
Abstract
Background
Ritlecitinib is an oral Janus kinase 3/tyrosine kinase expressed in hepatocellular carcinoma family inhibitor undergoing parallel clinical development for alopecia areata, vitiligo, ulcerative colitis, Crohn’s disease, and rheumatoid arthritis.
Objective
As studies read out simultaneously, strategic planning of population pharmacokinetic model development and evaluation is required to ensure timely decisions.
Methods
Data from healthy participants and patients from 12 clinical trials between December 2014 and July 2021 were included: seven phase I studies in healthy participants and organ impairment, five phase II/III studies in patients with rheumatoid arthritis, ulcerative colitis, alopecia areata, and vitiligo. Population pharmacokinetic models consisted of stepwise procedures to accommodate data availability and the model’s application to answering clinical development questions. At each iteration of the model update, parameters of the next model were re-estimated by leveraging previous information and new data.
Results
Three model development lifecycle iterations of the ritlecitinib population pharmacokinetic model were conducted to support alopecia areata, vitiligo, and ulcerative colitis study readouts. Initial structural modeling based on healthy participant data (and some rheumatoid arthritis and alopecia areata data) in iteration 1 provided a platform for comprehensive covariate testing during iteration 2, and model evaluation and implementation of the frequentist prior approach in iteration 3. The final model was a two-compartment model with first-order absorption and direct-response non-stationary clearance and bioavailability driven by concentrations in the peripheral compartment.
Conclusions
The present approach demonstrated the evolution of three population pharmacokinetic models with accumulating data, addressed clinical drug development questions related to systemic exposures of ritlecitinib, and informed the approved product label.
Clinical Trial Registration
NCT02309827, NCT02684760, NCT02958865, NCT02969044, NCT03232905, NCT03732807, NCT04016077, NCT03715829, NCT04037865, NCT04004663, NCT04634565, NCT02974868.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Ritlecitinib is undergoing parallel clinical drug development in multiple indications such that developing a full population pharmacokinetic model in preparation of phase III completion would have neglected support of concurrent phase II study completions in other indications. |
Using an iterative approach, three ritlecitinib population pharmacokinetic models were developed iteratively to evaluate newly available concentration data throughout the clinical development of ritlecitinib and to use a frequentist prior approach to update model parameter estimates. |
Ritlecitinib population pharmacokinetic models were used to timely address clinical drug development questions including dosing recommendations for adolescents, moderate hepatic impairment, and severe renal impairment populations. |
1 Introduction
Model development occurs through a series of iterative cycles. At each node of the model development lifecycle, new information is incorporated and, after assessment, the model becomes available for application in clinical drug development. As the drug moves from phase I through to phase III, the accruing data are used to evolve the population pharmacokinetic (PK) model in iterations to help with decisions needed for progressing the drug through each development phase. Availability of population PK models is important to address questions, inform dose selection and study designs, provide dose recommendations for specific subpopulations, and to provide individual-level exposure estimates for exposure-response models of efficacy and safety endpoints. However, at later stages of the clinical development cycle, sparse data predominate and the utility of methods that leverage prior information (either data or a prior model) to assist in the rapid analysis and evaluation of the newly available data becomes increasingly valuable. These strategies were implemented during consecutive updates of the population PK model for ritlecitinib.
Ritlecitinib (PF-06651600) is an oral Janus kinase 3/tyrosine kinase expressed in hepatocellular carcinoma family inhibitor [1, 2] that has completed clinical trials in autoimmune and inflammatory diseases. The plasma pharmacokinetics of ritlecitinib is approximately dose proportional with a rapid absorption of ≈ 89% (fa), an absolute oral bioavailability (F) of ≈ 64%, and an elimination half-life of ≈ 2 h. Ritlecitinib is metabolized by glutathione (GSH) conjugation and cytochrome P450 (CYP)-mediated oxidation with no single enzyme contributing > 25%. Ritlecitinib has demonstrated efficacy in alopecia areata (AA), rheumatoid arthritis (RA), vitiligo, and ulcerative colitis (UC) [3,4,5,6,7]. Patients with AA who received ritlecitinib had significant hair regrowth by week 24 compared with placebo [8]. Patients with non-segmental vitiligo who received ritlecitinib had a significantly greater change from baseline in the centrally read Facial Vitiligo Area Scoring Index at week 24 than those receiving placebo [3]. Ritlecitinib also has demonstrated a rapid onset of action, with a significant improvement in the partial Mayo Score for patients with UC after 2 weeks and in RA disease scores by 8 weeks [5, 7].
The development of population PK models for ritlecitinib consisted of stepwise procedures to accommodate data availability and the model’s application to timely answering clinical drug development questions (including confirming that expected ritlecitinib exposures were achieved in patient populations, generating individual PK parameters for exposure-response modeling of key efficacy and safety endpoints at the time of study completion, conducting simulations to assess formulation changes or pediatric extrapolation). There were three iterations in the model development lifecycle as described in Fig. 1. The objective of the base model was to define the structure for ritlecitinib pharmacokinetics using intense sampling from healthy participants (HP) and sparsely sampled data from patients with RA or AA. The objective of the updated model was to assess if the structure of the base model was adequate at describing ritlecitinib pharmacokinetics for data available from new HP, participants with moderate hepatic impairment, and patient (UC and vitiligo) populations (in addition to all base model data available) and perform stepwise covariate modeling. The objectives of the final model were to externally evaluate the final model’s ability to continue to describe newly available data intensively sampled from HP and participants with severe renal impairment (n = 17) and sparsely sampled from patients with AA (n = 584), to update structural parameters and estimate additional covariate effects using a frequentist prior approach.

