
Abstract
Introduction
SPI-62 is a small-molecule 11β-hydroxysteroid dehydrogenase type 1 (HSD-1) inhibitor exhibiting complicated nonlinear pharmacokinetics (PK) in human. Previously, we developed a target-mediated drug disposition (TMDD) model to characterize the substantial nonlinear PK of SPI-62.
Objective
The aim of the current analysis was to perform population PK/PD analysis to further link SPI-62 exposure (i.e., PK) with its response (i.e., inhibition of hepatic HSD-1 activity) to gain a quantitative understanding of the SPI-62 dose-exposure-response relationship.
Methods
PK and PD data from the first-in-human (FIH) clinical trials, including single ascending dose (SAD) and multiple ascending dose (MAD) studies, were used for model development. During the model development process, the final model selection was based on biological and physiological plausibility, goodness-of-fit plots, stability of parameter estimates, and objective function value. The nonlinear-mixed effect modeling (NONMEM) software was used for both the implementation of the PK/PD model and model simulation. SPI-62 plasma levels and hepatic HSD-1 inhibition over time following various dose regimens were simulated.
Results
The final model was a two-compartment TMDD model component for SPI-62 and an inhibitory Imax model component for hepatic HSD-1 activity. The TMDD-hepatic PD model that we established adequately characterized all remarkable PK and PD behaviors of SPI-62, such as extremely low plasma exposures following the first low doses, nonlinear PK turned into linear PK after repeated low doses, and substantial and long-lasting hepatic HSD-1 inhibition following low doses. SPI-62 was estimated to bind to the target with a second-order association rate constant (Kon) of 8.43 nM−1 h−1 and first-order dissociation rate constant (Koff) value of 0.229 h−1, indicating that SPI-62 binds rapidly to, and dissociates slowly from, its pharmacological target. The estimated target capacity (Rtot) of 5460 nmol corresponds to approximately 2.2 mg of SPI-62, which comports well with the dose range in which PK nonlinearity is prominent. Model simulation results reveal that a 6 mg once-daily regimen can lead to long-lasting and substantial hepatic HSD-1 inhibition.
Conclusions
A population TMDD-PD model that explains SPI-62 nonlinear PK and hepatic HSD-1 inhibition following different dose regimens in healthy adults was successfully established. Our simulation results provide a solid foundation for model-informed development of SPI-62.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A population model, including a two-compartment target-mediated drug disposition (TMDD) model with three transit absorption compartments for SPI-62 disposition and an inhibitory Imax model for hepatic 11β-hydroxysteroid dehydrogenase type 1 (HSD-1) activity, was established to describe SPI-62 nonlinear pharmacokinetics (PK) and substantial and durable hepatic HSD-1 inhibition in human. |
The TMDD-pharmacodynamic (PD) modeling and simulation results provide a strong foundation for model-informed development of SPI-62. To the extent that TMDD is a class effect of HSD-1 inhibitors, the TMDD-PD model that we developed for SPI-62 has potential to be utilized for other members of the class to facilitate interpretation of their PK and PD data as well as future clinical trial design. |
1 Introduction
The 11β-hydroxysteroid dehydrogenases (HSDs) regulate the intracellular levels of glucocorticoids. There are two isoforms: HSD-1 converts inactive cortisone to active cortisol, and HSD-2 converts cortisol to cortisone [1]. HSD-2 protects mineralocorticoid-sensitive tissues (e.g., distal tubule of the kidney) from acute cortisol toxicity. HSD-1 is abundantly expressed in tissues in which glucocorticoid signaling serves physiological functions (e.g., liver, fat, muscle, skin, eye, and brain) at normal levels and can have pathological effects when in excess. In addition to endogenous glucocorticoids, HSDs act similarly on the glucocorticoid medicines by interconverting their inactive (e.g. prednisone) and active (e.g. prednisolone) forms [2]. Chronic elevation of the endogenous glucocorticoid cortisol can result in Cushing’s syndrome and autonomous cortisol secretion (ACS), conditions associated with excess mortality and serious morbidity, including diabetes, cardiovascular disease, obesity, osteoporosis, mood disorders, and cognitive impairments [3,4,5]. Glucocorticoid medicines are associated with approximately 10% of all drg adverse events, including those that result in hospitalization [6]. The important role of HSD-1 in the biology of cortisol and the corticosteroid medicines makes it an attractive therapeutic target for the diseases and drug adverse effects mentioned above [7, 8].
