
Abstract
Background
Psychostimulants are considered first-line pharmacotherapy for youth with attention-deficit/hyperactivity disorder (ADHD), but questions remain regarding the comparative efficacy of amphetamine- and methylphenidate-based agents.
Objective
Our objective was to describe two acute randomized, double-blind, placebo-controlled, head-to-head studies of lisdexamfetamine dimesylate (LDX) and osmotic-release oral system methylphenidate (OROS-MPH) in adolescents with ADHD.
Methods
Adolescents (13–17 years) diagnosed with ADHD according to Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) criteria were enrolled in an 8-week flexible-dose study [LDX 30–70 mg/day (n = 186 randomized); OROS-MPH 18–72 mg/day (n = 185 randomized); placebo (n = 93 randomized)] or a 6-week forced-dose study [LDX 70 mg/day (n = 219 randomized); OROS-MPH 72 mg/day (n = 220 randomized); placebo (n = 110 randomized)]. Attention-Deficit/Hyperactivity Disorder Rating Scale IV (ADHD-RS-IV) total score changes from baseline (primary endpoint) at week 8 (flexible-dose study) or week 6 (forced-dose study) were assessed with mixed-effects models for repeated measures. Secondary endpoints included improvement on the dichotomized Clinical Global Impressions–Improvement scale (CGI-I; key secondary endpoint) and changes from baseline on the ADHD-RS-IV subscales. Safety assessments included treatment-emergent adverse events (TEAEs) and vital signs.
Results
Least squares (LS) mean ± standard error of the mean (SEM) ADHD-RS-IV total score changes from baseline to end of treatment were −17.0 ± 1.03 with placebo, −25.4 ± 0.74 with LDX, and −22.1 ± 0.73 with OROS-MPH in the forced-dose study and −13.4 ± 1.19 with placebo, −25.6 ± 0.82 with LDX, and −23.5 ± 0.80 with OROS-MPH in the flexible-dose study. LS mean ± SEM treatment difference for the change from baseline significantly favored LDX over OROS-MPH in the forced-dose [−3.4 ± 1.04, p = 0.0013, effect size (ES) −0.33] but not the flexible-dose (−2.1 ± 1.15, p = 0.0717, ES −0.20) study. The percentage of improved participants on the dichotomized CGI-I at end of treatment was significantly greater with LDX than with OROS-MPH in the forced-dose study (81.4 vs. 71.3%, p = 0.0188) but not the flexible-dose study (LDX 83.1%, OROS-MPH 81.0%, p = 0.6165). The LS mean ± SEM treatment differences for change from baseline on the ADHD-RS-IV hyperactivity/impulsivity and inattentiveness subscales nominally favored LDX in the forced-dose study (hyperactivity/impulsivity subscale −1.3 ± 0.49, nominal p = 0.0081, ES −0.27; inattentiveness subscale −2.0 ± 0.63, nominal p = 0.0013, ES −0.33), but there were no significant differences between active treatments in the flexible-dose study. In both studies, LDX and OROS-MPH were superior to placebo for all efficacy-related endpoints (all nominal p < 0.0001; ES range −0.43 to −1.16). The overall frequency of TEAEs for LDX and OROS-MPH, respectively, were 66.5 and 58.9% in the forced-dose study and 83.2 and 82.1% in the flexible-dose study. TEAEs occurring in ≥ 5% of participants that were also reported at two or more times the rate of placebo were decreased appetite, decreased weight, insomnia, initial insomnia, dry mouth, and nasopharyngitis (LDX and OROS-MPH), irritability and dizziness (LDX only), and increased heart rate (OROS-MPH only) in the forced-dose study and decreased appetite, decreased weight, insomnia, and dizziness (LDX and OROS-MPH) and dry mouth and upper abdominal pain (LDX only) in the flexible-dose study. Mean ± standard deviation (SD) increases from baseline in vital signs (systolic and diastolic blood pressure, pulse) were observed in the forced-dose study [LDX 1.6 ± 9.65 and 3.3 ± 8.11 mmHg, 6.7 ± 12.78 beats per minute (bpm); OROS-MPH 2.6 ± 10.15 and 3.3 ± 9.13 mmHg, 7.6 ± 12.47 bpm] and the flexible-dose study (LDX 2.4 ± 9.46 and 2.8 ± 8.41 mmHg, 4.7 ± 11.82 bpm; OROS-MPH 0.4 ± 9.90 and 2.2 ± 8.64 mmHg, 6.0 ± 10.52 bpm) at the last on-treatment assessment.
Conclusions
LDX was superior to OROS-MPH in adolescents with ADHD in the forced-dose but not the flexible-dose study. Safety and tolerability for both medications was consistent with previous studies. These findings underscore the robust acute efficacy of both psychostimulant classes in treating adolescents with ADHD.