Model development strategy
2 Methods
2.1 Study Population, Dosing, and Sampling Schedule
Combined data from HP and patients from 12 clinical trials between December 2014 and July 2021 were included (Table S1 of the Electronic Supplementary Material [ESM]): data from seven phase I studies in HP and organ impairment and five phase II/III studies in patients with RA, UC, AA, and vitiligo (NCT02309827, NCT02684760, NCT02958865, NCT02969044, NCT03232905, NCT03732807, NCT04016077, NCT03715829, NCT04037865, NCT04004663, NCT04634565, NCT02974868). Ritlecitinib dosing ranged from 5 to 800 mg/day. Details for dosing and plasma sampling are shown in Table S1 of the ESM. The study protocols were approved by the institutional review boards/ethics committees of the study sites and all participants provided written informed consent. The studies were conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and conducted according to the International Conference on Harmonization Guidelines for Good Clinical Practice.
2.2 Analytical Methods
Plasma concentrations of ritlecitinib were determined using validated, sensitive, and specific liquid chromatography-tandem mass spectrometric methods at York Bioanalytical Solutions and Covance Bioanalytical Laboratories (Shanghai, China). The lower limit of quantification for each study is shown in Table S1 of the ESM. Individuals who did not receive one or more doses of ritlecitinib or did not have one or more measurable concentration of ritlecitinib were excluded. No data imputations for missing or below the lower limit of quantification (BLQ) concentrations of ritlecitinib were performed. Sampled concentrations that were BLQ were excluded only during parameter estimation for all three iterations of population PK model development. Below the lower limit of quantification observations were available to assess model performance at representing the proportion of observations BLQ over time.
2.3 Base Model Development
One- and two-compartment disposition models with first-order oral absorption were evaluated as candidate structural models. Time- and/or concentration-dependent changes in clearance (CL) and F such as Michaelis–Menten elimination kinetics, and direct or indirect response models for autoinhibition were explored for their ability to describe non-linearity in ritlecitinib pharmacokinetics.
Structural covariates known to be highly influential a priori were built into the structural model. For example, allometric scaling on PK parameters using body weight (exponents of 0.75 and 1 for CL and volume parameters, respectively, referenced to 70 kg), effect of inflammatory disease burden on ritlecitinib CL (consistent with observations made with other Janus kinase inhibitors), and dose, formulation, and high-fat meal effects on absorption parameters [9,10,11,12,13].
Inter-individual variability (IIV) was added to structural model parameters to account for differences between individuals in the population and was assumed to be log-normally distributed:
where \({P}_{i}\) is the individual value for parameter (P) in the ith participant, \({\theta }_{P}\) is the population typical value for parameter P, and η is an independent random variable describing the variability in P among subjects with a mean of 0 and variance, ω2. Models with and without covariance between random effects were investigated.
A residual error model with a combination of additive and proportional effects was used to describe random unexplained variability (RUV) in ritlecitinib concentrations:
where \({C}_{ij}\) is the ritlecitinib concentration in the ith participant, at observation j, \(\hat{C}_{ij}\) is the model-predicted ritlecitinib concentration, \(\varepsilon_{ij}\) is a normally distributed random variable with a mean of 0 and variance of 1, \(\sigma_{pro}^{2}\) and \(\sigma_{add}^{2}\) are the estimated variance of proportional and additive error, respectively. Structural model selection was guided by changes in the Akaike information criterion, standard goodness-of-fit diagnostic plots, precision of parameter estimates, and η-shrinkage.
2.4 Updated Model Development
Model parameters of the base model were re-estimated using a combined dataset of previous and new data to provide a reference structural model for stepwise covariate modeling.
2.4.1 Covariate Model Descriptions
The effect of a categorical covariate on a parameter was represented as a discrete relationship proportional to the population parameter. For example, the effect of patient type (PTST) on a parameter (P) was described as:
where \(\theta_{{{\text{PTSTP}}}}\) is the estimable parameter for the effect of patient type on P.
The effect of a continuous covariate on a parameter was represented as a power model referenced to the median of the observed data. For example, the effect of age (AGE) on P was described as:
where \({\text{AGE}}_{i}\) is the age (years) in the ith participant, \({\text{AGE}}_{{{\text{ref}}}}\) is the median age in the observed population, and \(\theta_{{{\text{AGEP}}}}\) is the estimable parameter for the effect of age on P.
2.4.2 Covariate Selection
Stepwise covariate modeling approaches were conducted during the development of the updated model, and were used to identify key intrinsic and extrinsic factors that explained differences in ritlecitinib pharmacokinetics between individuals. Covariates for analysis included sex, age, baseline creatinine CL (Cockcroft–Gault), race, moderate hepatic impairment (based on Child–Pugh score), and formulation (tablets, capsules). Covariates were screened for a pairwise correlation. If a strong correlation existed, the more clinically relevant covariate continued to further analyses. Candidate covariates from screening procedures were independently added to the structural model to evaluate their individual significance in improving the fit of the model to the observed data. All covariates shown to be important from the univariable analyses were carried forward to the multivariable analyses.
