
Abstract
Background and Objective
The use of bedaquiline as a treatment option for drug-resistant tuberculosis meningitis (TBM) is of interest to address the increased prevalence of resistance to first-line antibiotics. To this end, we describe a whole-body physiologically based pharmacokinetic (PBPK) model for bedaquiline to predict central nervous system (CNS) exposure.
Methods
A whole-body PBPK model was developed for bedaquiline and its metabolite, M2. The model included compartments for brain and cerebrospinal fluid (CSF). Model predictions were evaluated by comparison to plasma PK time profiles following different dosing regimens and sparse CSF concentrations data from patients. Simulations were then conducted to compare CNS and lung exposures to plasma exposure at clinically relevant dosing schedules.
Results
The model appropriately described the observed plasma and CSF bedaquiline and M2 concentrations from patients with pulmonary tuberculosis (TB). The model predicted a high impact of tissue binding on target site drug concentrations in CNS. Predicted unbound exposures within brain interstitial exposures were comparable with unbound vascular plasma and unbound lung exposures. However, unbound brain intracellular exposures were predicted to be 7% of unbound vascular plasma and unbound lung intracellular exposures.
Conclusions
The whole-body PBPK model for bedaquiline and M2 predicted unbound concentrations in brain to be significantly lower than the unbound concentrations in the lung at clinically relevant doses. Our findings suggest that bedaquiline may result in relatively inferior efficacy against drug-resistant TBM when compared with efficacy against drug-resistant pulmonary TB.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
To date, there is no standard-of-care treatment regimen that is safe and efficacious for patients with drug-resistant TBM. Predictions of bedaquiline CNS exposures would provide insights into bedaquiline-containing dosing schedules as a treatment option for patients with drug-resistant TBM. |
A whole-body PBPK model including CNS distribution for bedaquiline and M2 was developed to predict drug exposures in CSF, brain interstitial, and brain intracellular compartments. |
Unbound bedaquiline and M2 concentrations at the CNS target site were predicted to be lower than unbound bedaquiline and M2 concentrations in lungs, suggesting that bedaquiline may provide inferior efficacy at the CNS site in patients with drug-resistant TBM as compared with the efficacy in lungs. |
1 Introduction
Tuberculosis meningitis (TBM) develops when Mycobacterium tuberculosis (Mtb) disseminates from the primary pulmonary site of infection to the central nervous system (CNS) including the brain [1]. TBM, the most severe form of Mtb infections, is associated with an approximately 42% mortality rate in hospitalized patients [2]. First-line treatment for patients with drug-susceptible TBM remains the same as that for patients with pulmonary tuberculosis, which includes a combination of rifampin, isoniazid, pyrazinamide, and ethambutol [3]. Second-line treatment for patients with TBM includes streptomycin, moxifloxacin, fluoroquinolones, cycloserine, linezolid, etc. Several first-line and second-line anti-tuberculosis (TB) drugs, including, rifampin, ethambutol, and streptomycin, penetrate poorly through the blood–brain barrier (BBB) [4,5,6]. Second-line drugs, such as moxifloxacin, fluoroquinolones, ethionamide, cycloserine, and linezolid, penetrate moderately through BBB [4]. Drug resistant TBM, i.e., Mtb resistant to rifampin and/or isoniazid, is challenging to diagnose and treat. Limited reporting on drug-resistant TBM is attributed to the rarity of the disease and the challenges associated with evaluation of resistance in cerebrospinal fluid (CSF). However, the prevalence of resistance aligns generally with Mtb resistance rates [7]. Currently, there are no standard treatment recommendations for drug-resistant TBM, and treatment approaches are generally selected by treating physicians based on individual patient factors often including extensive treatment with more than five antimicrobial agents. Concerns regarding safety issues of extensive treatments, as well as the high mortality rate (69–100%), among patients with drug-resistant TBM remain a clinical challenge [8].
Bedaquiline is one of the newer TB antibiotics and was the first novel anti-Mtb drug approved in over 40 years [9]. Bedaquiline has activity against Mtb strains resistant against several first-line and second-line TB therapeutics. Bedaquiline has now been evaluated in over 25 clinical trials as part of various combination regimens against Mtb infections [10]. A combination regimen containing bedaquiline, pretomanid, and linezolid (BPaL) is now recommended for the treatment of patients with rifampin-resistant and multidrug-resistant TB [11].
Given the efficacy of BPaL and other bedaquiline-containing combination regimens against drug-resistant pulmonary TB, bedaquiline-containing regimens are being evaluated for the treatment of drug-resistant TBM [12, 13]. Preclinical target site pharmacokinetic (PK) studies reported a brain-to-plasma exposure ratio ranging from 2 to 20% [14,15,16]; however, these studies do not differentiate between total and unbound exposures in the brain. In a preclinical efficacy study, combination therapy with BPaL showed significantly inferior efficacy compared with first-line anti-TB therapy in a mice TBM model following equivalent to human clinically relevant dosing for each drug [16]. On the other hand, comparable efficacy was observed following BPaL compared with first-line anti-TB therapy in a mice pulmonary model following equivalent to human clinically relevant dosing for each drug [17, 18]. In patients with pulmonary TB, bedaquiline and its active metabolite, M2, concentrations in CSF have been reported to be approximately equivalent to unbound plasma concentrations [13]. No clinical data on the efficacy of bedaquiline-containing regimens for the treatment of TBM are available [16, 19]. Overall, these contrary preclinical results and very limited clinical data are neither in favor of nor against the suitability of bedaquiline for the treatment of TBM.