ClinicalTrials.gov registry numbers
NCT01552915 and NCT01552902.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Lisdexamfetamine dimesylate (LDX) was superior to osmotic-release oral system methylphenidate (OROS-MPH) for improving attention-deficit/hyperactivity disorder (ADHD) symptoms in a forced-dose study but not in a flexible-dose study. |
Both LDX and OROS-MPH are highly efficacious in treating adolescents with ADHD and are generally well tolerated, demonstrating that either stimulant class can be used with confidence. This may be important in cases of inadequate response or poor tolerability to one of the stimulant classes (as has previously been reported), although the sequencing of treatment was not assessed in these studies. |
The overall safety and tolerability profiles of LDX and OROS-MPH were consistent with previous reports. |
LDX may have a somewhat higher effect size than OROS-MPH at US FDA-approved doses, though with perhaps slightly numerically higher rates of adverse events. |
1 Introduction
Although psychostimulants are considered first-line pharmacotherapy for youth with attention-deficit/hyperactivity disorder (ADHD) [1, 2], questions regarding the comparative efficacy of the newer, long-acting amphetamine-based versus methylphenidate-based agents [including lisdexamfetamine and osmotic-release oral system methylphenidate (OROS-MPH)] have not been fully resolved. Generally, amphetamine and methylphenidate are considered to have similar efficacy when dosed comparably, although individual differences in response and/or tolerability may be observed [3, 4]. Consistent with the above, US practice guidelines recommend that either amphetamine or methylphenidate be used as first-line therapy in children and adolescents and suggest switching to the other stimulant class or a nonstimulant if response or tolerability are suboptimal. The American Academy of Child and Adolescent Psychiatry (AACAP) and the UK National Institute for Health and Care Excellence (NICE) guidelines also note that nonstimulants (e.g., atomoxetine, immediate-release and extended-release guanfacine) may be used as first-line pharmacotherapy, but AACAP guidelines indicate that stimulants have a superior effect size (ES) [1, 5].
Multiple analyses have examined the comparative efficacy of amphetamine and methylphenidate [6,7,8,9,10]. A meta-analysis [6] that included studies of ≥ 2 weeks in duration found that amphetamine and methylphenidate were superior to placebo in children and adolescents with ADHD, with amphetamine having somewhat larger ESs. Another meta-analysis that included studies with durations of 2–12 weeks and examined the comparative efficacy of atomoxetine, bupropion, lisdexamfetamine (but not all amphetamine products), and methylphenidate (multiple formulations) in children and adolescents indicated that lisdexamfetamine and methylphenidate-based agents were superior to placebo and suggested that the ES was larger for lisdexamfetamine than for methylphenidate [7]. Although a combined systematic review and meta-analysis of studies ranging from 2 to 12 weeks in duration reported that a higher proportion of participants responded to lisdexamfetamine than methylphenidate-based agents [10], a network meta-analysis that included 190 trials ranging from 3 to 96 weeks in duration did not find evidence of statistically significant differences between amphetamine-based and methylphenidate-based stimulants [9].
There have been several head-to-head comparisons of amphetamine-based and methylphenidate-based agents, but the individual study populations were small (range 18–72 participants) and not all formulations were examined in any given study [3, 4, 11]. Results indicated that amphetamine and methylphenidate were superior to placebo in children and adolescents with ADHD; in some studies [11], but not all [3], amphetamine was modestly superior to methylphenidate. A post hoc analysis from a 7-week, double-blind, flexible-dose study in children and adolescents with ADHD reported that lisdexamfetamine was statistically superior to OROS-MPH on the ADHD Rating Scale IV (ADHD-RS-IV) total score and the Clinical Global Impressions–Improvement (CGI-I) scale [12]. However, following European regulatory guidelines, the maximum approved OROS-MPH dose used in that study was 54 mg/day. In the USA, the maximum approved OROS-MPH dose in adolescents is 72 mg/day, and whether a higher OROS-MPH dose might have yielded different results is unknown.
While the findings described above provide insight into the comparative efficacy of the two stimulant classes, they are limited by several factors. Meta-analyses are indirect by nature (i.e., the medications are each compared with placebo but not each other) and have not included all available formulations. Individual studies that can be used to assess potential differences between stimulant classes have used small sample sizes [3, 4, 11], medication doses that are below the maximum approved dose [12], and post hoc analyses to make inferences regarding comparative efficacy [12]. Consequently, well-powered head-to-head comparator trials of amphetamine and methylphenidate are a high priority.
This report describes two phase IV head-to-head studies (one flexible-dose study and one forced-dose study) of lisdexamfetamine and OROS-MPH in adolescents with ADHD using US FDA-approved dosing recommendations. The objectives were to evaluate the efficacy of lisdexamfetamine versus OROS-MPH, measured using the ADHD-RS-IV and CGI-I, and to evaluate the safety and tolerability of both medications. Following FDA guidelines, the maximum doses used were lisdexamfetamine 70 mg/day and OROS-MPH 72 mg/day. Based on previous publications [7, 10], it was hypothesized that the efficacy of lisdexamfetamine would be statistically superior to OROS-MPH. The current studies were not powered to assess differences in safety and tolerability between lisdexamfetamine and OROS-MPH; as such, there was no prespecified hypothesis regarding safety and tolerability.
2 Methods
2.1 Study Design and Treatment Regimens
Two randomized, double-blind, multicenter, parallel-group studies (one flexible-dose, one forced-dose) with placebo reference arms were conducted. Each study was conducted in accordance with the International Conference on Harmonisation of Good Clinical Practice and the Declaration of Helsinki. Study protocols and related information were approved by either a central review board or institution-specific review boards and appropriate regulatory agencies (US FDA, Therapeutic Product Directorate of Canada, Medical Products Agency of Sweden, Medical Research Council of Hungary, The Federal Institute for Drugs and Medical Devices [Bundesinstitut für Arzneimittel und Medizinprodukte] of Germany) before study initiation.
2.1.1 Flexible-Dose Study
The flexible-dose study [ClinicalTrials.gov: NCT01552915; Fig. S1A in the Electronic Supplementary Material (ESM)] was conducted at 70 US clinical sites between April 2012 and January 2014; it consisted of a screening and washout phase (up to 4 weeks), 8 weeks of double-blind (i.e., participants and investigators) treatment (dose optimization, 5 weeks; dose maintenance; 3 weeks), and a 1-week follow-up phase.
