Thomas RG, Rissman RA, Brewer JB, Rafii MS, van Dyck CH, et al. A phase 3 trial of IV immunoglobulin for Alzheimer disease. Neurology. 2017;88(18):1768–75.
Gómez-Isla T, Blesa R, Boada M, Clarimón J, Del Ser T, Domenech G, et al. A randomized, double-blind, placebo controlled-trial of triflusal in mild cognitive impairment: the TRIMCI study. Alzheimer Dis Assoc Disord. 2008;22(1):21–9.
Freund-Levi Y, Hjorth E, Lindberg C, Cederholm T, Faxen-Irving G, Vedin I, et al. Effects of omega-3 fatty acids on inflammatory markers in cerebrospinal fluid and plasma in Alzheimer’s disease: the OmegAD study. Dement Geriatr Cogn Disord. 2009;27(5):481–90.
Sakurai R, Koo B-K, Kaneda H, Bonneau HN, Nagai R. Cilostazol added to aspirin and clopidogrel reduces revascularization without increases in major adverse events in patients with drug-eluting stents: a meta-analysis of randomized controlled trials. Int J Cardiol. 2013;167(5):2250–8.
Sanchez-Ramos J, Cimino C, Avila R, Rowe A, Chen R, Whelan G, et al. Pilot study of granulocyte-colony stimulating factor for treatment of Alzheimer’s disease. J Alzheimers Dis. 2012;31(4):843–55.
Turner RS, Thomas RG, Craft S, van Dyck CH, Mintzer J, Reynolds BA, et al. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology. 2015;85(16):1383–91.
Acknowledgements
The authors thank Ildiko Racz and Cathy Widmann for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No outside funding was used for this article. The open access fee was covered by University of Bonn.
Conflicts of interest
MTH has a patent pending on nitration of amyloid β peptides (WO 2011006871 A1). GEL has a patent pending on an RXR agonist in Alzheimer’s disease (WO 2011006157 A2). PAL is an employee of Pfizer, Germany. AA-F, EWGMB, AB-S, BS, KC, CD, TD, GG, LH, AH, LI, SJ, SC-G, KK, NL, RMM, AP, KR, JMS-C, AT, AVdP, AV, CV, AW, PW, TSW, XX, and YY have no conflicts of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ardura-Fabregat, A., Boddeke, E.W.G.M., Boza-Serrano, A. et al. Targeting Neuroinflammation to Treat Alzheimer’s Disease. CNS Drugs 31, 1057–1082 (2017). https://doi.org/10.1007/s40263-017-0483-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-017-0483-3