
Abstract
Extracorporeal membrane oxygenation (ECMO) is an established advanced life support system, providing temporary cardiac and/or respiratory support in critically ill patients. Fungal infections are associated with increased mortality in patients on ECMO. Antifungal drug dosing for critically ill patients is highly challenging because of altered pharmacokinetics (PK). PK changes during critical illness; in particular, the drug volume of distribution (Vd) and clearance can be exacerbated by ECMO. This article discusses the available literature to inform adequate dosing of antifungals in this patient population. The number of antifungal PK studies in critically ill patients on ECMO is growing; currently available literature consists of case reports and studies with small sample sizes providing inconsistent findings, with scant or no data for some antifungals. Current data are insufficient to provide definitive empirical drug dosing guidance and use of dosing strategies derived from critically patients not on ECMO is reasonable. However, due to high PK variability, therapeutic drug monitoring should be considered where available in critically ill patients receiving ECMO to prevent subtherapeutic or toxic antifungal exposures.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Robust guidelines for dosing of antifungal drugs in critically ill adult patients receiving ECMO are lacking. |
Most ECMO pharmacokinetic studies are limited in that they do not include a non-ECMO comparator group although pharmacokinetic variability of antifungals is very high relative to other patient groups. |
Dosing should be optimised using therapeutic drug monitoring to avoid therapeutic failure as well as drug accumulation and toxicity. |
1 Introduction
Extracorporeal membrane oxygenation (ECMO) is an established advanced life support system which allows for prolonged temporary cardiopulmonary support in patients with life-threatening respiratory and/or cardiac failure refractory to maximal medical management [1, 2]. On its own, ECMO is not a disease-modifying intervention but a supportive therapy by providing temporary cardiorespiratory support while underlying pathologies, such as infection, are evaluated and treated. ECMO can also be used to bridge the patient to either organ recovery or lung or heart transplantation or to a long-term ventricular assist device. The two most common modalities of conventional ECMO are venoarterial (VA) or venovenous (VV) ECMO. In both modalities, the blood is drained from the venous system and oxygenated outside the body through an oxygenator. In VV ECMO, the blood is returned to the venous system driven by the patients’ own heart function, thereby maintaining pulsatile blood flow, providing respiratory support only, while in VA ECMO the blood is returned to the arterial system, bypassing both the heart and the lungs, hence supporting both respiratory and cardiac function.
ECMO is a highly invasive treatment with a number of associated complications, including infections, bleeding, thrombosis and renal failure [3]. Infection is a significant problem in this patient group and is associated with increased mortality and morbidity, especially in patients with sepsis [4,5,6]. Critically ill patients with ECMO support are at a higher risk of developing nosocomial infection [7,8,9], and this increases with prolonged duration of ECMO support [4, 5, 10]. The prevalence and incidence of hospital-acquired and bloodstream infections during ECMO ranges from 10–12% in registry data to 9–65% in single-centre studies [3, 4, 6, 7, 10,11,12,13,14]. These commonly are bacterial infections, including those caused by coagulase-negative staphylococci, Pseudomonas aeruginosa and Enterobacteriaceae, but also include life-threatening fungal infections caused by Aspergillus spp. and Candida spp. [15, 16]. Early work from the Extracorporeal Life Support Organisation registry found a higher mortality in neonates on ECMO with fungal as compared with bacterial infections (57.7 versus 30.6%, p = 0.007) [17]. Fungal infections, aspergillosis and candidaemia in patients on ECMO have been associated with increased mortality [15, 16, 18, 19].
Critically ill patients can manifest altered pharmacokinetics (PK) such as increased volume of distribution (Vd) and altered clearance (CL) with many drugs [20, 21]. ECMO may also further exacerbate these PK changes; ex vivo experiments have suggested that drugs which are lipophilic and are highly protein bound are likely to be sequestered within the ECMO circuit [22]. Acute kidney injury (AKI) during ECMO is associated with a poor outcome, with approximately 50% of patients requiring concomitant renal replacement therapy (RRT) [23]. In critically ill patients receiving RRT, no dosing adjustment is required for most antifungal agents commonly used to treat invasive fungal infections except for fluconazole, itraconazole and flucytosine [24]. Estimating antifungal PK changes in those patients receiving both ECMO and RRT support can be challenging. It is not simply additive, as the effects may cancel each other and are dependent on the drug’s physicochemical properties. It is crucial that critically ill ECMO patients receive the right antifungal dosing regimen; suboptimal dosing strategies can lead to both underdosing and supratherapeutic concentrations causing potential antifungal resistance and adverse drug effects, respectively [20]. Most antifungal drugs are lipophilic and/or highly protein bound, hence raising concerns regarding their ability to reach their target pharmacokinetic/pharmacodynamic (PK/PD) exposure which may in turn affect therapeutic outcomes in patients receiving ECMO.
The aims of this review are to discuss the potential impact of ECMO on the PK of antifungals and to describe how it may influence drug dosing requirements in critically ill adult patients receiving ECMO. Optimised antifungal dosing regimens in this patient population are also suggested based on available literature.
2 Methods
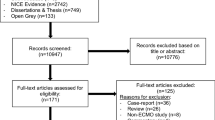
We performed a structured literature review by searching PubMed (1988–July 2022). Search terms included “pharmacokinetics AND/OR dosing”, “Extracorporeal Membrane Oxygenation AND/OR ECMO”, AND/OR “antifungal” AND/OR “fluconazole” AND/OR “itraconazole” AND/OR “isavuconazole” AND/OR “posaconazole” AND/OR “voriconazole” AND/OR anidulafungin” AND/OR caspofungin” AND/OR micafungin” AND/OR amphotericin”. Articles included in vitro and ex vivo studies, clinical PK studies and case reports. Animal studies and studies in languages other than English were excluded. References from searched articles and other research known to authors were also considered if they met the above criteria.