In univariable analyses, the effect of incorporating an additional covariate parameter compared with the structural model was assessed by the likelihood ratio test. The covariate model was considered significantly better than the structural model if p < 0.01. Candidate covariates also had to satisfy additional criteria: (1) 95% confidence interval (CI) of the covariate parameter estimate did not include zero; (2) addition of the covariate results in a reduction in IIV on the target population parameter; and (3) model diagnostic plots showed an improvement.
In multivariable analyses, the covariates identified in univariable analyses were added sequentially to the structural model in order of statistical significance to form the full model. The sequential addition of a covariate to the model had to continue to fulfill the requirements described by univariable analyses. Selection of the final model was conducted by backward elimination of covariates from the full model in order of highest to lowest p-values, where a covariate remained in the model if its removal resulted in a significant increase in objective function value as assessed by the likelihood ratio test (p < 0.001).
2.5 Final Model Development
2.5.1 Externally Evaluating the Updated Model
An external evaluation of the updated model was conducted to evaluate its ability to represent ritlecitinib concentrations available in an evaluation dataset (analysis population for the final model) before estimating population parameters. A combination of simulation-based (visual predictive check [VPC], prediction-corrected VPC, and assessment of normality of normalized prediction distribution errors) and empirical Bayes estimate (EBE)-based diagnostics (goodness-of-fit diagnostics and assessment of normality of conditional weighted residuals) were used. Model misspecification identified from this exercise guided covariate analysis of the final model. The structure of the updated model was not re-evaluated.
2.5.2 Frequentist Prior Update
The updated model was used as a “frequentist prior” (termed by Gisleskog et al. [14]) and the foundation of covariate analyses following external evaluation procedures. The PRIOR subroutine in NONMEM® applies a frequentist prior approach to inform population parameter estimates as opposed to fixing them. Here, a penalty is placed on the maximum likelihood (i.e., the objective function value), which accounts for the deviation of current model parameters from their prior estimate. This approach aims to reduce bias in parameter estimates where the reference population may be slightly different from the previous population used to develop the prior model [15].
The evaluation dataset for the final model predominantly consisted of sparse data from the phase II/III study in patients with AA (Table S1 of the ESM), such that it was anticipated that informative priors for all structural parameters adopted from the updated model were required to ensure adequate estimation of final model parameters. The final parameter estimates of the updated model were used as the prior during development of the final model and estimating parameters based on the evaluation dataset. The prior distributions of fixed-effect and random-effect (i.e., IIV) parameters from the updated model were assumed to be normally and normal-inverse Wishart distributed, respectively. Initially, the weight of priors on original model parameters were highly informative with low uncertainty (i.e., 10% relative standard error for the prior). Scenarios where fixed-effect parameters were uninformative (variance of parameter estimates set to 10,000) or highly informative (derived from the updated model’s variance-covariance matrix for parameter estimates) were examined. Scenarios where the prior variance of random-effect parameters for IIV were uninformative (dimensions of the variance-covariance matrix for IIV plus 1) or highly informative (dependent on the number of individuals used to build the prior model) were also examined [15]. Parameters quantifying the standard deviation of RUV and new covariates for assessment (such as age, severe renal impairment, and alopecia disease severity) did not include any prior information. The decision to proceed with a set of prior model parameters was based on model convergence (including successful minimization status), interpretation of final parameter estimates (i.e., consistent with expectations of the pharmacokinetics of ritlecitinib), and standard goodness-of-fit diagnostics.
2.6 Evaluation of Model Predictive Performance
The predictive performances of all models (base, updated, and final) were evaluated by VPCs stratified by candidate covariates and dose (not presented) and prediction-corrected VPCs based on 1000 simulations of their respective index datasets. The models’ abilities to adequately represent the observed proportion of BLQ concentrations were also evaluated.
2.7 Simulation Analyses for Evaluating the Impact of Covariates
Simulations were conducted to compare steady-state maximum plasma drug concentration (Cmax) and area under the concentration–time curve for the dosing interval (AUCτ) for ritlecitinib 50 mg once daily (QD) for 14 days for a series of covariate scenarios based on covariate effects in the updated and final models. The reference scenario was based on an HP, body weight 70 kg, fasted status, and administered the tablet formulation. For each covariate scenario, concentration–time profiles for 1000 trials of 118 randomly drawn individuals administered 50 mg QD for 14 days were simulated using the updated or final models and summarized by Cmax and AUCτ at steady state. The geometric mean ratios of Cmax or AUCτ for each covariate compared with the reference scenario were calculated for each trial.
2.8 Simulation Analyses for Assessment of Dose Proportionality and Accumulation
Simulations were conducted to compare steady-state Cmax and AUCτ for ritlecitinib 50 mg QD for 14 days versus (1) single ritlecitinib 50-mg dose Cmax and area under the concentration–time curve from time 0 to infinity (AUC0–∞), and (2) steady-state metrics for ritlecitinib 5–800 mg QD for 14 days for patients with AA with a body weight of 70 kg. For each dose scenario, concentration–time profiles for 1000 trials of 118 randomly drawn individuals administered ritlecitinib were simulated using the final model and summarized by Cmax and AUCτ on day 14 (or AUC0–∞ for a single-dose scenario). The geometric mean ratios of Cmax or AUCτ/AUC0–∞ for each dose scenario compared with the reference scenario were calculated for each trial. A steady-state accumulation ratio was assessed as the ratio of AUCτ divided by AUC0–∞.