SPI-62 (formerly known as ASP3662, CAS# 1204178-50-6), is a potent and selective small-molecule HSD-1 inhibitor with high affinity against human HSD-1 and minimal activity on HSD-2 [9]. Currently, SPI-62 is in clinical development for Cushing’s syndrome and ACS, and as adjunctive therapy to prednisolone in patients with polymyalgia rheumatica. As part of the clinical development of SPI-62, the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of SPI-62 in healthy adults were evaluated in a single ascending dose (SAD) trial as well as a multiple ascending dose (MAD) trial [9]. The PK results from these two phase I clinical trials showed that SPI-62 has substantial and complex nonlinear PK in humans, which appeared to be caused by the specific and saturable binding of a highly potent small molecule, SPI-62, to its low-capacity pharmacological target HSD-1, known as target-mediated drug disposition (TMDD) [10, 11]. The key nonlinear PK behaviors of SPI-62 are: (1) following single doses, SPI-62 demonstrated linear PK at high doses but pronounced nonlinear PK at low doses; and (2) following multiple doses, nonlinear PK observed following first low doses turned into linear PK with unusually high accumulation ratios for low doses that cannot be explained by SPI-62’s elimination half-life [9]. Based on the PK data from SAD and MAD trials, we established a TMDD model to characterize SPI-62 nonlinear PK in humans [12].
In MAD trial low-dose cohorts, in addition to PK, exploratory PD assessments were also performed. Cortisol and cortisone are converted by sequential action of 5α- and 5β-steroid reductases followed by 3-steroid reductase to form tetrahydrocortisol, allotetrahydrocortisol, and tetrahydrocortisone that are excreted in urine [13, 14]. The urinary ratio of cortisol metabolites (tetrahydrocortisol + allotetrahydrocortisol) to cortisone metabolite (tetrahydrocortisone) is commonly referred as the urinary HSD-1 ratio since it represents HSD-1 inhibition in liver. The urinary HSD-1 ratio data clearly showed that substantial HSD-1 inhibition was observed even after the first dose of 0.7 mg [9]. After repeated doses, full HSD-1 inhibition was achieved by all dose levels evaluated (0.2 mg with a 3 mg loading dose, 0.4 mg, 0.7 mg, and 2 mg). In addition, HSD-1 inhibition was long-lasting, as reflected by the incomplete return of the urinary HSD-1 ratio to baseline approximately 16 days after the last dose even with the lowest dose tested, i.e., 0.2 mg [9]. The data of the time course of the HSD-1 activity are informative and valuable—they not only serve as strong evidence of HSD-1-mediated nonlinear PK of SPI-62 (i.e. pharmacological TMDD) but also provide an opportunity to quantitatively evaluate the exposure-response relationship between SPI-62 and hepatic HSD-1 inhibition. As noted earlier, we previously developed a TMDD model to characterize the relationship between dose and exposure of SPI-62 in human. The aim of the current analysis was to perform population PK/PD analysis to further link SPI-62 exposure (i.e. PK) with its response (i.e. inhibition of hepatic HSD-1 activity) using the pooled PK data from the SAD and MAD trials and PD data from the MAD trial.
2 Methods
2.1 Data Source
The modeling work was conducted based on the key PK and PD data from two phase I clinical trials (SAD and MAD) conducted in healthy adults [9]. The data included in the analysis are summarized in Table 1 and are briefly described below.
In the SAD trial, 48 subjects were randomized into six dose cohorts: 1, 3, 6, 10, 30, and 60 mg. In each cohort, subjects were randomized to receive either placebo (n = 2) or SPI-62 oral administration (n = 6). Data used for the model development were from the 1, 3, 6, and 10 mg cohorts. For PK evaluation, serial blood samples were collected at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 18, 24, 36, 48, and 72 h after oral dose administration for the 1, 3, and 10 mg cohorts, while two additional blood samples were collected at 96 and 120 h after dose administration for the 6 mg cohort. SPI-62 plasma concentrations were determined by a validated high-performance liquid chromatography with tandem mass spectrometry (LC-MS/MS) method. The lower limit of quantification (LLOQ) was 0.1 ng/mL [9].
The MAD trial investigated high (10, 20, and 50 mg) and low (0.2, 0.4, 0.7, and 2 mg) oral doses separately. As high-dose data were not included in our analysis, here we only describe the low-dose design. Four subjects received a 3 mg loading dose on Day 1 and then 0.2 mg once-daily doses on Days 2–14. Four subjects received 0.4 mg once-daily doses on Days 1–14. Twelve subjects (six each at 0.7 and 2 mg doses) received a single dose of SPI-62 on Day 1, followed by a 6-day washout period, and then once-daily doses on days 7–20. For the PK assessment, serial blood samples were obtained at 0.5, 1, 1.5, 2, 3, 4, 8, 12, 16, and 24 h after the first dose, as well as 0.5, 1, 1.5, 2, 3, 4, 8, 12, 16, 24, 48, 72, 96, 120, 144 and 168 h after the last dose. Six additional PK samples were collected at 36, 48, 72, 96, 120, 144 h after the first dose of 0.7 or 2 mg. SPI-62 plasma concentrations were determined by a validated LC–MS/MS assay. The LLOQ was 4 pg/mL [9]. For PD assessment, urine samples from the first-morning void were collected at predose, and on Days 1, 2, 3, 4, 5, 6, 8, 10, 12, 14, 16, 18, 20, 22, 23, 24, 25, 26, and 27 for subjects administered 0.7 or 2 mg SPI-62. Urine samples from the first morning void were collected at predose, and on Days 1, 8, 10, 12, 14, 16, 18, 20, 21 for subjects administered 0.2 or 0.4 mg SPI-62 [9]. The urine samples for steroid analysis were analyzed using validated liquid chromatographic-tandem mass spectrometric methods [9, 15]. The hepatic activity of HSD-1 was calculated as the urinary ratio of (tetrahydrocortisol + allotetrahydrocortisol)/tetrahydrocortisone. While the amounts of tetrahydrocortisol, allotetrahydrocortisol, and tetrahydrocortisone vary (similarly to cortisol) between urine samples, the ratio is comparatively invariant.