3 Pharmacokinetic Determinants During ECMO
There are several patient factors related to critical illness and ECMO which can affect the PK of drugs, and these include renal or hepatic impairment, and alterations in fluid regulation [20]. The ECMO circuit consists of five main components: large arterial and venous cannulas, gas exchange membrane oxygenator, blood tubing, heat exchanger and a pump. The ECMO circuit is primed with combination of crystalloid, albumin and blood. Essentially, the addition of an ECMO circuit may further alter the PK in a critically ill patient, increasing the apparent Vd as a result of drug sequestration onto the ECMO circuit, and altering drug CL due to changes in renal and liver blood flow and altered plasma protein binding [25, 26].
3.1 Increased Apparent V d
3.1.1 Critical Illness-Related Physiological Changes
Patients with critical illness often have a number of physiological changes, including fluid shifts, altered blood pH, organ dysfunction and systemic inflammatory response syndrome (SIRS). These changes are associated with an apparent increase in the volume of distribution (Vd) of hydrophilic drugs [20]. Secondary to drug sequestration to the circuitry and haemodilution from the priming solution, the inclusion of an ECMO circuit could further increase the apparent Vd in critically ill patients, especially in neonates and young infants [25, 27, 28].
3.1.2 Drug Sequestration
Data suggests that certain drugs have the potential to be sequestered onto the ECMO circuit, likely due to the large surface area of the membrane oxygenator and tubing that drug can be adsorbed on [25, 26, 29]. The extent of drug loss or sequestration is dependent on both the physicochemical properties of the drug and factors related to the circuit itself [26, 29,30,31,32]. The degree to which a drug is sequestered can vary depending on several factors, such as the type of oxygenator [29, 33], tubing [29, 34] and pump used in the ECMO circuit [26], as well as the age of the circuit [35], and the priming solution used [36]. Hence, the same drug may exhibit different levels of sequestration under different conditions. In vitro and ex vivo experiments have shown lipophilic action, and highly protein-bound drugs are significantly sequestered to a greater degree in the circuit [31, 32]. As the binding sites on the circuit become saturated with drugs, the sequestration effect may diminish over time. However, it remains unclear whether the ECMO circuit may function as a drug reservoir, with drugs potentially leaching back into the circulation understood [37].
Importantly, over the years the improvement of ECMO circuits has decreased the complications associated with ECMO use, such as clotting. Also, the increasing use of other therapies that may result in drug sequestration such as concomitant cytokine absorption therapies, e.g., Cytosorb® and/or RRT with ECMO need to be studied to understand any impact on drug PK.
3.1.3 Haemodilution
Hydrophilic drugs with low Vd (e.g. fluconazole) are more affected by haemodilution than lipophilic drugs with a larger apparent Vd (e.g. posaconazole) with the effect being more significant in neonates and infants, as this volume of fluid required to be added to the circuit equates to a greater proportion of their circulating blood volume [38].
3.2 Altered Drug CL
While renal dysfunction is common in critically ill patients on ECMO support, the reasons are multifactorial but likely in part to be secondary to reduced renal perfusion and hypoxia prior to ECMO insertion. A recent meta-analysis showed no significant difference in the risk of developing AKI or severe AKI between those critically ill patients on VV and VA ECMO [23]. Additionally, the SIRS response can decrease the expression and function of drug metabolising enzymes, such as the cytochrome P450 (CYP450) enzymes [39, 40], and in combination with reduced hepatic perfusion and hypoxia, these phenomena can lead to decreased hepatic elimination of drugs.
4 Specific Antifungal Classes
Most of the reviewed data originated from ex vivo experiments, case reports and observational studies. Figure 1 summarises the interplay between antifungal drug, patient and fungi factors and their impact on antifungal PK and dosing in critical illness and ECMO.

Pharmacokinetic changes in critically ill patients receiving ECMO and alternative dosing strategies
4.1 Azoles
Azoles inhibit ergosterol synthesis, which is a major component of fungal cell membrane, affecting fungal cell growth and proliferation. All except fluconazole have antifungal activity against yeasts and moulds, with fluconazole being limited to yeasts. There is variability in PK amongst the azoles (Table 1) which in turn may influence both the choice of drug and dosing in critically ill patients. All are available as oral and intravenous (IV) formulations. Azole agents exhibit both concentration- and time-dependent anti-fungal activity with a prolonged post-antifungal effect (PAFE). As such, the ratio of the area under the concentration–time curve during a 24-h period to minimum inhibitory concentration (AUC0–24/MIC) is considered the predictive PK/PD index associated with maximal antifungal activity. Common adverse effects include liver toxicity and QT-prolongation (except with isavuconazole), and they all exhibit to different degrees drug–drug interactions via the cytochrome P450 enzyme system and/or P-glycoprotein transporters. Fluconazole [41, 42], isavuconazole [43, 44], posaconazole [45] and voriconazole [46, 47] PK have been studied in critically ill patients on ECMO support.
4.1.1 Fluconazole
Fluconazole is hydrophilic, has low protein binding with a Vd of 0.7L/kg and high oral bioavailability of > 90% (Table 1). It is mainly excreted unchanged via the kidneys, thereby necessitating dose reduction in patients with renal impairment. However, fluconazole CL is increased in those patients on RRT with subsequent increased dosing recommended [48, 49]. Significant inter-individual variability was seen in a heterogeneous cohort of critically ill patients [50], with mean (± SD) AUC0–24 of 359 ± 259 compared with 608 ± 118 reported in healthy volunteers [50], and 33% of patients not achieving the desired PK/PD index for maximal efficacy [51].
4.1.1.1 Ex Vivo
In an ex vivo study, Watt et al. studied fluconazole in three closed-loop paediatric circuits isolating the influence of the oxygenator, haemofilter and tubing on circuit sequestration. This study showed that fluconazole was not sequestered into the ECMO circuits; the recovery of fluconazole in the complete circuit was 97.8% at 4 h and 100.2% in the controls. In the circuits which ran up to 24 h, the recovery was high in both the ECMO circuits and controls, 95.2 and 100.6%, respectively, at 24 h [27]. These were similar to the findings in the ex vivo study reported by Shekar et al. using adult circuits, where mean recovery was 91% in the ECMO circuit and 102% in the controls [22].