Understanding of mechanisms behind the contradicting preclinical results can be useful in future preclinical and clinical study designs. Factors such as molecular weight, lipophilicity, protein binding, ionization, brain metabolism, and transporters play a role in drug distribution across BBB and blood–CSF–barrier (BCSFB) and the extent of unbound drug available to exert the effect. Physiologically based pharmacokinetic (PBPK) models integrate prior knowledge about physiological processes with the drug’s physicochemical and kinetic parameters to enable PK predictions of a drug within various tissue compartments. Thus, PBPK models are well suited to predict bedaquiline concentrations at the target sites of action, i.e., CSF, brain interstitial, and intracellular in patients with TBM to evaluate its potential for the treatment of patients with MDR TBM. However, a whole-body bedaquiline PBPK model with CNS compartments has not yet been developed. In this work, we aim to (1) develop and evaluate a whole-body PBPK model for bedaquiline and M2 including CSF compartment using plasma and CSF drug and M2 concentrations data from patients with TB and (2) simulate target site concentrations for bedaquiline and M2 for currently recommended bedaquiline dosing schedules in humans.
2 Methods
2.1 Data
Plasma PK data from patients with pulmonary TB were accessed through the Platform for Aggregation of Clinical TB Studies (TB-PACTS; https://c-path.org/programs/tb-pacts/). We first utilized bedaquiline and M2 physicochemical characteristics and plasma PK data from patients with pulmonary TB from a clinical study for the model development. Mean plasma bedaquiline and M2 concentrations by nominal time following bedaquiline doses of 400 mg on day 1, 300 mg on day 2, and 200 mg on day 3 through 14 (referred to as 400–300–200 QD hereinafter) from a phase 2 study were used for model fitting [20]. Next, the model was validated by comparing typical patient plasma PK predictions with observed data for four different dosing groups, including 200–100 mg QD, 500–400–300 mg QD, and 700–500–400 mg QD dosing in patients with TB, from a phase1 study, NCT01215110. Lastly, sparse steady-state individual (n = 7) plasma and CSF PK data, one sample per patient, for bedaquiline and M2 following bedaquiline 400 mg QD followed by 200 mg three times a week at week 24 were obtained from literature and were used for further validation of the CNS distribution component of the model [13].
2.2 Whole-Body PBPK Model Development
The standard PK-Sim whole-body PBPK structural model for small molecules was utilized to build a combined bedaquiline and M2 model [21,22,23]. The standard PK-Sim whole-body PBPK model consists of key tissues and organs, including, the brain, heart, lungs, liver, kidneys, GI tract, etc., connected through vascular and arterial blood circulation. Each compartment is divided into four subcompartments, i.e., vascular, blood cells, interstitial, and intracellular [22]. Physicochemical parameters for bedaquiline and M2 were obtained from literature (Table 1) [24]. Different values have been reported in literature for bedaquiline lipophilicity and fraction unbound; therefore, model evaluation using each of the reported values was conducted to select the lipophilicity and fraction unbound values that provide the best fit to bedaquiline plasma PK data [24, 25]. Bedaquiline oral absorption has previously been described as atypical with delay and double peaks [26,27,28]. The Weibull absorption model built within the PK-Sim software was selected due to its flexibility in describing atypical absorption profiles, and the parameters were estimated by fitting to the plasma PK data. Partition coefficients and cellular permeability parameters of bedaquiline and M2 in various tissues were calculated using the PK-Sim standard method [22, 29]. In PK-Sim, the standard calculation method uses lipophilicity and plasma protein binding parameters along with lipid, protein, and water fractions in each compartment and subcompartment to calculate partition coefficients. CYP3A4 enzyme is involved in the metabolism of bedaquiline to M2 [25]. Therefore, CYP3A4-mediated metabolism conversion from bedaquiline to M2 was modeled using the Michaelis–Menten equation. Experimental data also suggest contributions of CYP2C8 and CYP2C19 enzymes in the metabolism of bedaquiline to M2 [31] and, thus, were evaluated in the model using the Michaelis–Menten equation. Expression profiles for all three enzymes based on the RNA-sequencing (RNA-seq) method were obtained from the Bgee (https://www.bgee.org/) database accessible within PK-Sim [30]. The parameter Michaelis–Menten constant (Km) for the enzymatic reactions was obtained from literature from in vitro experiments [31]. Residual bedaquiline liver plasma clearance was obtained from literature [24]. Next, the model was simultaneously fitted to bedaquiline and M2 PK data following 400–300–200 QD dosing in patients with pulmonary TB to estimate Weibull absorption parameters, enzymatic reaction rates (Vmax), and M2 liver plasma clearance. The combined bedaquiline–M2 plasma PK model was validated by comparing the simulations versus observed plasma PK data for bedaquiline following 200–100 mg QD, 500–400–300 mg QD, and 700–500–400 mg QD dosing regimens (clinical trial: NCT01215110). M2 PK data for this study were not available.