At the start of double-blind treatment, participants were randomized (2:2:1) using an interactive web-response system (IWRS) to once-daily lisdexamfetamine, OROS-MPH, or placebo, respectively. To maintain blinding, treatments were identical in appearance; participants were also instructed to take one capsule from two separate bottles. Participants randomized to lisdexamfetamine started treatment at 30 mg/day; those randomized to OROS-MPH started treatment at 18 mg/day. Dosages could be raised or lowered by single dose levels at each weekly visit until week 5, when the optimal dose based on tolerability and clinical response (an ADHD-RS-IV total score reduction of ≥30% and CGI-I score of 1 or 2, with tolerable side effects) was established. After week 5, the dose could not be changed. This maintenance dose was continued for 3 weeks (weeks 6–8). Participants unable to tolerate active treatment after week 5 were discontinued. A follow-up telephone call was made 7 (+2) days after the last dose to assess adverse events (AEs) or serious AEs (SAEs), and concomitant medications. Follow-up continued until safety concerns were resolved or the participant returned to their baseline state.
2.1.2 Forced-Dose Study
The forced-dose study (ClinicalTrials.gov: NCT01552902; Fig S1B, in the ESM) was conducted at 77 clinical sites in the USA, Canada, and Europe between April 2012 and May 2014; it consisted of a screening and washout phase (up to 4 weeks), a 6-week double-blind treatment phase (forced titration, 4 weeks; dose maintenance, 2 weeks), and a 1-week follow-up period.
At the start of double-blind treatment, participants were randomized (2:2:1) using an IWRS to once-daily treatment with lisdexamfetamine 70 mg/day, OROS-MPH 72 mg/day, or placebo, respectively. To maintain blinding, treatments were over-encapsulated and identical in appearance; participants were also instructed to take one capsule from two separate bottles. The initial lisdexamfetamine dose (30 mg/day) was increased weekly in 10-mg increments up to 50 mg/day (week 1, 30 mg/day; week 2, 40 mg/day; week 3, 50 mg/day) and then in a 20-mg increment (week 4) to a maximum of 70 mg/day. The initial OROS-MPH dose (18 mg/day) was increased weekly in 18-mg increments to a maximum of 72 mg/day during weeks 2 through 4. For the remaining 2 weeks of treatment, doses were maintained at lisdexamfetamine 70 mg/day and OROS-MPH 72 mg/day. Participants could discontinue at any time because of unacceptable tolerability, but dose reductions were not allowed. Follow-up safety assessments occurred via telephone 7 (+2) days after the last dose of study drug and continued until safety concerns resolved or returned to their baseline state.
2.2 Study Population
Both studies enrolled adolescent males and nonpregnant, nonlactating females (13–17 years). Eligible participants were required to weigh > 79.5 lb, have a Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) primary ADHD diagnosis, a baseline ADHD-RS-IV total score ≥ 28, and screening and baseline blood pressure values that did not exceed the 90th percentile for age, sex, and height based on US Centers for Disease Control and Prevention (CDC) guidelines. If currently being treated, participants were required to not be completely satisfied with their current ADHD therapy, as judged by the investigator at the baseline assessment; this criterion reflects the ethical requirement to not discontinue treatment in individuals exhibiting a satisfactory response to their current therapy. The participant and the participant’s parent/legally authorized representative were required to provide informed assent and consent and to be willing and able to comply with study requirements.
Participants were excluded if they had a current controlled or uncontrolled comorbid Axis I or II psychiatric diagnosis (except oppositional defiant disorder, which was allowed), with significant symptoms requiring treatment or contraindicating lisdexamfetamine or OROS-MPH treatment. Comorbid psychiatric diagnoses were established using the Kiddie Schedule for Affective Disorders and Schizophrenia for School-Age Children–Present and Lifetime Version–Diagnostic Interview [13]. Participants were excluded if they were considered a suicide risk by the investigator, had previously made a suicide attempt, or were currently demonstrating active suicidal ideation. Additional exclusion criteria were underweight (less than the third percentile) or overweight (> 97th percentile) based on CDC body mass index (BMI) standards for age and sex; a medical or psychiatric condition that could confound safety assessments or increase participant risk; personal or family cardiovascular history or a clinically significant electrocardiogram (ECG) finding; history of suspected substance abuse or dependence (excluding nicotine) or a lifetime history of amphetamine, cocaine, or stimulant abuse and/or dependence; concomitant use of medications with central nervous system effects that could potentially affect study performance; documented allergy or hypersensitivity to amphetamine or methylphenidate; prior failed response to methylphenidate or amphetamine therapy (because it is not ethical to include individuals who do not have a reasonable expectation of deriving benefit from treatment); participation in another clinical study within 30 days before screening.
2.3 Efficacy Endpoints
Efficacy was assessed using the ADHD-RS-IV [14] and CGI-I [15]. The ADHD-RS-IV was administered to the parent (while the participant was present) at baseline and at all study visits by a clinician experienced in evaluating adolescent ADHD. The primary efficacy measure was the change in ADHD-RS-IV total score from baseline to end of study (EOS; week 8 in the flexible-dose study, week 6 in the forced-dose study).
ADHD severity was assessed using the CGI−Severity (CGI-S) scale, which rates symptoms from 1 (not ill) to 7 (extremely ill) [15]. The CGI-S was administered at baseline, week 5, and week 8/early termination (ET) in the flexible-dose study and at baseline and week 6/ET in the forced-dose study. Improvement in ADHD severity was assessed using the CGI-I, which rates change from baseline on a 7-point scale ranging from 1 (very much improved) to 7 (very much worse). The CGI-I (key secondary endpoint) was administered at all post-baseline visits.