Please note that during model development process, we initially used PK data from all dose groups, including those ≥ 20 mg dose groups where linear PK was demonstrated. However, although the model converged and generated similar parameter estimates as those obtained using the subset (doses up to 10 mg only) data, combining all data provided poor model fitting for those low-dose groups, which is not acceptable since those low doses are the target doses in patients. We would like to highlight that almost complete inhibition of hepatic HSD-1 was achieved with regimens as low as 0.2 mg once daily, the lowest dose tested; this indicated that data from those high-dose groups (≥ 20 mg) will not be informative, as those doses will not be considered in the patients.
In the population modeling analysis, there were 996 plasma concentrations, with 222 below LLOQ, and 279 observed baseline-corrected hepatic HSD-1 activity (%) data, with one outlier excluded. The PK and PD data were from 44 healthy subjects, including 24 subjects from the SAD study and 20 subjects from the low-dose cohort of the MAD study. Below the limit of quantification (BLQ) data for SPI-62 were replaced with LLOQ/2, considering that it has provided the best model fitting among seven different methods for BLQ inference that we have evaluated [12].
2.2 Pharmacokinetic (PK) and Pharmacodynamic (PD) Modeling
Simultaneous PK and PD modeling was performed using the nonlinear mixed-effect modeling approach with NONMEM (version 7.4.3; Icon Development Solutions, Ellicott City, MD, USA) using the first-order conditional estimation method with interaction (FOCEI) and a user-defined subroutine (ADVAN13). Simulations were performed in NONMEM (version 7.4.3; ICON Development Solutions) using the final PK/PD model. Data formatting, simulation dataset creation, and simulation postprocessing were performed using R software version 3.4.0. Graphical analyses were conducted using Sigmaplot 13.0 (Systat Software, San Jose, CA, USA) and R software (The R Foundation for Statistical Computing, Vienna, Austria).
We previously established a two-compartment TMDD model with three transit compartments to characterize nonlinear PK of SPI-62 in healthy adults. In the current analysis, SPI-62 plasma concentrations from the SAD (≤ 10 mg dose groups) and MAD (≤ 2 mg dose groups) trials, as well as HSD-1 activity data from the MAD trial, were pooled together for simultaneous fitting. The TMDD model structure is identical to the one we have published. Only a brief description of model structure with key equations for that reported TMDD model is provided here. In our reported model, SPI-62 in the depot was absorbed into the central compartment through three transit compartments, which were parameterized using four identical transit absorption rate constants (Ktr). SPI-62 was distributed between the central compartment and the peripheral compartment following a distribution flow (Q). Linear elimination of SPI-62 from the central compartment was characterized by clearance (CL). SPI-62 distributed in the central compartment could bind with HSD-1 to form a drug-target complex with a second-order association rate constant (kon), and in turn, the complex could dissociate to free drug and HSD-1 with a first-order dissociation rate constant (koff). The total amount of HSD-1 (Rtotal) was assumed to be a constant. No target synthesis or degradation processes were included in that model. Following are the equations used to characterize the PK data.
The equations for the depot and the three transit compartments were as follows (Eqs. 1–4):
where Adepot,SPI-62, Atran1,SPI-62, Atran2,SPI-62, and Atran3,SPI-62 represent the amount of SPI-62 in depot and transit compartments 1, 2, and 3, respectively; Ktr represents the transit absorption rate constant; and F represents the bioavailability of SPI-62.
The equation for the central compartment is shown below (Eq. 5):
The equation for the peripheral compartment is as follows (Eq. 6):
The equation for binding with the high-affinity/low-capacity site compartment is shown below (Eq. 7):
where Acen,SPI-62 and Ccen,SPI-62 represent the amount and concentration of SPI-62 in the central compartment, respectively; Aperi,SPI-62 represents the amount of SPI-62 in the peripheral compartment; Vcen,SPI-62 and Vperi,SPI-62 represent the volume of distribution of the central and peripheral compartments; CL and Q represent the clearance and distribution flow, respectively; Kon and Koff represent the association and dissociation rate constants, respectively; RC represents the amount of SPI-62: R complex; and Rtotal represents the total amount of HSD-1.