4.1.1.2 Clinical PK Studies/Case Reports
Watt et al. studied the PK of fluconazole in children on ECMO using fluconazole samples from three prospective trials; a total of 40 children were included, of which 21 were on ECMO. The difference in fluconazole concentrations in ECMO compared with those without was related to the increased Vd secondary to blood volume needed to prime the ECMO circuits in addition to the increased Vd due to critical illness [42].
In the Antibiotic, Sedative and Analgesic Pharmacokinetics during ECMO (ASAP ECMO) study, Shekar et al. described the PK of fluconazole, given at 400–800 mg per day dosing, in 10 critically ill patients receiving ECMO, of whom 8 were on concomitant RRT support. Marked variability [coefficient of variation (CV) of ≥ 30%] in fluconazole PK parameters was reported. In this study, patients who received RRT demonstrated lower fluconazole minimum blood plasma concentration (Cmin; 6.03 versus 20.15 mg/L), AUC0–24 (206.70 versus 592.14 mg·h/L) and higher CL (3.46 versus 1.08 L/h) when compared with those not on RRT. Although patients receiving VV ECMO support demonstrated lower AUC0–24 (95.45 versus 286.50 mg·h/L) when compared with VA ECMO patients, these observations could have also been influenced by concomitant RRT use, as all VV ECMO patients received continuous RRT during PK sampling. Only one patient did not achieve the PK/PD target in this analysis (AUC0–24/MIC ≥ 100 against C. albicans) [52].
Dhanani et al. reported the PK of IV fluconazole in an adult patient on VV ECMO. At the administered dose of fluconazole 6 mg/kg daily, the trough concentration and AUC0–24 met the PK/PD target for prophylaxis despite the 40% increase in Vd observed compared with critically ill adults not on ECMO [41].
4.1.2 Isavuconazole
Isavuconazole is the newest agent in this class, having extended spectrum of activity with yeast and moulds, including the zygomycetes family. Isavuconazonium sulphate is a water-soluble prodrug which undergoes rapid hydrolysis to the active moiety isavuconazole, which is highly lipophilic [octanol–water partition coefficient (LogP) of 3.6], and protein bound (> 98%) with a large Vd of 450 L indicating wide distribution throughout the body (Table 1). It is available in both IV and capsule formulations, having high bioavailability (> 98%). As the prodrug is water soluble, there is no requirement of solubilisation by a cyclodextrin vehicle, in contrast to itraconazole, voriconazole and posaconazole. Also, of note, it is both a substrate and moderate inhibitor of the CYP enzymes and has linear PK, and oral absorption is not influenced by food.
4.1.2.1 Ex-Vivo
There are no published ex vivo studies.
4.1.2.2 Clinical PK Studies/Case Reports
Isavuconazole concentrations were studied in a mixed cohort of patients with invasive fungal conditions; 33 courses in 32 patients were included, and 11 patients were on renal replacement therapy, of which 4 patients were additionally on extracorporeal treatments (3 on ECMO and 1 Cytosorb® adsorber therapy) [44]. The authors found that isavuconazole concentrations were 3.05 mg/L [interquartile range (IQR) 1.93–4.35] in patients without RRT or ECMO but were significantly lower in those patients with RRT and 0.91 mg/L (IQR 0.9–1.36) and in those with RRT and additional ECMO± Cytosorb®, 0.88 mg/L (IQR 0.71–1.21). However, the sample size was small, and the authors proposed the use of therapeutic drug monitoring (TDM) to guide dosing in this patient population. Subsequently, a prospective observational PK study by Kriegl et al. included seven adult critically ill patients who received isavuconazole—six for prophylaxis while on VV ECMO and one for treatment of invasive aspergillosis while on VA ECMO. Isavuconazole blood concentrations were taken at a number of timepoints from 2 h up to 168 h after the first dose was administered, with additional samples from three patients taken from the inflow and outflow lines to determine whether there were any direct effects from the ECMO. Median isavuconazole concentrations were > 1 mg/L after the first dose and > 2 mg/L at 96 h, suggesting that the higher Vd secondary to critical illness and ECMO may have contributed to the lower early concentrations. The steady-state concentrations pre- and post-membrane oxygenator in addition to plasma concentrations all correlated well; hence, isavuconazole was not altered by the ECMO oxygenator [43].
There are two case reports of isavuconazole PK during ECMO, one in VV ECMO for invasive aspergillosis and the other in conjunction with liposomal amphotericin B in VA ECMO for blastomycosis, which required a two-fold increase in dose to achieve target concentration of 2–4 and > 3 mg/L, respectively [53, 54]. Mertens et al. reported their mixed experience in four VV ECMO patients receiving isavuconazole; two of the patients achieved the target concentration of > 1 mg/L and two did not using standard maintenance dosing regimen of 200 mg daily [55].
4.1.3 Posaconazole
Posaconazole, like isavuconazole, is very lipophilic (LogP 5.5) and highly protein bound (> 98%) with a very large Vd of 1774 L (Table 1). It was originally available as a suspension formulation only, although it is now available as delayed-release oral tablets and IV formulations with improved PK. Like voriconazole, the IV formulation contains sulfobutylether-beta-cyclodextrin (SBECD) to facilitate solubilisation of the lipophilic drug. It is excreted unchanged in the faeces; hence, no renal and liver dose adjustments are required. It is not a substrate of the CYP but is of the P-glycoprotein transporter system. It is a potent inhibitor of the CYP3A4 enzymes, and therefore is associated with many drug–drug interactions.
4.1.3.1 Ex-Vivo
Lyster et al. conducted an ex vivo study investigating whether three antifungals were prone to circuit sequestration. The authors found that posaconazole exhibited significant sequestration in blood primed ECMO circuit of 63.6% compared with 11.4% (p < 0.005) in the controls at 24 h [56].