2.3 CNS PBPK Model Development
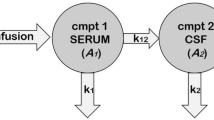
The standard PK-Sim whole-body PBPK structural model included the following brain subcompartments: plasma, blood cells, interstitial, and intracellular. Drug permeability across the blood–brain barrier (BBB) is empirically accounted for as half of the transmembrane permeability calculated from physicochemical properties to account for lipid bilayer [32]. Bedaquiline is not known to be a substrate of transporters located at BBB such as P-gp, BCRP, and MRPs [25]. Therefore, the contribution of such transporters in bedaquiline distribution to the brain was not incorporated. The model was then extended in Mobi to incorporate two CSF compartments, cranial and spinal CSF. Processes of drug distribution to and from CSF were adapted from literature and are illustrated in Fig. 1A [33, 34]. Flow rates to and from CSF were calculated based on the CSF production rate based on physiological knowledge as discussed in literature [33, 35] (Table 2). The same permeability parameter calculated from physicochemical properties was used for diffusion across BCSFB and BBB. The partition coefficient between plasma and CSF was calculated by incorporating fractions of water, lipid, and protein albumin in CSF as shown in Table 2 [29, 36, 37]. To validate the bedaquiline-M2 whole-body PBPK model, including CNS components, plasma and brain interstitial compartment predictions for bedaquiline and M2 were compared against the observed data [13].

Illustration of the bedaquiline-M2 whole-body PBPK model with CNS. The whole-body PBPK model for bedaquiline (bdq) and M2 was developed in PK-Sim. Enzymatic metabolism from bedaquiline to M2 is driven by CYP3A4 and CYP2C8. Additionally, bedaquiline is eliminated at clearance rate CLother–bdq and M2 is eliminated at rate CLM2. There are four routes for unbound drugs to distribute within CSF and brain: (1) from plasma into brain interstitial space, from brain interstitial space into brain intracellular space, (2) from plasma into cranial CSF, then into spinal CSF, (3) from cranial CSF to intracellular space, and (4) from cranial and spinal CSF into venous blood plasma. Drugs in interstitial and intracellular spaces can bind to the compartment lipid and protein content. The drug goes through mass transfer driven by CSF flow within cranial and spinal subcompartments of CSF and also to vascular blood. BBB, blood–brain barrier; BCSFB, blood–cerebrospinal fluid barrier; CLother–bdq, additional bedaquiline hepatic clearance (other than M2-relevant clearance); CLm2, M2 clearance; CSF, cerebrospinal fluid; PxSAB, permeability surface area product for BBB, PxSAC, permeability surface area product for BCSFB, PxSAE, permeability surface area product from brain intracellular to cranial CSF; Qbulk, mass transfer flow rate from intracellular to cranial CSF; Qcsink, CSF flow rate from cranial CSF to peripheral venous blood; Qsin, CSF flow rate from cranial CSF to spinal CSF; Qsout, CSF flow rate from spinal CSF to cranial CSF; Qssink, CSF flow rate from spinal CSF to peripheral venous blood. Figure generated using Biorender.com
Here, AlbCSF:plasma is the albumin CSF to plasma ratio, fup is the fraction unbound in plasma, Fwcsf is the fraction of water in CSF, Fwplasma is the fraction of water in plasma, and PCCSF is the plasma-to-CSF partition coefficient.
2.4 Sensitivity Analysis
A local sensitivity analysis was conducted to understand the impact of parameter uncertainty on bedaquiline and M2 plasma and brain intracellular exposure, AUC0–t. Lipophilicity and CNS distribution-related parameters, such as partition coefficients, permeability, and flow rates, were varied twofold. Fraction unbound for both bedaquiline and M2 was varied 1000-fold to cover the uncertainty range. Sensitivity analyses were run in Mobi for a typical subject following clinically relevant dosing, 400 mg QD for 14 days followed by 200 mg three times a week, and a sensitivity index was calculated.
2.5 Simulations
Simulations were conducted to predict target site, brain intracellular and interstitial, concentrations for bedaquiline, and M2 for clinically relevant bedaquiline dosing schedules in a typical patient with TBM. A typical virtual patient with TB was defined as an individual with 60 kg body weight, as well as 4.32 µmol/L and 2.56 µmol/L as reference concentrations of CYP4A4 and CYP2C8, respectively [20, 30]. Typical virtual patient simulations were conducted for the currently approved dosing regimen and an alternative dosing regimen that was suggested to provide an improved benefit–risk ratio in patients with pulmonary TB [38]. Thus, the following dosing regimens were simulated: (1) current standard bedaquiline dosing which is 400 mg QD followed by 200 mg three times a week, and (2) alternative 200 mg QD for 8 weeks followed by 100 mg QD. Longitudinal total and unbound bedaquiline and M2 concentrations in peripheral blood plasma, lung intracellular, brain intracellular, and spinal CSF were simulated.