2.4 Safety and Tolerability Endpoints
Adverse events were assessed at each visit and categorized by severity, relatedness to treatment, and seriousness. Vital sign assessments included systolic blood pressure (SBP), diastolic blood pressure (DBP), pulse, and weight. Blood pressure and pulse were assessed after the participant had been seated for ≥ 5 min. ECGs were performed after 5 min of rest. The Columbia-Suicide Severity Rating Scale (C-SSRS), a semistructured interview that assesses suicide-related thoughts and behaviors [16], was administered at screening and all post-screening visits.
2.5 Statistical Analyses
Statistical analysis of the primary and key secondary efficacy endpoints was conducted in the full analysis set (FAS: all participants taking at least one study drug dose and having at least one post-baseline primary efficacy assessment). Sample size estimates were based on the primary efficacy endpoint. To detect an ES of ≥ 0.35 between lisdexamfetamine and OROS-MPH (90% power at a two-sided significance level of 0.05 using a two-sample t test), a total of 456 participants (placebo 92; lisdexamfetamine 182; OROS-MPH 182) for the flexible-dose study and 542 participants (placebo 108; lisdexamfetamine 217; OROS-MPH 217) for the forced-dose study were planned for randomization after accounting for estimated discontinuation rates of 5 and 20%, respectively.
Change in ADHD-RS-IV total score from baseline to EOS was assessed using a linear mixed-effects model for repeated measures, without imputation for missing data. Treatment, visit, and the treatment × visit interaction were included as factors; baseline score was a covariate; the interaction of baseline score with visit was included in the model. The primary contrast was the comparison of ADHD-RS-IV total score in the lisdexamfetamine versus OROS-MPH groups at EOS [null hypothesis (H o): there is no difference between the effects of lisdexamfetamine and OROS-MPH on ADHD-RS-IV total score change from baseline]. For CGI-I analyses, scores were dichotomized as improved [very much improved (CGI-I = 1) or much improved (CGI-I = 2)] or not improved (minimally improved through very much worse; CGI-I = 3–7). The percentage of participants considered improved on the dichotomized CGI-I was assessed using a Cochran–Mantel–Haenszel test stratified by baseline CGI-S; missing data were imputed using last observation carried forward.
A fixed-sequence test procedure was applied to the primary and key secondary endpoints for the lisdexamfetamine versus OROS-MPH comparisons. If the H o for the primary efficacy endpoint was not rejected at the 0.05 significance level, the p-value from the key secondary efficacy analysis result could not be considered statistically significant. Because of this testing hierarchy, only nominal unadjusted p values are reported for assessments of active treatment versus placebo (active treatment–placebo); these values are intended for descriptive purposes only.
Safety and tolerability endpoints were assessed in the safety analysis set (all participants taking at least one study drug dose) and summarized descriptively. Conservative threshold criteria for vital sign outlier analyses, determined based on prior lisdexamfetamine studies and recommendations of professional organizations, were used to capture trends of potential clinical importance. CDC growth charts were used to determine z scores for weight and BMI changes.
3 Results
3.1 Disposition and Demographics
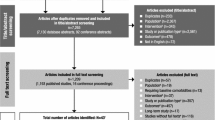
Participant disposition is summarized in Fig. 1. The safety analysis set and FAS, respectively, included 459 (placebo 91; lisdexamfetamine 184; OROS-MPH 184) and 452 (placebo 89; lisdexamfetamine 179; OROS-MPH 184) participants in the flexible-dose study and 547 (placebo 110; lisdexamfetamine 218; OROS-MPH 219) and 532 (placebo 106; lisdexamfetamine 210; OROS-MPH 216) participants in the forced-dose study. The most frequent reasons for discontinuation were lack of efficacy for placebo (both studies), AEs for lisdexamfetamine (both studies), “other” reasons for OROS-MPH (flexible-dose study), and AEs for OROS-MPH (forced-dose study).

Participant disposition. Flexible flexible-dose study, forced forced-dose study, LDX lisdexamfetamine dimesylate, OROS-MPH osmotic controlled-release methylphenidate
Baseline demographics were generally comparable across treatment groups in each study (Table 1). However, the percentage of participants with predominantly inattentive ADHD was numerically higher in the flexible-dose study than in the forced-dose study for placebo and lisdexamfetamine compared with OROS-MPH. Most participants had a history of prior ADHD medication use (Table S1 in the ESM).
3.2 Drug Exposure and Treatment Compliance
In the flexible-dose study, the mean ± standard deviation (SD) daily dose during double-blind treatment was lisdexamfetamine 50.15 ± 12.501 mg/day and OROS-MPH 44.47 ± 12.754 mg/day. The percentage of participants in the safety analysis set taking specific doses at the final treatment week was as follows: lisdexamfetamine 30 mg/day: 13/184 (7.1%); lisdexamfetamine 50 mg/day: 47/184 (25.5%); lisdexamfetamine 70 mg/day: 94/184 (51.1%); OROS-MPH 18 mg/day: 10/184 (5.4%); OROS-MPH 36 mg/day: 31/184 (16.8%); OROS-MPH 54 mg/day: 38/184 (20.7%); and OROS-MPH 72 mg/day: 76/184 (41.3%).