4.1.3.2 Clinical PK Studies/Case Reports
A prospective multicentre study to determine the PK and PK/PD target attainment of six adult patients who were treated with IV posaconazole while on ECMO found all trough concentrations were ≥ 0.7 mg/L and 11 of the 16 were ≥ 1 mg/L. The probability of target attainment analysis supported the observed results of prophylaxis, although less than 90% of simulated patients achieved the lower treatment threshold of 1 mg/L. The authors observed that ECMO did not influence posaconazole PK as much as had been hypothesised [45, 57]. They did find a gradual increase in posaconazole concentrations after multiple administrations, suggesting that it could be due to saturation of the ECMO circuit concluding that an a priori dose adjustment was not needed, but the dose should be guided by TDM.
4.1.4 Voriconazole
Voriconazole is lipophilic, (LogP 2.56) and moderately protein bound (58%), with a Vd of 4.6 L/kg (Table 1). It is available in both oral and IV formulations, with high oral bioavailability (> 90%). It is extensively metabolised in the liver; hence, no dosage adjustments are required in patients with renal dysfunction or receiving RRT [58]. The IV formulation contains SBECD to facilitate solubilisation of voriconazole. Originally its use was considered contraindicated in those patients with a creatinine clearance (CrCL) < 50 mL/min or on RRT, due to concerns of SBECD accumulation and subsequent kidney damage; however, this has shown not to be clinically relevant in both an animal [59] and human studies [60, 61]. It is both a substrate and potent inhibitor of the CYP3A4 enzymes; hence, drug interactions may be challenging. In addition, voriconazole exposure is affected by genetic polymorphism of CYP2C19 and exhibits nonlinear PK and high intra- and interpatient variability, and therefore, there is a clear recommendation for TDM when it is used in critically ill patients [62].
4.1.4.1 Ex-Vivo
There have been several ex vivo studies investigating potential voriconazole sequestration in ECMO circuits [33, 36, 56, 63, 64]. Mehta et al. reported a voriconazole loss of 71 versus 14.8% at 24 h using older style neonatal ECMO circuits with silicone and polyvinyl chloride materials involved [36]. Recently, Raffaeli et al. explored the extraction of voriconazole by the Xenios/Novalung ECMO circuits, finding the mean percentage recovery of 20% at 24 h, and Zhang et al. reported average recoveries of 60 and 101% [33]. Cies et al. demonstrated a significant loss of voriconazole in the ECMO circuits with an oxygenator in the circuit and no significant loss without one [63]. While Lyster et al. reported sequestration of 27% in the ECMO model versus 19.2% in controls [56], this is in contrast with previously published studies with results comparable to those reported by Cies et al. in the circuits without an oxygenator. The results suggest that differences may be attributable to differences in materials used for tubing and oxygenators.
4.1.4.2 Clinical PK Studies/Case Reports
A large multicentre retrospective study by van Daele et al. assessed voriconazole exposure in 69 patients with ECMO [46]. Of the 337 samples taken, there was no significant difference in subtherapeutic voriconazole concentrations (< 2 mg/L) during (57% of samples) and before/after (39% of samples) ECMO therapy. There was significant inter- and intra-patient PK variability, and no independent effect of the ECMO was observed [46]. However, Ye et al. conducted a large single-centre retrospective observational cohort study of 132 patients, of which 66 were on ECMO support. In this study, ECMO patients demonstrated significantly lower median trough concentrations when compared with non-ECMO patients (1.9 versus 4.4 mg/L, p < 0.001). Additionally, the proportion of sub-therapeutic concentrations (< 2 mg/L) was significantly higher in the ECMO group when compared with the non-ECMO group (51.5 versus 7.6%, p < 0.001) [47].
In the ASAP ECMO study, Shekar et al. described the PK of voriconazole, 200 mg twice daily, in a single patient on VV ECMO support, where the PK/PD target of Cmin ≥ 2 mg/L was not achieved [52]. Reduced voriconazole exposure requiring dosing escalation in ECMO patients has been documented in several case reports [37, 65,66,67,68,69]. Spriet et al. pre-emptively increased the voriconazole dosing in their two patients when commencing ECMO, as they anticipated a loss to the circuit. Trough levels > 10 mg/L were observed 2 days later, suggesting possible saturation of the binding sites on the ECMO circuits [37]. Lin et al. described a case of a patient who received voriconazole and had two PK profiles taken before and after initiation of ECMO; while the trough concentrations were slightly lower while on ECMO, the voriconazole exposure as determined by area under the curve (AUC) were similar on and off ECMO [70]. Peterson et al. reported decreased dosing requirement in a patient following discontinuation of ECMO therapy, suggesting in addition to TDM consideration of empiric dose reduction of 40–50% of voriconazole dose when coming off ECMO [65].
4.2 Echinocandins
Echinocandins exert their antifungal effect by inhibiting the synthesis of the polysaccharide 1,3 beta-d-glucan, causing fungal cell wall degradation and cell lysis. They have broad fungicidal activity against Candida spp. and fungistatic activity against most Aspergillus spp. While this antifungal class has varying lipophilicity characteristics, they all exhibit very high protein binding (Table 2), which raises concerns of circuit sequestration in patients receiving ECMO. They are available only as IV formulations. Their long half-life facilitates once-daily dosing, and they have very low excretion unchanged by the kidneys. They exhibit concentration-dependent fungicidal activity against Candida spp. and have prolonged PAFE, with both Cmax and AUC0–24/MIC associated with optimum activity.
4.2.1 Anidulafungin
4.2.1.1 Ex-Vivo
There are no published ex vivo studies.
4.2.1.2 Clinical PK Studies/Case Reports
Aguilar et al. reported the case of a single adult patient on ECMO and Novalung iLA ActivveTM treated with anidulafungin at usual maintenance dose of 100 mg daily. All PK parameters were comparable to published data in critically ill patients without extracorporeal support [71] supporting that ECMO had minimal impact on the PK of anidulafungin [72].