2.6 Software
Physiologically based PK modeling and simulation was performed in PK-Sim® and MOBI® (Open Systems Pharmacology Suite (OSPS), v11.0, www.open-systems-pharmacology.org) [39]. Statistical analysis and plots were generated in R (R for Windows, v4.1, https://www.r-project.org/) using RStudio (RStudio, v1-554, www.rstudio.com/).
3 Results
3.1 The Whole-Body PBPK Model Described the Observed PK Data from Plasma and CSF of Patients with TB
The final model contained the whole-body PBPK structure with an extended CNS distribution model for both bedaquiline and M2. Multiple literature-based values have been reported in the literature for bedaquiline lipophilicity and fraction unbound, and the values that provided the best fit to observed plasma PK data were retained in the final model (Table 1) [24, 25]. Enzymatic conversion of bedaquiline to M2 was first set up to be mediated by CYP3A4, CYP2C8, and CYP2C19; however, the Vmax for CYP2C19-mediated reaction was estimated to be very low and, thus, was not retained [31]. The CNS component of the model contained the brain compartment that included plasma, interstitial, cranial CSF, spinal CSF, and intracellular subcompartments. The parameters relevant to CNS distribution were described based on physiological and drug-specific knowledge (Fig. 1 and Table 2) [32,33,34,35].
The final bedaquiline-M2 whole-body PBPK model predictions agreed adequately with the observed plasma PK data for both bedaquiline and M2 following bedaquiline 400–300–200 QD dosing (Fig. 2A) [20], as well as four different bedaquiline dosing regimens (Fig. 2B). Additionally, simulated bedaquiline and M2 PK profiles agreed very well with the observed plasma PK data in patients following current clinically recommended bedaquiline dosing (Fig. 3). At week 24, predicted 24-h plasma average concentrations (Cavg) for bedaquiline and M2 were 718 ng/mL and 268 ng/mL, respectively. These results matched reasonably well with the reported 24-week plasma concentrations for both bedaquiline and M2 in literature following current clinically recommended bedaquiline dosing [median (IQR) concentrations 1264 (910–2244) ng/mL and 252 (34–290) ng/mL for bedaquiline and M2, respectively, based on data from 13 patients) [40]. At week 8, predicted peripheral vascular blood cell-to-plasma concentration ratios were 0.54 and 19.5 for bedaquiline and M2, respectively (Table 3). These results matched reasonably well with the reported peripheral blood mononuclear cells to plasma concentrations ratio in literature at week 8 (1.1 and 22.2 for bedaquiline and M2, respectively) [41].

Predicted and observed plasma PK profiles for A model fitting dataset, and B model validation dataset. The final bedaquiline–M2 whole-body PBPK model predictions agreed well with the observed plasma PK data for both bedaquiline and M2 following bedaquiline 400–300–200 QD dosing and four different bedaquiline dosing regimens from a phase 1 study. Dosing regimens were administered in increasing amounts for the first 3 days of the therapy. i.e., 400–300–200 mg QD represents 400 mg on day 1, 300 mg on day 2, and 200 mg QD on day 3 onwards

Bedaquiline and M2 observed versus predictions for plasma and spinal CSF compartments at steady-state after 9 weeks of treatment. The model reasonably predicted the observed data. Bedaquiline dosing was 400 mg once daily (QD) for 14 days followed by 200 mg three times a week up to 9 weeks. Steady-state concentrations were measured either 5, 7, or 10 h after the last 200 mg dose. The blue points represent the observed concentrations, and the black triangles represent typical patients’ predicted value
The model slightly underpredicted mean bedaquiline and M2 concentrations in spinal CSF; however, the CSF concentrations were within a standard deviation of the observed CSF concentrations (Fig. 3). Given that the CNS concentrations were bottom-up predictions based on physiological and drug-specific knowledge and that only sparse CSF observed data points (n = 7) are available to date, the model was deemed reliable for the objectives of this study. The final model files are available to download from Supplementary Material 3.
3.2 Local Sensitivity Analysis Elucidates the Influence of Uncertainty in Parameters on Drug Exposures
The overall impact of reported uncertainties in key physicochemical parameters, lipophilicity, and plasma protein binding, on vascular plasma and CNS drug exposure (AUC0–t) was evaluated by using local sensitivity analysis. A twofold decrease in bedaquiline lipophilicity (i.e., 5.12–2.5) was predicted to decrease brain intracellular unbound AUC0–t by 65.2% and vascular plasma AUC0–t by 24%. A twofold increase in the albumin CSF-to-plasma ratio (i.e., 0.008–0.016) was predicted to increase brain intracellular total and unbound AUC0–t by 0.4%. A 1000-fold increase (i.e., 0.0003–0.3) in bedaquiline fraction unbound was predicted to decrease brain intracellular unbound AUC0–t by 55.2% and vascular plasma AUC0–t by 40.6%.