2.2.1 PD Structural Model
Hepatic HSD-1 activity was measured as the urinary ratio of (tetrahydrocortisol + allotetrahydrocortisol) /tetrahydrocortisone. For modeling convenience, observed baseline-corrected HSD-1 activity was used and was calculated as shown in the following formula (Eq. 8):
Accordingly, the pharmacologic effect of SPI-62 on hepatic HSD-1 is calculated as follows (Eq. 9)
Exploratory graphical analysis was performed first to examine the relationship between the observed HSD-1 activity and SPI-62 exposure. Urine samples for PD assays and blood samples for the SPI-62 assay were not collected at the same times. Hence, time-matched SPI-62 plasma concentrations were predicted using the established TMDD model. Time-matched model-predicted SPI-62 concentrations were then plotted against the HSD-1 activity to better visualize the potential relation between each other. As a nonlinear relationship was observed, several different inhibitory Imax models were evaluated to describe the exposure-response relationship between SPI-62 concentrations and hepatic HSD-1 activities (electronic supplementary material [ESM] Table S1). The final PK/PD model contained a TMDD model with three transit absorption compartments for SPI-62 PK, and an inhibitory Imax model to link SPI-62 concentrations with HSD-1 activities. Hepatic HSD-1 activity (%) was modeled as follows (Eq. 10):
Inter-individual variability (IIV) was estimated using an exponential model that was assumed to be normally distributed, with a mean of 0 and a variance of ω2.
Additive and proportional models, and a combined proportional and additive residual variability (RV) model, were tested for both PK and PD structural models. RV was assumed to be normally distributed, with a mean of 0 and a variance of  .
.
After the structural PK/PD model was constructed, covariate analyses were conducted. Individual estimates of IIVs for several PK/PD parameters (Vcentral, CL, Ktr, Koff, Rtotal and IC50) were plotted against covariates including age (35.9 ± 9.65 years [mean ± SD]; distribution is shown in ESM Fig. S1), sex (33 males and 11 females), body weight (76.5 ± 12.1 kg [mean ± SD]; distribution is shown in ESM Fig. S1), and race (14 Black, 26 White, 3 Asian, and 1 Native) to visualize the potential relationships. Formal covariate analysis was to be performed using stepwise covariate modeling with an alpha level of 0.05 (ΔOFV >3.84, Δdf = 1) for forward addition and an alpha level of 0.001 (ΔOFV >10.83, Δdf = 1) for backward deletion.
2.2.2 Model Evaluation
The final model was selected based mainly on graphical diagnostics and objective function values. When models are nested, ΔOFV is nominally Chi-square distributed, and a difference of −3.84 corresponds approximately to a p value < 0.05 for 1 degree of freedom. Graphical diagnostics included standard goodness-of-fit plots (population-predicted concentrations [PRED] vs. observed concentrations [DV]; individual-predicted concentrations [IPRED] vs. DV; conditional weighted residuals [CWRES] vs. PRED; and CWRES vs. time), model fitting for time course PK and PD data, and prediction‐corrected visual predictive check (pcVPC).
2.2.3 Simulations
PK sampling timepoints included prior to dosing (i.e., 0) and 0.5, 1, 1.5, 2, 3, 4, 8, 12, 16, and 24 h after the first dose, as well as 0.5, 1, 1.5, 2, 3, 4, 8, 12, 16, 24, 48, 72, 96, 120, 144, and 168 h after the last dose. PD samples were scheduled to be collected immediately before each daily dosing (trough) and collected for 1 week after the last dose. Using the final model and its parameter estimates, 1000 virtual observations at each sampling time point were simulated. Variability in the simulation results was contributed by IIV in PK parameters. The 5th, 50th, and 95th percentiles of the simulated data were then plotted.
The following dosing scenarios were simulated:
Scenario 1: 0.4, 1, 2, 3, 6, 10 mg once-daily dose on days 1–14.
Scenario 2: 3 mg loading dose on day 1 followed by a 0.4 mg or 1 mg once-daily dose on days 2–14.
3 Results
3.1 Observed Data
Figure 1 shows the time course of hepatic HSD-1 inhibition, calculated based on the observed remaining HSD-1 activity, and time-matched SPI-62 concentrations predicted using the TMDD model established using PK data alone. As shown in Fig. 1, following low doses of SPI-62, substantial HSD-1 inhibition was observed, even though the plasma exposure of SPI-62 was extremely low. After the last dose of SPI-62, the HSD-1 inhibitory effect persisted for many days. In addition, there was no time delay between the maximum concentration (Cmax) of SPI-62 and maximal HSD-1 inhibition, indicating a direct relationship between SPI-62 exposure and response. Figure 2 shows the relationship between observed hepatic HSD-1 activity and time-matched SPI-62 concentrations predicted using the final TMDD model established using PK data alone. As shown in Fig. 2, a clear inhibitory nonlinear relationship was demonstrated.