4.2.2 Caspofungin
4.2.2.1 Ex-Vivo
In an ex vivo study, Shekar et al. reported a lower average caspofungin recovery at 24 h in ECMO circuits compared with controls (56 versus 99%, p = 0.001) [22]. Another ex vivo study reported a similar loss from blood primed ECMO circuits and controls of 80 and 61%, respectively [56]. However, contrary to these findings, Zhang et al. found no significant sequestration with average drug recovery at 24 h of 80 and 85% in the ECMO circuit and control group, respectively [64].
4.2.2.2 Clinical PK Studies/Case Reports
Wang et al. conducted a prospective single-centre study describing the PK of caspofungin in patients with or without ECMO during the postoperative period of lung transplantation [73]. Twelve ECMO and seven non-ECMO patients were enrolled. The PK of ECMO patients were compared with non-ECMO patients on the second dose of caspofungin. All 12 ECMO patients were weaned on the second day after surgery and became self-controls with PK data from the third dose. There were no significant differences in PK parameters between ECMO and non-ECMO groups [73].
In the ASAP ECMO study, Shekar et al. described the PK of caspofungin in nine critically ill patients receiving ECMO, of whom six were on concomitant RRT support. The authors found that caspofungin PK parameters were similar between VA and VV ECMO patients, and between those who received and did not receive RRT. Additionally, like the results with fluconazole, large variations (CV of ≥ 30%) in all PK parameters were observed. The authors reported that PK parameters were generally consistent with previously published data of critically ill patients without ECMO support. Only one patient did not achieve the PK/PD target in this analysis (AUC0–24/MIC ≥ 865 against C. albicans) [52].
The PK of caspofungin has been described in three earlier case reports with contradictory results [37, 66, 74]. Koch et al. described subtherapeutic caspofungin AUC and increased CL in an 11-month-old receiving VV ECMO for the treatment of Candida albicans and Candida tropicalis despite receiving a higher dosage of 78 mg/m2 daily compared with standard dosing of 50–70 mg/m2 daily [74]. Meanwhile, Spriet et al. reported two adult cases on ECMO, one of whom received caspofungin for the treatment of Candida albicans and achieved adequate caspofungin concentrations [37]. However, another adult case reported undetectable caspofungin blood concentrations using standard dosing of 50 mg daily (patient weighed 50 kg) [66].
4.2.3 Micafungin
4.2.3.1 Ex-Vivo
In an ex vivo study, Watt et al. separately studied micafungin in three closed-loop paediatric circuits isolating the influence of the oxygenator, haemofilter and tubing on circuit sequestration. This study showed that the loss was dependent on the time (4 and 24 h) as well as the circuit configuration, with 91% micafungin recovery at 4 h when there was no haemofilter compared with 46% with one in-line [27]. However, by 24 h micafungin recovery was 26–43% in all configurations and 57% recovery in the controls. In another ex vivo study, Zhang et al. demonstrated that the recovery of micafungin was higher, at 67% in the Sorin ECMO circuit and 99% in the control at 24 h [64].
4.2.3.2 Clinical PK Studies/Case Reports
Micafungin use has been studied in neonates and children. Autmizguine et al. reported a PK study with micafungin in 12 infants, 11 of whom received a prophylactic dose of 4 mg/kg/day and a treatment dose of 8 mg/kg/day. In this infant cohort, the PK model demonstrated a 20–90% higher Vd and a CL in the upper range of historical controls of infants not on ECMO [75]. To match adult exposure proven effective against Candida spp., the group recommended dosing of 2.5 mg and 5 mg/kg/day for prophylaxis and treatment, respectively [75].
Lopez-Sanchez et al. reported the PK of micafungin in 12 adult patients on ECMO. The PK values on day 1 and day 4 showed no difference between the samples taken before the membrane and those taken after, including Cmax, Cmin, Vd and CL [76]. Additionally, there was no significant difference with the AUC in samples taken before and after membrane, and on days 1 and 4.
In contrast, a post hoc analysis of the Empiricus randomised control trial [77], comparing the PK of first dose micafungin between those patients on ECMO and those without, showed that the AUC of micafungin was reduced by 23% in ICU patients on ECMO, suggesting a dose increase in this patient population [78].
A case report of micafungin described the successful treatment of a Candida glabrata fungaemia in an adult critically ill patient receiving VV ECMO and continuous renal replacement using the higher dose of 150 mg every 24 h [79].
4.2.4 Amphotericin
Amphotericin is a polyene antifungal agent with a broad spectrum of activity against yeast and moulds. It binds to ergosterol in the fungal cell membrane leading to disruption of the membrane structure, ultimately causing cell death. The original amphotericin deoxycholate formulation is associated with common nephrotoxicity and infusion-related reactions which led to the development of less toxic formulations such as liposomal amphotericin B (LAmB), allowing for higher dosage. It is only available as an IV formulation for systemic therapy, and no dosing adjustment is required for liver or renal dysfunction. LAmB is a concentration-dependent antifungal where Cmax/MIC > 40 has been linked to therapeutic effectiveness [80]. LAmB exhibits a nonlinear PK profile over the dosage range 7.5–15 mg/kg/day where the maximum serum concentration and AUC achieved an upper limit at 10 mg/kg/day. The AmBiLoad and AmbiZygo trials evaluated doses of 10 mg/kg/day; they observed no efficacy or mortality benefit but significantly increased incidence of nephrotoxicity [81, 82]. Amphotericin B deoxycholate is slightly lipophilic with a LogP 0.8, while lipid formulations such as LAmB are highly lipophilic; hence, there is potential for sequestration within ECMO circuit (Table 3).
4.2.4.1 Ex-Vivo
An early in vitro study suggested that, in the first 10–14 h on an ECMO circuit, more than 10% loss in liposomal amphotericin concentration occurs [83].
An ex vivo study found no loss of amphotericin B deoxycholate at 4 or 12 h in the complete ECMO circuit configuration, with recovery of 98.9% (88.7, 109.1) and 99.2% (88.5, 110.1), respectively [84].