3.3 PBPK Simulations Predicted Significantly Lower Bedaquiline Brain Concentrations in the Intracellular Compartment Compared with Peripheral Blood and Lung
Unbound bedaquiline and M2 concentrations in lung interstitial and intracellular, brain interstitial, and CSF were predicted to be comparable with unbound plasma concentrations. However, unbound bedaquiline and M2 concentrations in brain intracellular were predicted to be significantly lower than unbound plasma and unbound lung intracellular concentrations (ratio ~0.07 compared with unbound concentrations in plasma) (Fig. 4 and Table 3). Additionally, our simulations suggested that 200 mg QD for 8 weeks followed by 100 mg QD would achieve relatively increased plasma concentrations for a longer duration of time when compared with 400 mg QD for 14 days followed by 200 mg three times a week (Fig. 4).

Typical patient simulations of bedaquiline and M2 concentrations within various compartments following the current and alternative bedaquiline dosing regimens. The simulations suggested limited availability of unbound bedaquiline and M2 in brain intracellular compartment. The model suggested relatively higher concentrations for longer duration for all compartments following the alternative dosing regimen, 200 mg QD for 8 weeks then 100 mg QD, than the current dosing regimen, 400 mg QD for 14 days, followed by 200 mg three times a week
4 Discussion
We developed a whole-body PBPK model for bedaquiline and M2, coupled with CSF and brain compartments using clinical data. We then used the model to predict bedaquiline and M2 exposures in CNS compartments, including the brain intracellular compartment.
Our PBPK model enabled predictions of bedaquiline and M2 drug concentration-time profiles at target sites-of-action, brain interstitial and intracellular, for patients with TBM. The model predicted a total bedaquiline tissue-to-plasma concentration ratio of 27% for brain intracellular. This predicted brain intracellular to plasma ratio is relatively comparable to the findings from preclinical studies that reported brain parenchyma to plasma concentrations ratios ranging from 2 to 20% following oral bedaquiline dosing suggesting the reliability of our model [14, 15]. However, only unbound drug at site-of-action is assumed to be available to exert an antibacterial effect. The model-predicted unbound concentrations within brain intracellular compartment were drastically lower as compared with unbound concentrations in plasma and lungs (ratio ~0.07 compared with unbound concentrations in plasma) (Fig. 4). We predicted that suboptimal drug concentrations would be available in brain intracellular to exert effect following bedaquiline at clinically relevant dosing in patients with TBM. These predictions may support the experimental findings of inferior efficacy of BPaL combination with that of standard first-line anti-TB therapy in the TBM mouse model following dosing equivalent to human efficacious dosing for each drug [16]. Prospective studies comparing the efficacy of various combination regimens with and without bedaquiline in drug-resistant TBM models are suggested. Our model predicted no delay or nonlinearity between plasma, brain, and lung tissues (Supplementary Material 2). This suggests that plasma drug concentrations may be considered as surrogate for tissue concentrations considering relative ratio.
Sensitivity analysis allowed estimation of the impact of known uncertainty in protein binding and lipophilicity parameters on predictions of vascular plasma and CNS concentrations. However, it should be noted that our model was calibrated using observed vascular plasma concentrations and described observed CSF concentrations well. Thus, the estimated parameters of our model would be correlated with protein binding and lipophilicity parameters used in the model development. Therefore, it is unlikely that protein binding and lipophilicity parameters different than those used in our model development would lead to very different predictions of plasma or target site exposures.
We focused on typical TB patient simulations and did not simulate with interindividual variability (IIV), because direct application of the IIV estimates from PopPK models to the PBPK model often does not appropriately characterize observed variability in plasma PK profiles. Previously, high IIV (> 80%) in absorption-related parameters and moderate IIV (~50%) in distribution and clearance-related parameters have been estimated for bedaquiline using empirical population PK models [26, 27]. These IIV estimates translate into moderate IIV in steady-state exposure (AUCss–weekly interquartile range ~90–250 ug × h/mL). Thus, the impact of observed IIV on overall conclusions would be negligible given the fold difference between predicted total and unbound drug concentrations at the site of action for patients with TBM.
Our model described plasma bedaquiline and M2 drug concentration data well for various dosing scenarios. Our typical patient CSF concentration predictions were slightly lower than the observed mean concentrations for both bedaquiline and M2 but were within the observed standard deviation range. Some uncertainty exists in observed CSF concentrations due to the small sample size (n = 7, one sample for each patient) and bioanalytical challenges due to the high protein binding nature of bedaquiline. The impact of the slight underpredictions of mean CSF concentrations on overall predictions of unbound drug concentrations within the brain intracellular compartment seems negligible given the fold difference between unbound brain intracellular and CSF drug concentrations.