2.9 Software
NONMEM version VII, level 4.3 (ICON Development Solutions, Ellicott City, MD, USA) was used for development of the base and updated models. NONMEM version VII, level 5.0 (ICON Development Solutions) was used for the development of the final model. In all models, parameter estimation used the first-order conditional estimation method with interaction algorithm and individual parameters were obtained from EBE. The ADVAN13 subroutine with TOL = 9 was used. Statistical and graphical output were generated using the R programming and statistical language (R version 3.6.1) [16].
3 Results
3.1 Analysis Population Summary
Characteristics of the individuals in ritlecitinib analysis populations are presented in Table 1. For the base, updated, and final models, the PK analysis datasets consisted of 186 individuals and 2174 ritlecitinib concentrations, 668 individuals and 5187 ritlecitinib concentrations, and 601 individuals and 2944 ritlecitinib concentrations, respectively. The proportion of BLQ observations summarized as time after dose for phase I studies and phase II/III studies are depicted in Fig. 2a and b. Owing to the relatively short half-life of ritlecitinib and QD dosing, greater than 60% of trough concentrations (determined as 24 hours post-dose) were BLQ (Fig. 2).

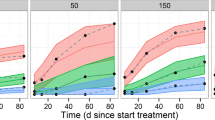
Prediction-corrected visual predictive checks for updated and final models. Prediction-corrected visual predictive checks for (a) the updated model and (b) the final model. Top: the prediction-corrected observed data against time after dose represented by blue circles and dashed black lines (median, 5th and 95th percentiles). The prediction-corrected simulated ritlecitinib concentrations, based on 1000 simulations of the index population, are represented by the red lines and red shaded ribbons (median and 95% prediction interval of the median, respectively) and the blue lines and blue shaded ribbons (median and 95% prediction interval of the 5th and 95th percentiles, respectively). Bottom: the black solid lines represent the proportion of observed below the limit of quantification concentrations over time after a dose. The green solid line and green shaded ribbons are the median and 95% prediction intervals, respectively, of simulated concentrations below the limit of quantification for 1000 simulations of the index population. Yellow indicators in the x-axis represent the time bins for summarizing the data
3.2 Structure of Base Model
Ritlecitinib pharmacokinetics was described by a two-compartment model with first-order absorption, IIV on CL/F and Vc/F, proportional RUV, with direct-response non-stationary CL and F driven by concentrations in the peripheral compartment (Fig. 3, Table S2 and NONMEM code in the ESM). The model semi-mechanistically quantifies the decreases in the rate of elimination and/or first-pass effect resulting in accumulation of ritlecitinib following multiple doses.

Model schematic of ritlecitinib pharmacokinetics. ka is the first-order absorption rate constant for transfer of ritlecitinib from the depot to the central compartment (h-1), Vc is the volume of the central compartment (L), Vp is the volume of the peripheral compartment (L), Q is the inter-compartmental CL between central and peripheral compartments (L/h), and CL is the CL from the central compartment. In the structural model, concentrations in the peripheral compartment (PERIC) have a direct effect on CL and the fraction of the dose bioavailable to the systemic circulation (F). The degree of non-linearity is described by an Emax relationship parameterized with Imax (maximum degree of inhibition proportional to initial CL) and IC50 (peripheral concentration at which 50% of Imax is achieved). INHCL and INHF are the overall non-stationary effects on CL and F, respectively. It is assumed the same mechanisms that impact CL inversely impact F. Equations for model parameters are presented in the Electronic Supplementary Material (NONMEM code)
Structural covariates incorporated into the base model included the effects of patients with RA and AA on CL/F, patients with RA and AA on standard deviation in IIV on CL/F and Vc/F, patients with RA and AA on standard deviation in RUV, high-fat meal on the first-order absorption rate constant (ka), and 800-mg dose on ka. Final parameter estimates for the base model are provided in Table 2.
3.3 Updated Model and Stepwise Covariate Analysis
Covariate effects for UC and vitiligo populations on CL/F were considered structural in nature and were implemented in the structural model prior to a stepwise covariate analysis (see ESM for variables and NONMEM control stream). Covariates that were carried forward for multivariable analyses included the effects of capsule formulation on ka, patients with UC on F, and moderate hepatic impairment on F (Table S3 of the ESM). After forward inclusion and backward elimination procedures, the full model was deemed to be the final model. Final parameter estimates and goodness-of-fit diagnostics for the final model are provided in Table 2 and Fig. S1 of the ESM, respectively. Figure 2a depicts the predictive performance of the final model. The predictions of the final model overlay the observed data with good agreement and adequately reproduce the proportion of BLQ observations for phase I, and some underprediction for phase II and III studies.
3.4 Frequentist Prior Final Model
Both simulation- and EBE-based diagnostics demonstrated the updated model was acceptable at describing the observed concentrations in the evaluation dataset (data not shown). Therefore, the updated model was considered acceptable to serve as a frequentist prior for updating model parameters with new available data from HP, participants with severe renal impairment, and patients with AA.
Informative priors were required for a structural parameter estimation given that the majority of the newly available data was from the phase IIb/III AA population and was sparsely sampled (see ESM for variables and NONMEM control stream). The effect of severe renal impairment F was included in the model. The covariate effect’s addition reduced IIV for the phase I populations in CL/F and Vc/F by 1.59% and 10.1%, respectively, and amended bias in η versus covariate diagnostics. Final parameter estimates and goodness-of-fit diagnostics for the final model are in Table 2 and Fig. S2 of the ESM, respectively. Figure 2b depicts the predictive performance of the final model. It should be noted that differences in the proportion of BLQ observations between phase I and phase II/III studies are because (1) the assays used in phase I and phase II/III studies are different (Table S1 of the ESM), (2) the doses used are different (Table S1 of the ESM), (3) the differences in sampling schedules between phase I and phase II/III studies and the time bins for the VPCs are different, and (4) overall pharmacokinetics are different (i.e., patients with inflammatory disease have lower CL relative to HP, but phase I also includes a combination of HP and participants with severe renal impairment).