Time courses of mean observed hepatic HSD-1 inhibition and mean individual model-predicted SPI-62 plasma concentrations following 0.2, 0.4, 0.7, and 2 mg oral multiple doses of SPI-62. Vertical black lines represent the start and stop of multiple doses. For subjects who received 0.2 mg SPI-62, a loading dose of 3 mg was administered. For subjects who received 0.7 mg or 2 mg, a single dose of SPI-62 was followed by a 6-day washout before the start of the QD dose regimens. QD once daily

Log-scale scatter plot of the relationship between time-matched SPI-62 plasma concentrations predicted using the established TMDD model and the percentage of the observed baseline-corrected hepatic HSD-1 activity. TMDD target-mediated drug disposition, LLOQ lower limit of quantification
3.2 Final Model Structure
During the model development phase, the TMDD model component for the PK part and the different types of Imax model component for the PD part were explored. In addition to the structural model, we also optimized the statistical model component by testing IIV on different parameters as well as error models. Among the different models tested, the TMDD model with three transit absorption compartments for PK and an inhibitory Imax model \((100 \times \left(1- \frac{{I}_{\text{max}}\times {C}^{\text{r}}}{{{\text{IC}}_{50}}^{\text{r}}+{C}^{\text{r}}}\right))\) with IIV on IC50 for PD was found to be the best PK/PD model structure (Fig. 3). Exploratory covariate analysis showed that there was no meaningful impact of age, sex, body weight or race on any model parameters (p > 0.05 for all plots) [ESM Fig. S2]. Accordingly, formal standard forward addition and backward elimination covariate testing was not performed. Since there were no covariates, the final PK/PD model was identical to the base PK/PD model.

Final TMDD model describing the pharmacokinetics and pharmacodynamics of SPI-62. The absorption process of SPI-62 was characterized by four sequential first-order transit absorption rate constants (Ktr). SPI-62 in the central compartment (Ccentral, Vcentral) can distribute to the peripheral compartment (Cperipheral, Vperipheral) by distribution flow (Q) and be eliminated by a linear elimination pathway that is characterized by CL/V. SPI-62 can bind with HSD-1 (i.e., R) with second-order association rate constant (Kon) to form SPI-62:R complexes. SPI-62:R can dissociate back to free drug and free HSD-1 target with the first-order dissociation rate constant (Koff). The total amount of HSD-1 in human (Rmax) is assumed to be constant. The relation between HSD-1 inhibition versus SPI-62 plasma concentrations was described by a sigmoid Imax model. In the sigmoid Imax model, the sigmoid character of the curve is determined by the power coefficient γ. Imax maximum drug inhibition effect, IC50 SPI-62 plasma concentration at 50% of Imax, TMDD target-mediated drug disposition
3.3 Parameter Estimates
The parameter estimates for the final simultaneous PK/PD model are presented in Table 2. SPI-62 bound to its target with a Kon of 8.43 nM−1 h−1, and dissociated from the SPI-62:R complex with a Koff of 0.229 h−1. These PK parameters estimated using combined PK and PD data are very close to those estimated using the PK data alone. The capacity of the target (Rmax) was estimated to be 5460 nmol, which is similar to the value estimated using PK data alone (6070 nmol). The IC50 of SPI-62 on hepatic HSD-1 inhibition was estimated to be 0.0787 nM (i.e., 0.0334 ng/mL), within twofold of the average Cmax of a single dose of 0.7 mg SPI-62 (0.0185 ng/mL). The estimated IC50 is close to the estimated Kd (0.0272 nM), calculated as the ratio of Koff and Kon, and is far less than the Ki (5.3 nM) value measured in vitro. As shown in Table 2, the standard errors of the estimated typical structural PK/PD parameters were all estimated with sufficient precision, as suggested by low relative standard errors.
3.4 Model Evaluation
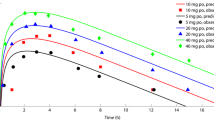
The time courses of mean observed versus population predicted plasma concentrations of SPI-62 after single- or multiple-dose in healthy adults are presented in Fig. 4. Concentration–time profiles of SPI-62 after various single-dose level (1–10 mg) administrations were adequately captured by the final model. This model also provided a good fit for SPI-62 PK profiles in the multiple-dose study, except for a slight overprediction for concentrations measured on day 1 of the 0.7 mg dose group.

Time courses of mean observed (symbols) and population model-predicted (lines) SPI-62 plasma concentrations following 1, 3, 6, and 10 mg single oral doses of SPI-62, as well as 0.2, 0.4, 0.7, and 2 mg oral QD doses of SPI-62. Subjects who received 0.2 mg SPI-62 also received a loading dose of 3 mg. Subjects who received 0.7 mg or 2 mg SPI-62 were administered a single dose of SPI-62, followed by a 6-day washout before the start of 14-day QD dosing. QD once daily
The time course of mean observed versus population predicted SPI-62 concentrations and HSD-1 activity after 0.2, 0.4, 0.7, and 2 mg multiple doses in healthy adults are presented in Fig. 5. The final model adequately characterized the HSD-1 activity time profiles after multiple-dose administrations. There is good agreement between the observed and predicted data, except for slight overprediction for HSD-1 activity on day 1 of both the 0.7 and 2 mg dose groups.

Time courses of mean observed (symbols) and population model-predicted (lines) SPI-62 plasma concentrations and hepatic HSD-1 activity following 0.2, 0.4, 0.7, and 2 mg oral QD doses of SPI-62. Subjects who received 0.2 mg SPI-62 also received a loading dose of 3 mg. Subjects who received 0.7 mg or 2 mg SPI-62 were administered a single dose of SPI-62, followed by a 6-day washout before the start of 14-day QD dosing. QD once daily
Additional goodness-of-fit plots, presented in ESM Fig. S3, further confirmed that the final model described SPI-62 PK and PD adequately at both the population and individual levels. The pcVPC plots, which show the adequacy of the final TMDD-liver PD model, are shown in Fig. 6.