We did not evaluate the impact of TBM disease on bedaquiline and M2 concentrations in CNS. In our model, parameters relevant to drug penetration in CNS were calculated based on physicochemical and physiological parameters, including the albumin plasma-to-CSF ratio, obtained from healthy subjects [37]. Our typical patient CSF concentration predictions were compared against the observed CSF concentrations data from patients with pulmonary TB [13]. Our sensitivity analysis suggested a small impact of albumin plasma-to-CSF ratio on brain concentrations of bedaquiline. However, higher CSF protein levels are associated with increased the partition coefficient for rifampin [42]. Future studies should evaluate the impact of TBM disease on bedaquiline PK.
PBPK models including the transporters involved in BBB and BCSFB have been reported in the literature. These models included active transport processes. Key apical efflux transporters involved in blood–brain barrier or blood–CSF barrier are P-gp, BCRP, and MRPs [33]. Bedaquiline is not known to be a substrate of any of these transporters [25]; therefore, we did not explicitly include brain transporters in this work.
We present a whole-body PBPK model for bedaquiline and its active metabolite coupled with an expanded CNS distribution model, including CSF and brain subcompartments. The model appropriately described observed plasma and CSF bedaquiline and M2 concentrations from patients with TB. However, unbound concentrations in brain intracellular were predicted to be much lower than the predicted unbound concentrations in plasma and lungs. These results can be useful in designing experiments comparing the efficacy of various combination regimens with and without bedaquiline in drug-resistant TBM models.
References
Seddon JA, Wilkinson R, van Crevel R, et al. Knowledge gaps and research priorities in tuberculous meningitis. Wellcome Open Res. 2019;4:1–18. https://doi.org/10.12688/wellcomeopenres.15573.1.
Navarro-Flores A, Fernandez-Chinguel JE, Pacheco-Barrios N, Soriano-Moreno DR, Pacheco-Barrios K. Global morbidity and mortality of central nervous system tuberculosis: a systematic review and meta-analysis. J Neurol. 2022;269(7):3482–94. https://doi.org/10.1007/s00415-022-11052-8.
Nahid P, Dorman SE, Alipanah N, et al. Executive Summary: Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: treatment of drug-susceptible tuberculosis. Clin Infect Dis. 2016;63(7):853–67. https://doi.org/10.1093/cid/ciw566.
Ramachandran R, Muniyandi M. Towards improved treatment outcomes for tuberculosis meningitis—rethinking the regimen. Open Access J Neurol Neurosurg. 2018. https://doi.org/10.19080/OAJNN.2018.08.555734.
Ruslami R, Ganiem AR, Dian S, Apriani L, Achmad TH, van der Ven AJ, Borm G, Aarnoutse RE, van Crevel R. Intensified regimen containing rifampicin and moxifloxacin for tuberculous meningitis: an open-label, randomised controlled phase 2 trial. Lancet Infect Dis. 2013;13(1):27–35. https://doi.org/10.1016/S1473-3099(12)70264-5.
Maranchick NF, Alshaer MH, Smith AGC, Avaliani T, Gujabidze M, Bakuradze T, Sabanadze S, Avaliani Z, Kipiani M, Peloquin CA, Kempker RR. Cerebrospinal fluid concentrations of fluoroquinolones and carbapenems in tuberculosis meningitis. Front Pharmacol. 2022;12(13):1048653. https://doi.org/10.3389/fphar.2022.1048653.
Heemskerk AD, Nguyen MTH, Dang HTM, Vinh Nguyen CV, Nguyen LH, Do TDA, Nguyen TTT, Wolbers M, Day J, Le TTP, Nguyen BD, Caws M, Thwaites GE. Clinical outcomes of patients with drug-resistant tuberculous meningitis treated with an intensified antituberculosis regimen. Clin Infect Dis. 2017;65(1):20–8. https://doi.org/10.1093/cid/cix230.
Evans EE, Avaliani T, Gujabidze M, et al. Long term outcomes of patients with tuberculous meningitis: the impact of drug resistance. PLoS One. 2022;17(6): e0270201. https://doi.org/10.1371/journal.pone.0270201.
Khoshnood S, Goudarzi M, Taki E, et al. Bedaquiline: current status and future perspectives. J Glob Antimicrob Res. 2021;25:48–59. https://doi.org/10.1016/j.jgar.2021.02.017.
Andries K, Verhasselt P, Guillemont J, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307(5707):223–7. https://doi.org/10.1126/science.1106753.
World Health Organization. Consolidated operational guidelines on handbook tuberculosis. 2020. https://www.who.int/publications/i/item/9789240063129.
Akkerman OW, Odish OFF, Bolhuis MS, et al. Pharmacokinetics of bedaquiline in cerebrospinal fluid and serum in multidrug-resistant tuberculous meningitis. Clin Infect Dis. 2015;62(4):523–4. https://doi.org/10.1093/cid/civ921.