3.5 Impact of Covariates on Ritlecitinib Exposure
The impact of intrinsic (patient type, body weight, hepatic, and renal function) and extrinsic (high-fat meal status, formulation) factors on steady-state AUCτ and Cmax based on geometric mean ratios compared with the reference scenario are shown in Fig. 4. Patient type (RA, UC, AA, and vitiligo), organ impairment (moderate hepatic and severe renal), and lower body weight resulted in higher ritlecitinib exposure. The effects are quantified relative to the steady-state exposure of HP weighing 70 kg who received 50 mg QD of the tablet formulation under fasted conditions. The ratios depicted in Fig. 4 are independent of other intrinsic and extrinsic factors that may modify ritlecitinib exposure.

(a) Ratios of area under the concentration–time curve for the dosing interval [AUCτ] and (b) maximum plasma drug concentration [Cmax] on day 14 after ritlecitinib 50 mg once daily (QD) for covariate scenarios. For each covariate scenario on the left y-axis, concentration–time profiles for 1000 trials of 118 randomly drawn individuals administered 50 mg QD for 14 days were simulated and summarized by a AUCτ and b Cmax. The geometric mean ratio of AUCτ or Cmax on day 14 compared to the reference scenario (70 kg, fasted, healthy participant) was calculated for each trial. The gray density distributions represent the geometric mean ratios across all trials; the red numbers are the proportion of trials with ratios less than 0.8 (left) or greater than 1.25 (right). Black numbers on the right y-axis are the median (5th and 95th percentiles) of ratios for the covariate scenario. The blue shaded region is the range of geometric mean ratios from 0.8 to 1.25, and the black vertical dashed line is a geometric mean ratio of 1. Reference low and high body weights are the 5th and 95th percentiles of the analysis population. Scenarios for patients with alopecia areata, severe renal impairment, and body weight were simulated using the final model. Scenarios for patients with rheumatoid arthritis, ulcerative colitis, vitiligo, moderate hepatic impairment, high-fat meal, and the capsule formulation were simulated using the updated model
3.6 Dose Proportionality and Accumulation Assessments
The population PK model addresses greater than proportional increases in ritlecitinib exposure or non-stationary pharmacokinetics at steady state by incorporating concentration-dependent changes in CL/F and F. These effects on steady-state AUCτ and Cmax based on geometric mean ratios compared with the reference scenario are shown in Fig. 5. The steady-state accumulation ratio for 50 mg QD for 14 days was 1.45 (90% PI: 1.26–1.64).

Dose proportionality and accumulation assessment of area under the concentration–time curve [AUC] (a) and maximum plasma drug concentration [Cmax] (b). For each dose scenario on the left y-axis, concentration time-profiles for 1000 trials of 118 randomly drawn individuals administered ritlecitinib as a single dose or once daily (QD) for 14 days were simulated and summarized by a AUC for the dosing interval [AUCτ] (or from time 0 to infinity [AUC0–∞] for single-dose scenario) and b Cmax on day 14. The geometric mean ratio of AUCτ or Cmax compared to the reference scenario (patients with alopecia areata, 70 kg, fasted) was calculated for each trial. The gray density distributions represent the geometric mean ratios across all trials; the red numbers are the proportion of trials with ratios less than 0.8 (left) or greater than 1.25 (right). Black numbers on the right y-axis are the median (5th and 95th percentiles) of ratios for the dosing scenario. The blue shaded region is the range of geometric mean ratios from 0.8 to 1.25, and the black vertical dashed line is a geometric mean ratio of 1
4 Discussion and Conclusions
The iterative approach was efficient at assessing emerging concentration data throughout the clinical drug development of ritlecitinib. Population PK models are evolving platforms describing the concentration–time relationship for a drug. However, updating model parameter estimates with all available data or every time new information becomes available is time and resource consuming under accelerated drug development paradigms. Ritlecitinib is undergoing parallel clinical development in multiple indications (AA, vitiligo, UC, CD, and RA), such that developing a full population PK model in preparation of phase III completion would have neglected support of concurrent phase II study completions in other indications (i.e., supporting end-of-phase II regulatory interactions, phase III dose selection and rationale, pediatric extrapolation, providing exposure metrics for subsequent efficacy and safety models) [3,4,5,6,7].
The three ritlecitinib population PK models presented were all available to timely address clinical development questions, and EBEs for individual patients were used in subsequent exposure-response modeling analyses for safety and efficacy endpoints. The structural elements of the population PK model (particularly fixed allometric scaling exponents and parameterization of non-stationary CL) were retained in each iteration and parameter estimates were comparable between each model (Table 2). The use of concentrations in the peripheral compartment to drive non-stationary characteristics hypothesizes that ritlecitinib distributing out of the systemic circulation, such as to the site of metabolism, is either inhibiting metabolism or non-productively binding to metabolizing enzymes to reduce overall CL (systemic and first-pass effect). With increasing doses, there were greater than proportional increases in exposure. However, doses greater than 100 mg approach maximum inhibition on CL/F and F is achieved and there are no apparent changes in CL/F and F with further dose increases as depicted in the PK summary metrics of clinical relevance such as AUCτ and Cmax (Fig. 5). A similar structural model has been previously applied to clarithromycin, which experiences CYP3A4 autoinhibition [17]. External evaluation and a frequentist prior approach to updating the model upon completion of the phase III AA study was performed owing to a previous iteration of the model being available based on phase II AA concentration data.