4.2.4.2 Clinical PK Studies/Case Reports
The use of LAmB in ECMO has been described in a few case reports with contradictory findings. Hertzog et al. reported the use of conventional amphotericin deoxycholate in a 15-year old patient receiving ECMO with acute respiratory distress syndrome secondary to pulmonary blastomycosis. Amphotericin concentrations were measured prior to dose and 1 h post-completion of IV infusion and when there was a circuit change. The authors found no significant changes in the PK of amphotericin secondary to the ECMO circuit, with concentrations remaining within the therapeutic range using standard dosing of 1 mg/kg daily [85].
Subsequent reports have used LAmB which has different properties and PK from conventional amphotericin B, in particular in that it is more lipophilic. Case reports of LAmB for the treatment of invasive aspergillus in patients on ECMO have suggested that the PK was not altered, with concentrations observed in the therapeutic range [66], and that PK parameters were like those in critically ill patients not on ECMO [86]. In contrast, other case reports have demonstrated significant loss of LAmB to the ECMO circuit, necessitating increased dosing to 10 mg/kg daily in one [54] and switching to conventional amphotericin B deoxycholate in another [87], both in the management of blastomycosis in adult patients on ECMO.
5 Therapeutic Drug Monitoring (TDM)
As most drug dosing studies are conducted in healthy volunteers, extrapolating these recommendations to other patient groups such as critically ill patients on ECMO does not take into consideration pathophysiological and PK differences. Another challenge seen is lower pathogen susceptibility in the critical care setting, with higher MICs influencing PK/PD target attainment. TDM was traditionally used to minimise drug adverse effects, e.g., when using anti-infective agents with narrow therapeutic windows, but its use has now expanded to optimise efficacy. Individualised dosing using TDM is a tool which can be used individually or together with dosing software. In a recent conference report and expert panel position paper, antifungal TDM in critically patients has been shown to be of clinical benefit in those where a therapeutic target has been established, such as voriconazole [62]. Although the panel’s recommendation is to “neither recommend or discourage” for several other antifungal drugs [62], due to significant PK/PD variability in this population, routine TDM would be advantageous, where available, to guide dosing to prevent sub-optimal exposure and to minimise the likelihood of adverse events during ECMO. The need for antifungal dose optimisation using TDM in critically ill patients is well described in a review by Baracaldo-Santamaria et al. [88].
6 Conclusions and Future Directions
There is still a lack of well-established guidelines for dosing antifungal drugs in critically ill adults who are on ECMO. This emphasises the need for a comprehensive approach that includes additional clinical pharmacokinetic studies to identify all the covariates contributing to the variability seen. Most observational studies are limited in that they do not include a non-ECMO comparator group, with comparison based on PK studies in other populations, not on ECMO support. Another limitation in the studies is total concentrations are measured; often unbound drug concentrations are not feasible, as protein binding can change over time in critically ill patients and between individuals, assuming a protein binding may translate into error. The PK studies did not report antifungal drug-related adverse effects; these are scarcely reported in and investigated in critically ill, including ECMO, patients, and should be included in future studies. The challenge of establishing appropriate dosing is exacerbated by the physiological changes that accompany critical illness, including inflammation and hepatic and renal dysfunction (including the use of RRT). It is crucial to understand the complex interactions between these factors.
The physicochemical properties of antifungal drugs can be used to anticipate PK changes: ex vivo studies have shown that drugs with high protein binding (e.g., > 70%) and that are highly lipophilic (e.g., Log P > 2) are likely to be sequestered on the circuit; hence, increased loading or maintenance dosing may be required [89]. In contrast, recent comprehensive reviews of clinical PK studies of antimicrobial dosing in ECMO concluded that most PK changes are more reflective of critical illness rather than the ECMO device. The ASAP ECMO study, authors have shown that the high PK variability in this patient group (CV ± 30%) may result in patients receiving sub-therapeutic antifungal drug exposures, the clinical consequence of which is uncertain. Therefore, it is acceptable to use dosing approaches that are based on critically ill adult patients who are not receiving ECMO. However, it is important to include the organism variable PD susceptibility and optimise these approaches through TDM to avoid treatment failure, drug accumulation and potential toxicity.
References
Bartlett RH. Extracorporeal life support: history and new directions. ASAIO J. 2005;51(5):487–9.
Bartlett RH, Gattinoni L. Current status of extracorporeal life support (ECMO) for cardiopulmonary failure. Minerva Anestesiol. 2010;76(7):534–40.
Zangrillo A, Landoni G, Biondi-Zoccai G, Greco M, Greco T, Frati G, et al. A meta-analysis of complications and mortality of extracorporeal membrane oxygenation. Crit Care Resusc. 2013;15(3):172–8.
Bizzarro MJ, Conrad SA, Kaufman DA, Rycus P. Infections acquired during extracorporeal membrane oxygenation in neonates, children, and adults. Pediatr Crit Care Med. 2011;12(3):277–81.
Vogel AM, Lew DF, Kao LS, Lally KP. Defining risk for infectious complications on extracorporeal life support. J Pediatr Surg. 2011;46(12):2260–4.
Biffi S, Di Bella S, Scaravilli V, Peri AM, Grasselli G, Alagna L, et al. Infections during extracorporeal membrane oxygenation: epidemiology, risk factors, pathogenesis and prevention. Int J Antimicrob Agents. 2017;50(1):9–16.
Burket JS, Bartlett RH, Vander Hyde K, Chenoweth CE. Nosocomial infections in adult patients undergoing extracorporeal membrane oxygenation. Clin Infect Dis. 1999;28(4):828–33.
Quintana MT, Mazzeffi M, Galvagno SM, Herrera D, Boyajian GP, Hays NM, et al. A retrospective study of infection in patients requiring extracorporeal membrane oxygenation support. Ann Thorac Surg. 2021;112(4):1168–75.
Sun G, Li B, Lan H, Wang J, Lu L, Feng X, et al. Risk factors for nosocomial infections in patients receiving extracorporeal membrane oxygenation supportive therapy. Med Clin (Barc). 2017;149(10):423–8.