Upton CM, Steele CI, Maartens G, Diacon AH, Wiesner L, Dooley KE. Pharmacokinetics of bedaquiline in cerebrospinal fluid (CSF) in patients with pulmonary tuberculosis (TB). J Antimicrob Chemother. 2022;77(6):1720–4. https://doi.org/10.1093/jac/dkac067.
Ordonez AA, Carroll LS, Abhishek S, et al. Radiosynthesis and PET bioimaging of 76Br-bedaquiline in a murine model of tuberculosis. ACS Infect Dis. 2019;5(12):1996–2002. https://doi.org/10.1021/acsinfecdis.9b00207.
Pamreddy A, Baijnath S, Naicker T, et al. Bedaquiline has potential for targeting tuberculosis reservoirs in the central nervous system. RSC Adv. 2018;8(22):11902–7. https://doi.org/10.1039/c8ra00984h.
Mota F, Ruiz-Bedoya CA, Tucker EW, et al. Dynamic 18F-Pretomanid PET imaging in animal models of TB meningitis and human studies. Nat Commun. 2022;13(1):7974. https://doi.org/10.1038/s41467-022-35730-3.
Tasneen R, Betoudji F, Tyagi S, et al. Contribution of oxazolidinones to the efficacy of novel regimens containing bedaquiline and pretomanid in a mouse model of tuberculosis. Antimicrob Agents Chemother. 2016;60(1):270–7. https://doi.org/10.1128/AAC.01691-15.
Food and Drug Administration (FDA). FDA Briefing Document Pretomanid Tablet, 200 mg Meeting of the Antimicrobial Drugs Advisory Committee (AMDAC). Published online 2019. https://www.fda.gov/media/127592/download.
Smith AGC, Gujabidze M, Avaliani T, et al. Clinical outcomes among patients with tuberculous meningitis receiving intensified treatment regimens. Int J Tuberc Lung Dis. 2021;25(8):632–9. https://doi.org/10.5588/ijtld.21.0159.
Diacon AH, Dawson R, Von Groote-Bidlingmaier F, et al. Bactericidal activity of pyrazinamide and clofazimine alone and in combinations with pretomanid and bedaquiline. Am J Respir Critical Care Med. 2015;191(8):943–53. https://doi.org/10.1164/rccm.201410-1801OC.
Kuepfer L, Niederalt C, Wendl T, et al. Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacometrics Syst Pharmacol. 2016;5(10):516–31. https://doi.org/10.1002/psp4.12134.
Willmann S, Höhn K, Edginton A, et al. Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn. 2007;34(3):401–31. https://doi.org/10.1007/s10928-007-9053-5.
Willmann S, Schmitt W, Keldenich J, Dressman JB. A physiologic model for simulating gastrointestinal flow and drug absorption in rats. Pharm Res. 2003;20(11):1766–71. https://doi.org/10.1023/B:PHAM.0000003373.72652.c0.
Gaohua L, Wedagedera J, Small BG, et al. Development of a multicompartment permeability-limited lung PBPK model and its application in predicting pulmonary pharmacokinetics of antituberculosis drugs. CPT Pharmacometrics Syst Pharmacol. 2015;4(10):605–13. https://doi.org/10.1002/psp4.12034.
van Heeswijk RPG, Dannemann B, Hoetelmans RMW. Bedaquiline: a review of human pharmacokinetics and drug–drug interactions. J Antimicrob Chemother. 2014;69(9):2310–8. https://doi.org/10.1093/jac/dku171.
Svensson EM, Dosne AG, Karlsson MO. Population pharmacokinetics of bedaquiline and metabolite m2 in patients with drug-resistant tuberculosis: the effect of time-varying weight and albumin. CPT Pharmacometrics Syst Pharmacol. 2016;5(12):682–91. https://doi.org/10.1002/psp4.12147.
McLeay SC, Vis P, Van Heeswijk RPG, Green B. Population pharmacokinetics of bedaquiline (TMC207), a novel antituberculosis drug. Antimicrob Agents Chemother. 2014;58(9):5315–24. https://doi.org/10.1128/AAC.01418-13.
Mehta K, Guo T, van der Graaf PH, van Hasselt JGC. Predictions of bedaquiline and pretomanid target attainment in lung lesions of tuberculosis patients using translational minimal physiologically based pharmacokinetic modeling. Clin Pharmacokinet. 2023;62(3):519–32. https://doi.org/10.1007/s40262-023-01217-7.
Schmitt W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol In Vitro. 2008;22(2):457–67. https://doi.org/10.1016/j.tiv.2007.09.010.
Cordes H, Rapp H. Gene expression databases for physiologically based pharmacokinetic modeling of humans and animal species. CPT Pharmacometrics Syst Pharmacol. 2023;12(3):311–9. https://doi.org/10.1002/psp4.12904.
Liu K, Li F, Lu J, et al. Bedaquiline metabolism: enzymes and novel metabolites. Drug Metab Dispos. 2014;42(5):863–6. https://doi.org/10.1124/dmd.113.056119.