The structural model was not re-developed with each iteration and was considered to appropriately capture ritlecitinib pharmacokinetics based on HP data from phase I studies and known routes of metabolism and elimination. Rather, prior to each iteration, the structural model was evaluated based on goodness-of-fit diagnostics, VPCs, and changes in estimates for key structural parameters. Covariate modeling proceeded if the previous structural model was considered adequate. Sparse sampling obtained from phase II/III studies offered challenges in evaluating the model’s ability to describe PK data from patients with inflammatory disease owing to the high proportion of trough concentrations that were BLQ, and that terminal concentrations are the most informative at determining differences in CL. Pharmacokinetic sampling from patients was limited to pre-dose and no longer than 4 h after the dose to reduce the duration of clinic visits for patients. Considering the structural model adequately described the proportion BLQ for the phase I populations, censoring approaches for handling data BLQ were not implemented. It is not likely that ignoring BLQ observations contributed to misspecification of the structural model (i.e., one- vs two- vs three-compartment models) as informed by densely sampled, phase I data and demonstrated adequate representation of the proportion BLQ for these studies. The high proportion of BLQ observations were attributed to the short half-life of ritlecitinib and QD dosing, on average, ritlecitinib is expected to be 97% eliminated after five half-lives (10 h) [2].
Covariate analyses identified several predictors of differences in ritlecitinib exposure. Patients with inflammatory disease (i.e., RA, AA, UC, and vitiligo) have lower baseline CL/F compared with HP. With respect to steady-state AUCτ, patients with RA, AA, UC, and vitiligo experienced 2.19-, 1.56-, 1.92-, and 1.31-fold increases, respectively, in exposure relative to HP administered the same dose and of the same weight. It has been suggested that infectious and inflammatory disease states alter the activities and expression of drug transporters and drug-metabolizing enzymes, such as CYP [9, 11, 13, 18]. For a drug metabolized by multiple CYP and glutathione-S-transferase enzymes, it is realistic to suspect that systemic inflammation is sufficiently high in patients with inflammatory disease to drive down-regulation of metabolic pathways. Ritlecitinib is metabolized by glutathione-S-transferase, an enzyme that catalyzes the conjugation of GSH with ritlecitinib. Glutathione in its reduced form possesses antioxidant effects by acting as an electron donor to protect cells from reactive oxygen species. Inflammatory or immunological diseases and redox balance are closely related, and the pathogenesis of these diseases has been suspected of being associated with increased reactive oxygen species formation and reduced GSH levels [19]. Therefore, GSH depletion may potentially contribute to the decreased CL observed in patient populations with inflammation or immunological disease. In the absence of individual-level GSH information, only patient population effects can be tested on PK parameters. As ritlecitinib is also metabolized by multiple CYP enzymes, down-regulation of these pathways in the setting of inflammation could also attribute to the lower apparent CL observed in patient populations [20].
Scaling parameters were also used to quantify the magnitude of unexplained variability in the pharmacokinetics between HP and patient populations. It was anticipated that phase II studies would demonstrate greater RUV and IIV owing to heterogeneity in dose administration conditions and the sample collection of which there is less regulation than procedures in phase I studies. However, to evaluate the model structure’s ability to describe apparent non-stationary pharmacokinetics, parameters scaling the degree of variability in the patient population compared to HP were implemented. Variability in disease activity between subjects or patient types, and therefore availability of GSH for conjugation, could potentially explain the higher variability in pharmacokinetics observed patients relative to HP.
Patients with moderate-to-severe UC demonstrated a 22.4% reduced F relative to other patient populations and HP. Despite the largest decreases in CL/F relative to other populations, overall exposure as summarized by AUCτ did not exceed that of patients with RA.
Moderate hepatic impairment significantly increases Cmax and AUCτ by 1.28-fold and 1.32-fold, respectively. The effect on F suggests that first-pass metabolism in these patients is impaired, resulting in more ritlecitinib available to the systemic circulation and decreasing apparent CL. A phase I ritlecitinib study (NCT04016077) observed AUCτ ratios of 1.18 for moderate hepatically impaired subjects, and no change in Cmax [21]. The final model provides a comprehensive evaluation of the magnitude of hepatic impairment on ritlecitinib exposures in the context of the entire analysis population.
Severe renal impairment participants demonstrated higher F relative to HP resulting in an average 47% increase in AUCτ and 41% increase in Cmax at steady state. These results are consistent with a phase I study in severe renal impairment participants (NCT04037865) that demonstrated 55% and 44% increases in steady-state AUCτ and Cmax, respectively, relative to a shared cohort from another ritlecitinib phase I study (NCT04016077) of an age- and weight-matched normal renal function population [21].
Chronic kidney disease can impact the pharmacokinetics of drugs that are not predominantly renally excreted, such as ritlecitinib. Physiological processes involved in absorption and hepatic metabolism, such as CYP3A4 metabolism, can be affected by chronic kidney disease resulting in reduced first-pass metabolism and increased F [22,23,24]. As ritlecitinib is approximately 60% bioavailable with > 80% fraction absorbed, the ceiling for increases in F due to severe renal impairment is low.