pcVPC of SPI-62 PK and PD. The observed prediction-correct data are represented by blue circles, and the median of prediction-corrected observed and simulated data are represented by the solid red and black lines. The semi-transparent red field represents a simulation-based 95% CI for the median. The observed and simulated 5% and 95% percentiles are represented by dashed red and black lines. The 95% CIs for the corresponding model-predicted percentiles are shown as semi-transparent blue fields. pcVPC prediction-corrected visual predicted check, PK pharmacokinetic, PD pharmacodynamic, CI confidence interval
3.5 Simulation Profiles
To better understand the PK/PD relationship, simulations for PK and PD (HSD-1 activity) under different dose regimens were conducted. The results are shown in Figs. 7 and 8.

Time course of the median, 5th and 95th percentiles of the simulated SPI-62 plasma concentration and hepatic HSD-1 activity in 1000 virtual subjects receiving 0.4, 1, 2, 3, 6, or 10 mg SPI-62 once daily on days 1–14. PK pharmacokinetic, PD pharmacodynamic

Time course of the median, 5th and 95th percentiles of the simulated SPI-62 plasma concentration and hepatic HSD-1 activity in 1000 virtual subjects receiving a 3 mg loading dose on day 1 followed by 0.4 mg or 1 mg SPI-62 QD on days 2–14. QD once daily
Substantial HSD-1 inhibition was predicted for all simulated doses, with full inhibition being achieved when SPI-62 ≥ 2 mg once daily is administered. The inhibitory effect of SPI-62 on hepatic HSD-1 is long lasting, as reflected by the substantial inhibition through 7 days after the last dose. The time to reach maximum PD effect is dose-dependent. For example, it took around 10 days to reach steady-state after a 0.4 once-daily dose and only about 1 day to reach steady-state after a 4 mg once-daily dose. Administration of a loading dose, as shown in Fig. 8, could bring HSD-1 inhibition to its maximum level within the first dose interval. The variability of simulated HSD-1 activity became smaller as SPI-62 dose increased.
4 Discussion
For small molecules exhibiting TMDD with targets located in tissues, although the TMDD model has been developed for several of them [16,17,18], reports on the TMDD-PD model are quite limited, possibly due to the lack of time-course PD data. Extensive time-course hepatic HSD-1 activity data, together with the unusual and complex nonlinear PK of SPI-62, enabled construction of a population TMDD-PD model that adequately captured SPI-62 nonlinear PK and hepatic HSD-1 inhibition following different dose regimens in healthy adults. The final model structure contains a two-compartment TMDD model with three transit absorption compartments for SPI-62 disposition and an inhibitory Imax model for hepatic HSD-1 activity. SPI-62 was estimated to bind to the target with a second-order association rate constant (Kon) of 8.43 nM−1h−1, and dissociate back to free enzyme and drug with a Koff value of 0.229 h−1, indicating a rapid binding and slow dissociation of SPI-62 to its pharmacological target, HSD-1. The estimated target capacity (Rtot) of 5460 nmol corresponds to approximately 2.2 mg of SPI-62, which comports well with the dose range in which PK nonlinearity is prominent. Both the estimated dissociation equilibrium constant (Koff/Kon) of 27.2 pM from the TMDD model component and estimated IC50 of 78.7 pM from the PD model component are consistent with very high SPI-62 potency and observed achievement of substantial HSD-1 inhibition at steady state, with SPI-62 daily doses substantially lower than would be predicted from a measured Ki of 5.3 nM.
Our results provide a compelling example showing that TMDD, mediated by its pharmacological target, does occur in small-molecule compounds. TMDD is a term to describe nonlinear PK that is caused by the interaction between a drug and its pharmacological target [10]. Although TMDD in large-molecule compounds is well accepted, its occurrence in small molecules has not been widely recognized [11, 19, 20]. One reason for this oversight is that mechanistic studies or PD investigation are often not conducted for many small molecules that exhibit TMDD. As a result, whether the observed nonlinear PK are truly caused by the interaction with the drug’s pharmacological target might remain unverified. For small molecules exhibiting TMDD with target located in tissues, they demonstrate linear PK at high doses and substantial nonlinear PK following single low doses, as reflected by unusually low plasma exposures [19, 21]. The explanation for this nonlinear PK behavior is that after a first low dose, a large fraction of the dose is trapped to a high-affinity/low-capacity site so that only a small amount of drug molecules is present in systemic circulation, resulting in extremely low plasma exposures [21]. If this ‘site’ is truly the drug’s pharmacological target, a significant PD effect should be observed following the first dose no matter how low the plasma exposure is. In addition, the PD effect would last long after the last dose since drug molecules bind tightly to the pharmacological target. That is exactly what was observed in SPI-62 in these two phase I clinical trials. For example, more than 40% of hepatic HSD-1 inhibition was observed following a single dose of 0.7 mg SPI-62, when the average Cmax of SPI-62 was only 0.02 ng/mL and concentrations were undetectable at many time points [9]. In addition, inhibition of hepatic HSD-1 persisted even after a 6-day wash out, with > 30% inhibition being observed on day 6. Similarly, full HSD-1 inhibition was observed after repeated doses in all low dose groups (0.2–2 mg), and the inhibition lasted many days after the last dose, even with the lowest dose tested, i.e. 0.2 mg [9]. The TMDD-hepatic PD model that we established adequately characterized all remarkable PK and PD behaviors of SPI-62, such as extremely low plasma exposures following the first low doses, nonlinear PK turned into linear PK after repeated low doses, as well as substantial and long-lasting hepatic HSD-1 inhibition following low doses.