Sun HY, Ko WJ, Tsai PR, Sun CC, Chang YY, Lee CW, et al. Infections occurring during extracorporeal membrane oxygenation use in adult patients. J Thorac Cardiovasc Surg. 2010;140(5):1125-32.e2.
Aubron C, Cheng AC, Pilcher D, Leong T, Magrin G, Cooper DJ, et al. Infections acquired by adults who receive extracorporeal membrane oxygenation: risk factors and outcome. Infect Control Hosp Epidemiol. 2013;34(1):24–30.
Hsu MS, Chiu KM, Huang YT, Kao KL, Chu SH, Liao CH. Risk factors for nosocomial infection during extracorporeal membrane oxygenation. J Hosp Infect. 2009;73(3):210–6.
Pieri M, Agracheva N, Fumagalli L, Greco T, De Bonis M, Calabrese MC, et al. Infections occurring in adult patients receiving mechanical circulatory support: the two-year experience of an Italian National Referral Tertiary Care Center. Med Intensiva. 2013;37(7):468–75.
Schmidt M, Bréchot N, Hariri S, Guiguet M, Luyt CE, Makri R, et al. Nosocomial infections in adult cardiogenic shock patients supported by venoarterial extracorporeal membrane oxygenation. Clin Infect Dis. 2012;55(12):1633–41.
Cavayas YA, Yusuff H, Porter R. Fungal infections in adult patients on extracorporeal life support. Crit Care. 2018;22(1):98.
Pluim T, Halasa N, Phillips SE, Fleming G. The morbidity and mortality of patients with fungal infections before and during extracorporeal membrane oxygenation support. Pediatr Crit Care Med. 2012;13(5):e288–93.
Douglass BH, Keenan AL, Purohit DM. Bacterial and fungal infection in neonates undergoing venoarterial extracorporeal membrane oxygenation: an analysis of the registry data of the extracorporeal life support organization. Artif Organs. 1996;20(3):202–8.
Poth JM, Schewe JC, Putensen C, Ehrentraut SF. Impact of invasive fungal diseases on survival under veno-venous extracorporeal membrane oxygenation for ARDS. J Clin Med. 2022;11(7):1940.
Rosas MM, Sobieszczyk MJ, Warren W, Mason P, Walter RJ, Marcus JE. Outcomes of fungemia in patients receiving extracorporeal membrane oxygenation. Open Forum Infect Dis. 2022;9(8):ofac374.
Roberts JA, Aziz MHA, Lipman J, Mouton JW, Vinks AA, Felton TW, et al. Challenges and potential solutions—individualised antibiotic dosing at the bedside for critically ill patients: a structured review. Lancet Infect Dis. 2014;14(6):498–509.
Varghese JM, Roberts JA, Lipman J. Pharmacokinetics and pharmacodynamics in critically ill patients. Curr Opin Anaesthesiol. 2010;23(4):472–8.
Shekar K, Roberts JA, McDonald CI, Ghassabian S, Anstey C, Wallis SC, et al. Protein-bound drugs are prone to sequestration in the extracorporeal membrane oxygenation circuit: results from an ex vivo study. Crit Care. 2015;19(1):164.
Mou Z, He J, Guan T, Chen L. Acute kidney injury during extracorporeal membrane oxygenation: VA ECMO versus VV ECMO. J Intensive Care Med. 2022;37(6):743–52.
Roger C, Sasso M, Lefrant JY, Muller L. Antifungal dosing considerations in patients undergoing continuous renal replacement therapy. Curr Fungal Infect Rep. 2018;12:1–11.
Shekar K, Roberts JA, McDonald CI, Fisquet S, Barnett AG, Mullany DV, et al. Sequestration of drugs in the circuit may lead to therapeutic failure during extracorporeal membrane oxygenation. Crit Care. 2012;16(5):R194.
Wildschut ED, Ahsman MJ, Allegaert K, Mathot RA, Tibboel D. Determinants of drug absorption in different ECMO circuits. Intensive Care Med. 2010;36(12):2109–16.
Watt KM, Cohen-Wolkowiez M, Williams DC, Bonadonna DK, Cheifetz IM, Thakker D, et al. Antifungal extraction by the extracorporeal membrane oxygenation circuit. J Extra Corpor Technol. 2017;49(3):150–9.
Raffaeli G, Pokorna P, Allegaert K, Mosca F, Cavallaro G, Wildschut ED, et al. Drug disposition and pharmacotherapy in neonatal ECMO: from fragmented data to integrated knowledge. Front Pediatr. 2019;7:360.
Park J, Shin DA, Lee S, Cho YJ, Jheon S, Lee JC, et al. Investigation of Key circuit constituents affecting drug sequestration during extracorporeal membrane oxygenation treatment. Asaio J. 2017;63(3):293–8.
Raffaeli G, Pokorna P, Allegaert K, Mosca F, Cavallaro G, Wildschut ED, et al. Drug disposition and pharmacotherapy in neonatal ECMO: from fragmented data to integrated knowledge. Front Pediatr. 2019;7:360.
Lemaitre F, Hasni N, Leprince P, Corvol E, Belhabib G, Fillatre P, et al. Propofol, midazolam, vancomycin and cyclosporine therapeutic drug monitoring in extracorporeal membrane oxygenation circuits primed with whole human blood. Crit Care. 2015;19(1):40.
Shekar K, Roberts JA, Barnett AG, Diab S, Wallis SC, Fung YL, et al. Can physicochemical properties of antimicrobials be used to predict their pharmacokinetics during extracorporeal membrane oxygenation? Illustrative data from ovine models. Crit Care. 2015;19:437.
Raffaeli G, Cavallaro G, Allegaert K, Koch BCP, Mosca F, Tibboel D, et al. Sequestration of voriconazole and vancomycin into contemporary extracorporeal membrane oxygenation circuits: an in vitro study. Front Pediatr. 2020;8:468.
Preston TJ, Ratliff TM, Gomez D, Olshove VE Jr, Nicol KK, Sargel CL, et al. Modified surface coatings and their effect on drug adsorption within the extracorporeal life support circuit. J Extra Corpor Technol. 2010;42(3):199–202.