3.3 Efficacy
3.3.1 ADHD-RS-IV Total Score
ADHD-RS-IV total scores across treatments decreased during each study (Table 2; Fig. 2). Least squares (LS) mean ± standard error of the mean (SEM) treatment differences for ADHD-RS-IV total score change from baseline at EOS statistically favored lisdexamfetamine in the forced-dose study (p = 0.0013; ES −0.33) but not in the flexible-dose study (p = 0.0717; ES −0.20). The LS mean ± SEM treatment differences for change from baseline ADHD-RS-IV total scores at EOS for lisdexamfetamine and OROS-MPH were nominally greater than placebo in the flexible-dose study (lisdexamfetamine: ES −1.16; OROS-MPH: ES −0.97; both nominal p < 0.0001) and in the forced-dose study (lisdexamfetamine: ES −0.82; OROS-MPH: ES −0.50; both nominal p < 0.0001).

ADHD-RS-IV total score by treatment week, full analysis set, in the flexible-dose study (A) and the forced-dose study (B). ADHD-RS-IV Attention-Deficit/Hyperactivity Disorder Rating Scale IV, LDX lisdexamfetamine dimesylate, OROS-MPH osmotic-release oral system methylphenidate, SD standard deviation
3.3.2 Dichotomized CGI-I Scores
Improvement on the dichotomized CGI-I at EOS was significantly greater with lisdexamfetamine than with OROS-MPH in the forced-dose study (p = 0.0188) but not in the flexible-dose study (p = 0.6165; Table 2). In both studies, improvement on the dichotomized CGI-I at EOS with lisdexamfetamine and OROS-MPH was nominally greater than with placebo (all nominal p < 0.001).
3.3.3 ADHD-RS-IV Subscale Scores
In both studies, ADHD-RS-IV hyperactivity/impulsivity and inattentive subscale scores were decreased relative to baseline for all treatments at EOS. LS mean ± SEM treatment differences for change from baseline on the ADHD-RS-IV subscales nominally favored lisdexamfetamine in the forced-dose study (hyperactivity/impulsivity: p = 0.0081, ES −0.27; inattentiveness: p = 0.0013, ES −0.33); there were no significant differences between active treatments in the flexible-dose study (Table 2). In both studies, LS mean ± SEM treatment differences for change from baseline at EOS favored lisdexamfetamine and OROS-MPH over placebo on both subscales (all nominal p < 0.001).
3.4 Safety and Tolerability
3.4.1 Adverse Events
In both studies, treatment-emergent AEs (TEAEs) occurred more frequently with lisdexamfetamine and OROS-MPH than with placebo (Table 3). Most TEAEs were rated as mild to moderate in intensity. Severe TEAEs in the flexible-dose and forced-dose studies, respectively, were reported in 2.2 and 0.9% of participants receiving placebo, 5.4 and 1.4% of those receiving lisdexamfetamine, and 3.8 and 2.7% of those receiving OROS-MPH. Serious TEAEs were reported in one participant receiving placebo (forced-dose study: psychotic disorder), two participants receiving lisdexamfetamine [flexible-dose study: suicidal ideation (n = 1); forced-dose study: suicidal ideation (n = 1)], and two participants receiving OROS-MPH [flexible-dose study: renal cyst (n = 1); forced-dose study: appendicitis (n = 1)]. TEAEs leading to study discontinuation in two or more participants with any treatment were decreased appetite (n = 3), suicidal ideation (n = 2), and irritability (n = 2) with lisdexamfetamine in the flexible-dose study; suicidal ideation, dizziness, and palpitations (all n = 2) with lisdexamfetamine in the forced-dose study; and insomnia, irritability, headache, nausea, vomiting, and decreased appetite (all n = 2) with OROS-MPH in the forced-dose study. TEAEs occurring in ≥ 5% of participants that were also reported at two or more times the rate of placebo were decreased appetite, decreased weight, insomnia, initial insomnia, dry mouth, and nasopharyngitis (lisdexamfetamine and OROS-MPH), irritability and dizziness (lisdexamfetamine only), and increased heart rate (OROS-MPH only) in the forced-dose study and decreased appetite, decreased weight, insomnia, and dizziness (lisdexamfetamine and OROS-MPH), and dry mouth and upper abdominal pain (lisdexamfetamine only) in the flexible-dose study (Table 3). No deaths occurred in either study.
3.4.2 Vital Signs
At the last on-treatment assessment, mean SBP and DBP increased relative to baseline with lisdexamfetamine and OROS-MPH and decreased with placebo in both studies (Table 4). Mean increases in pulse from baseline were greater with lisdexamfetamine and OROS-MPH than with placebo in both studies. Vital sign outlier analyses are summarized in Table 4.
At the last on-treatment assessment, mean decreases in weight and BMI were observed with lisdexamfetamine and OROS-MPH, and mean increases were observed with placebo (Table 4). Median z scores were positive for all treatments, indicating that weight and BMI in > 50% of participants exceeded the mean for the CDC age- and sex-matched population. Bodyweight outlier analyses are summarized in Table 4.
3.4.3 Columbia-Suicide Severity Rating Scale
On the C-SSRS, no episodes of suicidal behavior were reported during double-blind treatment or follow-up in either study. Three participants reported nonsuicidal self-injurious behavior (flexible-dose study: OROS-MPH, n = 1 each at weeks 1 and 4; forced-dose study: lisdexamfetamine, n = 1 at week 5), and three participants reported active suicidal ideation with any method (flexible-dose study: lisdexamfetamine, n = 1 at week 2; forced-dose study: placebo, n = 1 at week 5; lisdexamfetamine, n = 1 at week 3 that resulted in discontinuation) during double-blind treatment. The relationship of these events to treatment was not assessed.