The nonlinear PK behavior of SPI-62 in human is not unique. Other HSD-1 inhibitors, including ABT-384 [17], MK-0916 [22], and BMS-823778 [23], showed strikingly similar nonlinear PK patterns in SAD and MAD trials when drug PK across a wide dose range, starting from a very low dose, were evaluated in healthy adults. For ABT-384 nonlinear PK, we previously performed population PK modeling. The best model was found to be a two-compartment TMDD model with three transit absorption compartments [17], a model structure that is identical to the one we established for SPI-62 in the current analysis. In the ABT-384 MAD trial, daily doses of 1–100 mg were evaluated, but HSD-1 activity was evaluated only prior to the first dose (Day−1) and on the last day of dosing (Day 7 or 14). Although not as informative as time-course HSD-1 activity data available for SPI-62, ABT-384 PD results clearly showed that full hepatic HSD-1 inhibition was achieved following the lowest daily dose of 1 mg ABT-384 [15]. That observation is consistent with the SPI-62 results. These interesting PK and PD behaviors of HSD-1 inhibitors indicate that small-molecule TMDD might be a class effect. The nonlinear PK observed at those low doses should not be ignored as they provide a strong sign of target engagement.
After the final TMDD-PD model was established, we performed comprehensive simulation to predict SPI-62 PK and time course of hepatic HSD-1 activity following various dose regimens, including those that have not been evaluated in the clinical trials. The simulation results showed substantial and long-lasting HSD-1 inhibition in all dose groups evaluated, including the lowest presented daily dose of 0.4 mg; full hepatic HSD-1 inhibition can be achieved following administration of ≥ 2 mg once daily. The time needed to reach maximum HSD-1 inhibition is dose-dependent. For example, it takes around 10 days to reach PD steady state with 0.4 mg SPI-62 once daily, but only 2 days with 2 mg SPI-62 once daily. To rapidly reach maximal HSD-1 inhibition, a loading dose as shown in Fig. 8 should be considered. It is worth pointing out that the simulated work only predicts hepatic HSD-1 inhibition and cannot necessarily be extrapolated to target inhibition of HSD-1 located in other tissues. Work to establish PK/PD models for HSD-1 inhibition by SPI-62 in human brain and adipose is ongoing. When available, the results of the three models together will inform dose selection for future SPI-62 clinical trials. The TMDD-PD modeling and simulation results provide a strong foundation for model-informed development of SPI-62. In addition, since TMDD has been reported to be a class effect of HSD-1 inhibitors [24], the TMDD-PD model that we developed for SPI-62 has potential to be utilized for other members of the class to facilitate interpretation of their PK and PD data as well as future clinical trial design.
5 Conclusions
Due to the complex nonlinear PK and the disconnect between plasma exposure and PD effect, as reflected by the significant HSD-1 inhibition with extremely low or undetectable drug concentrations in plasma, pharmacometrics modeling is important to evaluate the dose-exposure-response relationship. In the current analysis, we established a TMDD-PD model for SPI-62 that adequately captured the complex nonlinear PK of SPI-62 and substantial and durable hepatic HSD-1 inhibition in human.
References
Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, et al. 11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev. 2004;25(5):831–66.
Diederich S, Eigendorff E, Burkhardt P, Quinkler M, Bumke-Vogt C, Rochel M, et al. 11beta-hydroxysteroid dehydrogenase types 1 and 2: an important pharmacokinetic determinant for the activity of synthetic mineralo- and glucocorticoids. J Clin Endocrinol Metab. 2002;87(12):5695–701.
Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006;367(9522):1605–17.
Wang M. The role of glucocorticoid action in the pathophysiology of the metabolic syndrome. Nutr Metab (Lond). 2005;2(1):3.
Sandeep TC, Walker BR. Pathophysiology of modulation of local glucocorticoid levels by 11beta-hydroxysteroid dehydrogenases. Trends Endocrinol Metab. 2001;12(10):446–53.
Weiss AJ, Elixhauser A, Bae J, Encinosa W. Origin of adverse drug events in us hospitals, 2011. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. 2013. https://www.ncbi.nlm.nih.gov/books/NBK169247. Accessed 28 Jul 2022.
Wamil M, Seckl JR. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 as a promising therapeutic target. Drug Discov Today. 2007;12(13–14):504–20.