Yamamoto Y, Välitalo PA, van den Berg DJ, et al. A generic multi-compartmental CNS distribution model structure for 9 drugs allows prediction of human brain target site concentrations. Pharm Res. 2017;34(2):333–51. https://doi.org/10.1007/s11095-016-2065-3.
Gaohua L, Neuhoff S, Johnson TN, Rostami-Hodjegan A, Jamei M. Development of a permeability-limited model of the human brain and cerebrospinal fluid (CSF) to integrate known physiological and biological knowledge: estimating time varying CSF drug concentrations and their variability using in vitro data. Drug Metab Pharmacokinet. 2016;31(3):224–33. https://doi.org/10.1016/j.dmpk.2016.03.005.
Saleh MAA, Loo CF, Elassaiss-Schaap J, De Lange ECM. Lumbar cerebrospinal fluid-to-brain extracellular fluid surrogacy is context-specific: insights from LeiCNS-PK3.0 simulations. J Pharmacokinet Pharmacodyn. 2021;48(5):725–41. https://doi.org/10.1007/s10928-021-09768-7.
Verscheijden LFM, Koenderink JB, de Wildt SN, Russel FGM. Development of a physiologically-based pharmacokinetic pediatric brain model for prediction of cerebrospinal fluid drug concentrations and the influence of meningitis. PLoS Comput Biol. 2019;15(6): e1007117. https://doi.org/10.1371/journal.pcbi.1007117.
Utsey K, Gastonguay MS, Russell S, Freling R, Riggs MM, Elmokadem A. Quantification of the impact of partition coefficient prediction methods on physiologically based pharmacokinetic model output using a standardized tissue composition. Drug Metab Dispos. 2020;48(10):903–16. https://doi.org/10.1124/DMD.120.090498.
Koch S, Donarski N, Goetze K, et al. Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res. 2001;42(7):1143–51.
Conradie F, Bagdasaryan TR, Borisov S, et al. Bedaquiline–pretomanid–linezolid regimens for drug-resistant tuberculosis. N Engl J Med. 2022;387(9):810–23. https://doi.org/10.1056/nejmoa2119430.
Lippert J, Burghaus R, Edginton A, et al. Open systems pharmacology community—an open access, open source, open science approach to modeling and simulation in pharmaceutical sciences. CPT Pharmacomet Syst Pharmacol. 2019;8(12):878–82. https://doi.org/10.1002/psp4.12473.
Perrineau S, Lachâtre M, Lê MP, Rioux C, Loubet P, Fréchet-Jachym M, Gonzales MC, Grall N, Bouvet E, Veziris N, Yazdanpanah Y, Peytavin G. Long-term plasma pharmacokinetics of bedaquiline for multidrug- and extensively drug-resistant tuberculosis. Int J Tuberc Lung Dis. 2019;23(1):99–104. https://doi.org/10.5588/ijtld.18.0042.
Ngwalero P, Brust JCM, van Beek SW, Wasserman S, Maartens G, Meintjes G, Joubert A, Norman J, Castel S, Gandhi NR, Denti P, McIlleron H, Svensson EM, Wiesner L. Relationship between plasma and intracellular concentrations of bedaquiline and its M2 metabolite in South African patients with rifampin-resistant tuberculosis. Antimicrob Agents Chemother. 2021;65(11): e0239920. https://doi.org/10.1128/AAC.02399-20.
Svensson EM, Dian S, Te Brake L, Ganiem AR, Yunivita V, van Laarhoven A, Van Crevel R, Ruslami R, Aarnoutse RE. Model-based meta-analysis of rifampicin exposure and mortality in indonesian tuberculous meningitis trials. Clin Infect Dis. 2020;71(8):1817–23. https://doi.org/10.1093/cid/ciz1071.
Van Essen DC, Donahue CJ, Glasser MF. Development and evolution of cerebral and cerebellar cortex. Brain Behav Evol. 2018;91(3):158–69. https://doi.org/10.1159/000489943.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for this work.
Conflict of Interest
The other authors declare no conflicts of interest.
Ethics Approval
Not applicable
Consent to Participate
Not applicable
Consent for Publication
All authors provided consent for publication.
Availability of Data and Material
All data used in the analysis were either obtained from literature (as cited) or were obtained from the TB-PACTS database (https://c-path.org/programs/tb-pacts/).
Code Availability
Final model codes are provided in the Supplementary Materials.
Author Contributions
All authors contributed to the study conception and design. The analysis was performed by K.M. The first draft of the manuscript was written by K.M. All authors reviewed and edited the manuscript. All authors reviewed and approved the final manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Mehta, K., Balazki, P., van der Graaf, P.H. et al. Predictions of Bedaquiline Central Nervous System Exposure in Patients with Tuberculosis Meningitis Using Physiologically based Pharmacokinetic Modeling. Clin Pharmacokinet 63, 657–668 (2024). https://doi.org/10.1007/s40262-024-01363-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-024-01363-6