4 Discussion
This report describes two large, randomized, double-blind, placebo-controlled studies that directly compared the efficacy, safety, and tolerability of lisdexamfetamine and OROS-MPH in adolescents with ADHD. To our knowledge, these studies constitute the largest head-to-head comparison trials of amphetamine and methylphenidate conducted to date. As measured by ADHD-RS-IV total score and dichotomized CGI-I, lisdexamfetamine was statistically superior to OROS-MPH in the forced-dose study but not the flexible-dose study, though there was a numerically larger ES for lisdexamfetamine in that study as well. As previously reported in individual trials [17,18,19], reviews [4], and meta-analyses [6,7,8], lisdexamfetamine and OROS-MPH both exhibited superior efficacy versus placebo, supporting the assay sensitivity of the studies and the use of amphetamine or methylphenidate classes for the treatment of ADHD.
The primary efficacy findings reported here partially support previous indirect comparisons of lisdexamfetamine and OROS-MPH [7, 10] in particular, and of the amphetamine and methylphenidate stimulant classes in general [11], which have suggested that amphetamine-based stimulants (e.g., lisdexamfetamine) may have superior efficacy to methylphenidate-based stimulants (e.g., OROS-MPH) at the dosages used in these studies [7, 10, 11]. However, the ES for the differences between lisdexamfetamine and OROS-MPH (−0.33 and −0.20 for ADHD-RS-IV total score change from baseline in the forced-dose and flexible-dose studies, respectively) was lower than the estimated 0.35 for which the studies were powered. This ES, which would represent a large difference between two active medications, fell short of significance in the flexible-dose study. This finding is perhaps not surprising because forced-dose titration is generally considered better able to detect differences between active medications than flexible-dose titration studies, which rely on subjective clinical judgment and do not necessarily offer optimal doses to all participants.
These results also partially (but not fully) confirm findings from a post hoc analysis comparing lisdexamfetamine and OROS-MPH in a 7-week, double-blind, flexible-dose study in children and adolescents with ADHD, which found that lisdexamfetamine was statistically superior to OROS-MPH in reducing ADHD-RS-IV total score (ES 0.54) and in producing global improvement on the CGI-I [12]. However, as indicated earlier, the latter study was conducted in Europe and used a maximum OROS-MPH dose of 54 mg/day; it also enrolled children and adolescents rather than adolescents only. The different findings seen between the prior European study and the current studies could be partially accounted for by the lower OROS-MPH dose used in the European study, the focus on adolescents in the current studies, and/or other issues related to study design and implementation.
The safety and tolerability profiles of lisdexamfetamine and OROS-MPH were consistent with findings from published placebo-controlled studies in children and/or adolescents with ADHD [17,18,19]. In the flexible-dose study, the overall frequency of TEAEs was comparable between active treatments, and the frequencies of severe TEAEs and TEAEs leading to discontinuation were numerically higher with lisdexamfetamine than with OROS-MPH (note that the AE data in the two studies are based solely on descriptive statistics and there was no significance testing). In the forced-dose study, frequencies of any TEAEs and TEAEs leading to discontinuation were numerically higher with lisdexamfetamine, but the frequency of severe TEAEs was numerically lower with lisdexamfetamine. Heart rate increases were numerically larger with OROS-MPH than with lisdexamfetamine and weight decreases were numerically larger with lisdexamfetamine than with OROS-MPH in both studies. Blood pressure changes for lisdexamfetamine versus OROS-MPH were less consistent across studies; increases from baseline in SBP and DBP were numerically greater with lisdexamfetamine in the flexible-dose study, and increases in SBP were numerically greater with OROS-MPH in the forced-dose study.
What is the clinical take-home message of these studies? The primary findings reported here (small but inconsistent ESs favoring lisdexamfetamine over OROS-MPH and slightly higher rates of TEAEs and TEAEs leading to discontinuation with lisdexamfetamine) are consistent with the conclusion that lisdexamfetamine may be somewhat more efficacious than OROS-MPH at clinically approved doses, though with perhaps slightly higher AE rates (which may either reflect characteristics of the compound or the relatively higher potency of approved doses). Taking a broader view, and considering the robust improvement over placebo and high degree of tolerability seen with both medications, one can conclude that both lisdexamfetamine and OROS-MPH are highly efficacious in treating adolescents with ADHD and are generally well tolerated. For clinicians in countries where amphetamine use has historically been less common, or where amphetamine formulations such as lisdexamfetamine are now first being approved, this is important because it demonstrates that either stimulant class can be used with confidence (as is the current US practice). It is also important for patients who do not exhibit a sufficient response to or who do not tolerate treatment with a given stimulant class (either amphetamine or methylphenidate), because switching such patients to the alternative stimulant class may produce a more robust and/or better tolerated clinical response. Because these studies were not designed as crossover studies, information on individual response to amphetamine versus methylphenidate cannot be inferred. However, taken together with findings from other studies reporting that a fairly large proportion of patients have a preferential response to amphetamine or methylphenidate [3], with current US treatment guidelines from AACAP, and with the NICE guidelines, these results suggest that trying different stimulant classes to optimize response and tolerability is reasonable and important.