Intrinsic patient factors that did not demonstrate a statistically or a clinically significant impact on ritlecitinib pharmacokinetics include age, sex, and race, suggesting that inclusion of body weight via allometric scaling adequately accounted for differences between categories within these covariates. The pilot capsule formulation (phase I ritlecitinib study [NCT04004663]) demonstrated a 14% lower Cmax relative to the reference tablet formulation and approximately no difference in AUCτ. These model estimates of the impact of the capsule formulation on ritlecitinib pharmacokinetics are consistent with the results of the phase I study (NCT04004663) and were considered preliminary prior to the results of the pivotal bioequivalence study (NCT04390776) with the to-be-marketed capsule formulation.
The updated and final population PK models were used to underwrite the final approved product label for Litfulo® (ritlecitinib) 50 mg QD for the treatment of severe AA in both adults and adolescents aged 12 years and older [25]. Intrinsic and extrinsic factors that contributed to 0.5-fold to two-fold changes from the mean in ritlecitinib exposure (Cmax and/or AUC) were considered clinically relevant[21]. Incorporation of body weight via allometric scaling principles accounted for differences in exposure, and the effect was not considered to achieve a magnitude of clinical relevance to warrant weight-based dose adjustment for the adolescent population (Fig. 4). Population PK modeling approaches confirmed the expectations of the impact of moderate hepatic impairment and severe renal impairment on ritlecitinib exposure, and co-administration with a high-fat meal, such that dose adjustments were also not necessary for these populations (Fig. 4) [21]. No other significant intrinsic covariates within the AA population were identified (such as age and alopecia disease severity), which justified recommendations of no dose adjustments based on ritlecitinib exposure for these groups.
In summary, three ritlecitinib population PK models were developed iteratively to evaluate newly available data and highlighted the application of a frequentist prior approach to update model parameter estimates in the context of sparsely sampled data from a phase II/III population. Presentation of the three iterations demonstrated how the understanding of ritlecitinib pharmacokinetics evolved throughout clinical drug development from quantitatively describing the differences in RA, UC, AA, and vitiligo patient populations on CL/F relative to each other and HP, to generating hypotheses related to ritlecitinib accumulation following repeated dosing based on structural model identification and confirmation.
References
Xu H, Jesson MI, Seneviratne UI, Lin TH, Sharif MN, Xue L, et al. PF-06651600, a dual JAK3/TEC family kinase inhibitor. ACS Chem Biol. 2019;14(6):1235–42. https://doi.org/10.1021/acschembio.9b00188.
Telliez JB, Dowty ME, Wang L, Jussif J, Lin T, Li L, et al. Discovery of a JAK3-selective inhibitor: functional differentiation of JAK3-selective inhibition over pan-JAK or JAK1-selective inhibition. ACS Chem Biol. 2016;11(12):3442–51. https://doi.org/10.1021/acschembio.6b00677.
Ezzedine K, Peeva E, Yamaguchi Y, Cox LA, Banerjee A, Han G, et al. Efficacy and safety of oral ritlecitinib for the treatment of active nonsegmental vitiligo: a randomized phase 2b clinical trial. J Am Acad Dermatol. 2023;88(2):395–403. https://doi.org/10.1016/j.jaad.2022.11.005.
King B, Guttman-Yassky E, Peeva E, Banerjee A, Sinclair R, Pavel AB, et al. A phase 2a randomized, placebo-controlled study to evaluate the efficacy and safety of the oral Janus kinase inhibitors ritlecitinib and brepocitinib in alopecia areata: 24-week results. J Am Acad Dermatol. 2021;85(2):379–87. https://doi.org/10.1016/j.jaad.2021.03.050.
Robinson MF, Damjanov N, Stamenkovic B, Radunovic G, Kivitz A, Cox L, et al. Efficacy and safety of PF-06651600 (ritlecitinib), a novel JAK3/TEC inhibitor, in patients with moderate-to-severe rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. 2020;72(10):1621–31. https://doi.org/10.1002/art.41316.
Guttman-Yassky E, Pavel AB, Diaz A, Zhang N, Del Duca E, Estrada Y, et al. Ritlecitinib and brepocitinib demonstrate significant improvement in scalp alopecia areata biomarkers. J Allergy Clin Immunol. 2022;149(4):1318–28. https://doi.org/10.1016/j.jaci.2021.10.036.
Sandborn WJ, Danese S, Leszczyszyn J, Romatowski J, Altintas E, Peeva E, et al. Oral ritlecitinib and brepocitinib for moderate-to-severe ulcerative colitis: results from a randomized, phase 2b study. Clin Gastroenterol Hepatol. 2023;S1542–3565(23):00007–11. https://doi.org/10.1016/j.cgh.2022.12.029.
King B, Zhang X, Harcha WG, Szepietowski JC, Shapiro J, Lynde C, et al. Efficacy and safety of ritlecitinib in adults and adolescents with alopecia areata: a randomised, double-blind, multicentre, phase 2b–3 trial. Lancet. 2023;401(10387):1518–29. https://doi.org/10.1016/S0140-6736(23)00222-2.
Wojciechowski J, Malhotra BK, Wang X, Fostvedt L, Valdez H, Nicholas T. Population pharmacokinetics of abrocitinib in healthy individuals and patients with psoriasis or atopic dermatitis. Clin Pharmacokinet. 2022;61(5):709–23. https://doi.org/10.1007/s40262-021-01104-z.