Sandeep TC, Yau JL, MacLullich AM, Noble J, Deary IJ, Walker BR, et al. 11Beta-hydroxysteroid dehydrogenase inhibition improves cognitive function in healthy elderly men and type 2 diabetics. Proc Natl Acad Sci USA. 2004;101(17):6734–9.
Bellaire S, Walzer M, Wang T, Krauwinkel W, Yuan N, Marek GJ. Safety, pharmacokinetics, and pharmacodynamics of ASP3662, a novel 11beta-hydroxysteroid dehydrogenase type 1 inhibitor, in healthy young and elderly subjects. Clin Transl Sci. 2019;12(3):291–301.
Levy G. Pharmacologic target-mediated drug disposition. Clin Pharmacol Ther. 1994;56(3):248–52.
An G. Concept of pharmacologic target-mediated drug disposition in large-molecule and small-molecule compounds. J Clin Pharmacol. 2020;60(2):149–63.
Wu N, Katz DA, An G. A Target-mediated drug disposition model to explain nonlinear pharmacokinetics of the 11beta-hydroxysteroid dehydrogenase type 1 inhibitor SPI-62 in healthy adults. J Clin Pharmacol. 2021;61(11):1442–53.
Choi KB. Hypertensive hypokalemic disorders. Electrolyte Blood Press. 2007;5(1):34–41.
Finken MJ, Andrews RC, Andrew R, Walker BR. Cortisol metabolism in healthy young adults: sexual dimorphism in activities of A-ring reductases, but not 11beta-hydroxysteroid dehydrogenases. J Clin Endocrinol Metab. 1999;84(9):3316–21.
Liu W, Katz DA, Locke C, Daszkowski D, Wang Y, Rieser MJ, et al. Clinical safety, pharmacokinetics, and pharmacodynamics of the 11beta-hydroxysteroid dehydrogenase type 1 inhibitor ABT-384 in healthy volunteers and elderly adults. Clin Pharmacol Drug Dev. 2013;2(2):133–51.
Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507–32.
An G, Liu W, Katz DA, Marek GJ, Awni W, Dutta S. Population pharmacokinetics of the 11beta-hydroxysteroid dehydrogenase type 1 inhibitor ABT-384 in healthy volunteers following single and multiple dose regimens. Biopharm Drug Dispos. 2014;35(7):417–29.
Wu N, Hammock BD, Lee KSS, An G. Simultaneous target-mediated drug disposition model for two small-molecule compounds competing for their pharmacological target: soluble epoxide hydrolase. J Pharmacol Exp Ther. 2020;374(1):223–32.
An G. Small-molecule compounds exhibiting target-mediated drug disposition (TMDD): a minireview. J Clin Pharmacol. 2017;57(2):137–50.
van Waterschoot RAB, Parrott NJ, Olivares-Morales A, Lave T, Rowland M, Smith DA. Impact of target interactions on small-molecule drug disposition: an overlooked area. Nat Rev Drug Discov. 2018;17(4):299.
Bach T, Jiang Y, Zhang X, An G. General pharmacokinetic features of small-molecule compounds exhibiting target-mediated drug disposition (TMDD): a simulation-based study. J Clin Pharmacol. 2019;59(3):394–405.
Wright DH, Stone JA, Crumley TM, Wenning L, Zheng W, Yan K, et al. Pharmacokinetic-pharmacodynamic studies of the 11beta-hydroxysteroid dehydrogenase type 1 inhibitor MK-0916 in healthy subjects. Br J Clin Pharmacol. 2013;76(6):917–31.
Gong J, Hansen L, Iacono L. Clinical pharmacokinetics and the impact of genetic polymorphism on a CYP2C19 substrate, BMS-823778, in healthy subjects. Drug Metab Dispos Biol Fate Chem. 2018;46(3):316–25.
An G, Katz DA. Importance of target-mediated drug disposition (TMDD) of small-molecule compounds and its impact on drug development-example of the class effect of HSD-1 inhibitors. J Clin Pharmacol. 2023;63(5):526–38.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The work was supported by Sparrow Pharmaceuticals, Inc.
Conflicts of interest
David A. Katz is a Sparrow Pharmaceuticals employee, officer, and shareholder, and Guohua An is a consultant of Sparrow Pharmaceuticals, Inc. Nan Wu declares no potential conflicts of interest that might be relevant to the contents of this manuscript.
Authors’ contributions
Conception and design of the work: GA, DAK. Analysis of the data: NW, GA. Interpretation of the data: GA, NW, DAK. Drafting and critical revision of the manuscript: GA, NW, DAK.
Ethics approval
Not applicable as the current work was a secondary data analysis.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Data availability statement
The datasets generated and analyzed during the current study are proprietary and will not be made available to other investigators.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Wu, N., Katz, D.A. & An, G. Population Target-Mediated Pharmacokinetic/Pharmacodynamic Modeling to Evaluate SPI-62 Exposure and Hepatic 11β-Hydroxysteroid Dehydrogenase Type 1 (HSD-1) Inhibition in Healthy Adults. Clin Pharmacokinet 62, 1275–1288 (2023). https://doi.org/10.1007/s40262-023-01278-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01278-8