These findings should be considered in light of several limitations that could impact their generalizability. (1) It is not possible to draw inferences regarding the long-term effectiveness of lisdexamfetamine versus OROS-MPH on ADHD symptomatology or functional outcomes, or on differences in long-term safety and tolerability of either medication based on these 6-week and 8-week studies. (2) Both studies excluded individuals with comorbid Axis I and II psychiatric conditions (except oppositional defiant disorder). Because comorbid psychiatric conditions are often observed in adolescents with ADHD, the generalizability of these findings to the broader population is unclear. (3) These studies only examined the comparative efficacy of lisdexamfetamine and OROS-MPH, so the findings cannot be extrapolated to other amphetamine or methylphenidate formulations. (4) These studies only included individuals who were not satisfied with their current ADHD therapy and who had not previously failed an adequate course of treatment with amphetamine-based or methylphenidate-based agents. Therefore, the findings may overestimate the magnitude of response and/or tolerability of either or both treatments. (5) Lisdexamfetamine was not available in some countries when the forced-dose study was conducted. As such, it is theoretically possible that selection bias could have resulted in the preferential enrollment of a population that did not respond optimally to methylphenidate. (6) The recommended upper dose limit of OROS-MPH is 90 mg/day in adolescents in the Canadian ADHD practice guidelines [20], and it is theoretically possible that the use of higher OROS-MPH doses in these studies may have resulted in a larger treatment response (though it is also possible that this might have also resulted in higher rates of AEs). However, this approach would involve dosing above the FDA-recommended maximum in the USA (72 mg/day) and the maximum European approved dose (54 mg/day). The doses of the two medications used in these trials were based on FDA guidelines, and we can only comment here on the doses studied. (7) Comparisons of the safety and tolerability findings between lisdexamfetamine and OROS-MPH should be considered with caution because the studies were not powered for statistical comparison of these endpoints, and the reported data are descriptive.
5 Conclusions
Lisdexamfetamine was statistically superior to OROS-MPH in adolescents with ADHD in a forced-dose design study (lisdexamfetamine 70 mg/day; OROS-MPH 72 mg/day), as measured by ADHD-RS-IV total score and dichotomized CGI-I score, but not in a flexible-dose design study. Confirming assay sensitivity, both lisdexamfetamine and OROS-MPH were statistically superior to placebo in reducing ADHD symptoms and improving overall ADHD severity in both studies. The safety and tolerability of lisdexamfetamine and OROS-MPH were generally consistent with previous reports. Findings from these acute treatment studies underscore the robust efficacy of lisdexamfetamine and OROS-MPH in particular, and of the amphetamine and methylphenidate stimulant classes in general, in adolescents with ADHD, and support professional guidelines that advise that both classes can be used as first-line treatments in this population.
References
Atkinson M, Hollis C. NICE guideline: attention deficit hyperactivity disorder. Arch Dis Child Educ Pract Ed. 2010;95(1):24–7.
Subcommittee on Attention-Deficit/Hyperactivity Disorder Steering Committee on Quality Improvement and Management. ADHD: clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Pediatrics. 2011;128(5):1007–22.
Stein MA, Waldman ID, Charney E, et al. Dose effects and comparative effectiveness of extended release dexmethylphenidate and mixed amphetamine salts. J Child Adolesc Psychopharmacol. 2011;21(6):581–8.
Arnold LE. Methylphenidate vs. amphetamine: Comparative review. J Atten Disord. 2000;3(4):200–2011.
Pliszka S. AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894–921.
Faraone SV, Buitelaar J. Comparing the efficacy of stimulants for ADHD in children and adolescents using meta-analysis. Eur Child Adolesc Psychiatry. 2010;19(4):353–64.
Stuhec M, Munda B, Svab V, et al. Comparative efficacy and acceptability of atomoxetine, lisdexamfetamine, bupropion and methylphenidate in treatment of attention deficit hyperactivity disorder in children and adolescents: a meta-analysis with focus on bupropion. J Affect Disord. 2015;178:149–59.
Joseph A, Ayyagari R, Xie M, et al. Comparative efficacy and safety of attention-deficit/hyperactivity disorder pharmacotherapies, including guanfacine extended release: a mixed treatment comparison. Eur Child Adolesc Psychiatry. 2017;26(8):875–97.
Catala-Lopez F, Hutton B, Nunez-Beltran A, et al. The pharmacological and non-pharmacological treatment of attention deficit hyperactivity disorder in children and adolescents: a systematic review with network meta-analyses of randomised trials. PLoS One. 2017;12(7):e0180355.
Roskell NS, Setyawan J, Zimovetz EA, et al. Systematic evidence synthesis of treatments for ADHD in children and adolescents: indirect treatment comparisons of lisdexamfetamine with methylphenidate and atomoxetine. Curr Med Res Opin. 2014;30(8):1673–85.
Pliszka SR, Browne RG, Olvera RL, et al. A double-blind, placebo-controlled study of Adderall and methylphenidate in the treatment of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2000;39(5):619–26.
Soutullo C, Banaschewski T, Lecendreux M, et al. A post hoc comparison of the effects of lisdexamfetamine dimesylate and osmotic-release oral system methylphenidate on symptoms of attention-deficit hyperactivity disorder in children and adolescents. CNS Drugs. 2013;27(9):743–51.
Kaufman J, Birmaher B, Brent D, et al. Schedule for affective disorders and schizophrenia for school-age children-present and lifetime version (K-SADS-PL): initial reliability and validity data. J Am Acad Child Adolesc Psychiatry. 1997;36(7):980–8.
DuPaul GJ, Power TJ, Anastopoulos AD, et al. ADHD rating scale-IV: checklists, norms, and clinical interpretation. New York: Guilford Press; 1998.
Guy W. Clinical Global Impressions. Rockville, MD: US Department of Health, Education, and Welfare; Public Health Service; Alcohol, Drug Abuse and Mental Health Administration; NIMH Psychopharmacology Research Branch; 1976.
Posner K, Brent D, Lucas C, et al. Columbia-suicide severity rating scale. http://www.cssrs.columbia.edu/scale_versions.html. Accessed 7 June 2016.
Findling RL, Childress AC, Cutler AJ, et al. Efficacy and safety of lisdexamfetamine dimesylate in adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2011;50(4):395–405.