Svensson RJ, Jonsson EN. Efficient and relevant stepwise covariate model building for pharmacometrics. CPT Pharmacometrics Syst Pharmacol. 2022;11(9):1210–22. https://doi.org/10.1002/psp4.12838.
Ma G, Xie R, Strober B, Langley R, Ito K, Krishnaswami S, et al. Pharmacokinetic characteristics of tofacitinib in adult patients with moderate to severe chronic plaque psoriasis. Clin Pharmacol Drug Dev. 2018;7(6):587–96. https://doi.org/10.1002/cpdd.471.
Klünder B, Mohamed MF, Othman AA. Population pharmacokinetics of upadacitinib in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I and II clinical trials. Clin Pharmacokinet. 2018;57(8):977–88. https://doi.org/10.1007/s40262-017-0605-6.
Nader A, Stodtmann S, Friedel A, Mohamed MF, Othman AA. Pharmacokinetics of upadacitinib in healthy subjects and subjects with rheumatoid arthritis, Crohn’s disease, ulcerative colitis, or atopic dermatitis: population analyses of phase 1 and 2 clinical trials. J Clin Pharmacol. 2020;60(4):528–39. https://doi.org/10.1002/jcph.1550.
Gisleskog PO, Karlsson MO, Beal SL. Use of prior information to stabilize a population data analysis. J Pharmacokinet Pharmacodyn. 2002;29(5–6):473–505. https://doi.org/10.1023/a:1022972420004.
Chan Kwong AHP, Calvier EAM, Fabre D, Gattacceca F, Khier S. Prior information for population pharmacokinetic and pharmacokinetic/pharmacodynamic analysis: overview and guidance with a focus on the NONMEM PRIOR subroutine. J Pharmacokinet Pharmacodyn. 2020;47(5):431–46. https://doi.org/10.1007/s10928-020-09695-z.
R: A language and environment for statistical computing. R Foundation for Statistical Computing. 2019. https://www.R-project.org/. Accessed 14 Aug 2023.
Abduljalil K, Kinzig M, Bulitta J, Horkovics-Kovats S, Sörgel F, Rodamer M, et al. Modeling the autoinhibition of clarithromycin metabolism during repeated oral administration. Antimicrob Agents Chemother. 2009;53(7):2892–901. https://doi.org/10.1128/AAC.01193-08.
Morgan ET. Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther. 2009;85(4):434–8. https://doi.org/10.1038/clpt.2008.302.
Perricone C, De Carolis C, Perricone R. Glutathione: a key player in autoimmunity. Autoimmun Rev. 2009;8(8):697–701. https://doi.org/10.1016/j.autrev.2009.02.020.
Schmitt C, Kuhn B, Zhang X, Kivitz AJ, Grange S. Disease-drug-drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther. 2011;89(5):735–40. https://doi.org/10.1038/clpt.2011.35.
Purohit V, Huh Y, Wojciechowski J, Plotka A, Salts S, Antinew J, et al. Leveraging prior healthy participant pharmacokinetic data to evaluate the impact of renal and hepatic impairment on ritlecitinib pharmacokinetics. AAPS J. 2023;25(3):32. https://doi.org/10.1208/s12248-023-00792-8.
Lea-Henry TN, Carland JE, Stocker SL, Sevastos J, Roberts DM. Clinical pharmacokinetics in kidney disease: fundamental principles. Clin J Am Soc Nephrol. 2018;13(7):1085–95. https://doi.org/10.2215/CJN.00340118.
Olyaei AJ, Steffl JL. A quantitative approach to drug dosing in chronic kidney disease. Blood Purif. 2011;31(1–3):138–45. https://doi.org/10.1159/000321857.
Nolin TD. A synopsis of clinical pharmacokinetic alterations in advanced CKD. Semin Dial. 2015;28(4):325–9. https://doi.org/10.1111/sdi.12374.
Litfulo (ritlecitinib). Prescribing information. Pfizer. 2023. https://labeling.pfizer.com/ShowLabeling.aspx?id=19638. Accessed 14 Aug 2023.
Acknowledgements
Writing and scientific support was funded by Pfizer LLC. Third-party medical editing assistance provided by Denise Kenski, of Health Interactions, Inc. was funded by Pfizer LLC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Pfizer Inc.
Conflicts of Interest/Competing Interests
Jessica Wojciechowski, Vivek Purohit, Yeamin Huh, Christopher Banfield, and Timothy Nicholas are employees of and hold stock in Pfizer.
Ethics Approval
The study protocols were approved by the Institutional Review Boards/Ethics Committees of the study sites. The studies were conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and conducted according to the International Conference on Harmonization Guidelines for Good Clinical Practice.
Consent to Participate
All participants provided written informed consent.
Availability of Data and Material
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Code Availability
Not applicable.
Authors’ Contributions
Wrote the manuscript: JW. Designed the research: JW, VSP, YH, CB, TN. Performed the research: JW, VSP, YH, CB. Analyzed the data: JW, VSP, YH, CB, TN.
Consent for Publication
Consent to Publish. Patients signed informed consent regarding publishing their data.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Wojciechowski, J., S Purohit, V., Huh, Y. et al. Evolution of Ritlecitinib Population Pharmacokinetic Models During Clinical Drug Development. Clin Pharmacokinet 62, 1765–1779 (2023). https://doi.org/10.1007/s40262-023-01318-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01318-3