Newcorn JH, Kratochvil CJ, Allen AJ, et al. Atomoxetine and osmotically released methylphenidate for the treatment of attention deficit hyperactivity disorder: acute comparison and differential response. Am J Psychiatry. 2008;165(6):721–30.
Wolraich ML, Greenhill LL, Pelham W, et al. Randomized, controlled trial of oros methylphenidate once a day in children with attention-deficit/hyperactivity disorder. Pediatrics. 2001;108(4):883–92.
Canadian Attention Deficit Hyperactivity Disorder Resource Alliance (CADDRA). Canadian ADHD Practice Guidelines, Third Edition. Toronto, ON, Canada: CADDRA; 2011.
Acknowledgements
Although the sponsor was involved in the study design, collection, management, analysis, interpretation, and fact checking of the data, the decision to submit for publication in CNS Drugs was made by the authors. Jeffrey Newcorn, the lead author and primary investigator, had full access to all study data and takes responsibility for the integrity of the data and the accuracy of the data analysis. The authors would like to acknowledge Lisa Politza, BS (a former employee of Shire), for her insightful comments during the development of this manuscript. Under the direction of the authors, Stefan Kolata, PhD (a former employee of Complete Healthcare Communications, LLC [CHC], an ICON plc Company; West Chester, PA), and Craig Slawecki, PhD (a current employee of CHC), provided writing assistance for this manuscript. Editorial assistance in formatting, proofreading, copyediting, and fact checking was also provided by CHC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The clinical research described in this paper was funded by Shire Development LLC (Lexington, MA, USA). Shire Development LLC also provided funding to CHC for support in writing and editing this manuscript and provided the funding for the open access fee for this paper. The sponsor, Shire Development LLC, was involved in the study design, data collection and analysis, and data interpretation. The sponsor was also involved in the writing of the manuscript and in the decision to submit the article for publication, but the final content and decision to submit the manuscript to CNS Drugs was made by the authors.
Conflicts of interest
Jeffrey Newcorn has been an advisor and/or consultant to Arbor, Akili Interactive, Alcobra, Enzymotec, Ironshore, KemPharm, Lundbeck, Medici, Neos, NLS, Pearson, Shire, Sunovion, and Supernus and has received research support from Enzymotec, Lundbeck, and Shire. Peter Nagy has served on an advisory board for Lilly Hungaria and Medice and has received research support from the Tourette Syndrome Association of the USA, the Hungarian Ministry of Education, the National Development Agency of Hungary, Otsuka, and Shire Pharmaceuticals. Ann Childress has been a consultant for Arbor, Ironshore, Neos, NextWave Pharmaceuticals, Novartis Pharmaceutical Corporation, Rhodes, and Shire Pharmaceuticals; has served as a speaker for Arbor, Bristol-Myers Squibb, Novartis Pharmaceutical Corporation, Pfizer, Shire Pharmaceuticals, and Shionogi; has received research support from Arbor, Bristol-Myers Squibb, Forest Research Institute, Ironshore, Johnson & Johnson Pharmaceutical Research & Development, Lilly USA, LLC, Medgenics, Neos, Neurovance, NextWave Pharmaceuticals, Novartis Pharmaceutical Corporation, Noven, Otsuka, Pfizer, Purdue, Rhodes, Sepracor Inc, Shire Pharmaceuticals, Shionogi, Sunovion, Theravance, and Tris; and has served on advisory boards for Arbor, Ironshore, Neos, Pfizer¸ Rhodes, and Shionogi. Glen Frick is a former employee of Shire Development LLC and holds stock/stock options in Shire. Brian Yan is an employee of Shire Development LLC and holds stock/stock options in Shire. Steven Pliszka has received research support from Ironshore, Shire, and Purdue University; has received consulting fees or an honorarium from Ironshore; and has served as an expert witness for Janssen.
Ethical approval and informed consent
Each study was conducted in accordance with the International Conference on Harmonisation of Good Clinical Practice and the Declaration of Helsinki. Study protocols and related information were approved by either a central review board or institution specific review boards and appropriate regulatory agencies (US FDA, Therapeutic Product Directorate of Canada, Medical Products Agency of Sweden, Medical Research Council of Hungary, The Federal Institute for Drugs and Medical Devices [Bundesinstitut für Arzneimittel und Medizinprodukte] of Germany) before study initiation. The participant’s parent or legally authorized representative must have provided informed consent and been willing and able to comply with all study requirements.
Electronic supplementary material
Below is the link to the electronic supplementary material.
40263_2017_468_MOESM1_ESM.tiff
Fig. S1 Study Design: Flexible Dose (A) and Forced Dose (B). EOS=end of study; LDX=lisdexamfetamine dimesylate; OROS-MPH=osmotic controlled-release methylphenidate; V=visit. (TIFF 2958 kb)
40263_2017_468_MOESM2_ESM.docx
Table S1ADHD Medication History, Safety Analysis Set. ADHD=attention-deficit/hyperactivity disorder; AMP=amphetamine; HCl=hydrochloride; LDX=lisdexamfetamine dimesylate; MPH=methylphenidate; OROS-MPH=osmotic controlled-release methylphenidate. aTerms are derived from the World Health Organization Drug Dictionary. (DOCX 48 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Newcorn, J.H., Nagy, P., Childress, A.C. et al. Randomized, Double-Blind, Placebo-Controlled Acute Comparator Trials of Lisdexamfetamine and Extended-Release Methylphenidate in Adolescents With Attention-Deficit/Hyperactivity Disorder. CNS Drugs 31, 999–1014 (2017). https://doi.org/10.1007/s40263-017-0468-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-017-0468-2