Bhatt-Meht V, Annich G. Sedative clearance during extracorporeal membrane oxygenation. Perfusion. 2005;20(6):309–15.
Mehta NM, Halwick DR, Dodson BL, Thompson JE, Arnold JH. Potential drug sequestration during extracorporeal membrane oxygenation: results from an ex vivo experiment. Intensive Care Med. 2007;33(6):1018–24.
Spriet I, Annaert P, Meersseman P, Hermans G, Meersseman W, Verbesselt R, et al. Pharmacokinetics of caspofungin and voriconazole in critically ill patients during extracorporeal membrane oxygenation. J Antimicrob Chemother. 2009;63(4):767–70.
Watt KM, Benjamin DK Jr, Cheifetz IM, Moorthy G, Wade KC, Smith PB, et al. Pharmacokinetics and safety of fluconazole in young infants supported with extracorporeal membrane oxygenation. Pediatr Infect Dis J. 2012;31(10):1042–7.
Rivory LP, Slaviero KA, Clarke SJ. Hepatic cytochrome P450 3A drug metabolism is reduced in cancer patients who have an acute-phase response. Br J Cancer. 2002;87(3):277–80.
Stanke-Labesque F, Gautier-Veyret E, Chhun S, Guilhaumou R. Inflammation is a major regulator of drug metabolizing enzymes and transporters: consequences for the personalization of drug treatment. Pharmacol Ther. 2020;215: 107627.
Dhanani JA, Lipman J, Pincus J, Townsend S, Livermore A, Wallis SC, et al. Pharmacokinetics of fluconazole and ganciclovir as combination antimicrobial chemotherapy on ECMO: a case report. Int J Antimicrob Agents. 2021;58(5): 106431.
Watt KM, Gonzalez D, Benjamin DK, Brouwer KLR, Wade KC, Capparelli E, et al. Fluconazole population pharmacokinetics and dosing for prevention and treatment of invasive candidiasis in children supported with extracorporeal membrane oxygenation. Antimicrob Agents Chemother. 2015;59(7):3935–43.
Kriegl L, Hatzl S, Zurl C, Reisinger AC, Schilcher G, Eller P, et al. Isavuconazole plasma concentrations in critically ill patients during extracorporeal membrane oxygenation. J Antimicrob Chemother. 2022;77(9):2500–05.
Zurl C, Waller M, Schwameis F, Muhr T, Bauer N, Zollner-Schwetz I, et al. Isavuconazole treatment in a mixed patient cohort with invasive fungal infections: outcome, tolerability and clinical implications of isavuconazole plasma concentrations. J Fungi (Basel). 2020;6(2):90.
Van Daele R, Brüggemann RJ, Dreesen E, Depuydt P, Rijnders B, Cotton F, et al. Pharmacokinetics and target attainment of intravenous posaconazole in critically ill patients during extracorporeal membrane oxygenation. J Antimicrob Chemother. 2021;76(5):1234–41.
Van Daele R, Bekkers B, Lindfors M, Broman LM, Schauwvlieghe A, Rijnders B, et al. A large retrospective assessment of voriconazole exposure in patients treated with extracorporeal membrane oxygenation. Microorganisms. 2021;9(7):1543.
Ye Q, Yu X, Chen W, Li M, Gu S, Huang L, et al. Impact of extracorporeal membrane oxygenation on voriconazole plasma concentrations: a retrospective study. Front Pharmacol. 2022;13: 972585.
Muhl E, Martens T, Iven H, Rob P, Bruch HP. Influence of continuous veno-venous haemodiafiltration and continuous veno-venous haemofiltration on the pharmacokinetics of fluconazole. Eur J Clin Pharmacol. 2000;56(9–10):671–8.
Yagasaki K, Gando S, Matsuda N, Kameue T, Ishitani T, Hirano T, et al. Pharmacokinetics and the most suitable dosing regimen of fluconazole in critically ill patients receiving continuous hemodiafiltration. Intensive Care Med. 2003;29(10):1844–8.
Sobue S, Tan K, Layton G, Eve M, Sanderson JB. Pharmacokinetics of fosfluconazole and fluconazole following multiple intravenous administration of fosfluconazole in healthy male volunteers. Br J Clin Pharmacol. 2004;58(1):20–5.
Sinnollareddy MG, Roberts JA, Lipman J, Akova M, Bassetti M, De Waele JJ, et al. Pharmacokinetic variability and exposures of fluconazole, anidulafungin, and caspofungin in intensive care unit patients: data from multinational Defining Antibiotic Levels in Intensive care unit (DALI) patients study. Crit Care. 2015;19:33.
Shekar K, Abdul-Aziz MH, Cheng V, Burrows F, Buscher H, Cho YJ, et al. Antimicrobial exposures in critically ill patients receiving extracorporeal membrane oxygenation. Am J Respir Crit Care Med. 2023;207(6):704–20.
Miller M, Kludjian G, Mohrien K, Morita K. Decreased isavuconazole trough concentrations in the treatment of invasive aspergillosis in an adult patient receiving extracorporeal membrane oxygenation support. Am J Health Syst Pharm. 2022;79(15):1245–9.
Zhao Y, Seelhammer TG, Barreto EF, Wilson JW. Altered pharmacokinetics and dosing of liposomal amphotericin B and isavuconazole during extracorporeal membrane oxygenation. Pharmacotherapy. 2020;40(1):89–95.
Mertens B, Wauters J, Debaveye Y, Van Regenmortel N, Degezelle K, Meersseman P, et al. The impact of extracorporeal membrane oxygenation on the exposure to isavuconazole: a plea for thorough pharmacokinetic evaluation. Crit Care. 2022;26(1):227.
Lyster H, Pitt T, Maunz O, Diamond S, Roberts J, Brown D, et al. Variable sequestration of antifungals in an extracorporeal membrane oxygenation circuit. ASAIO J. 2022.
Cornely OA, Duarte RF, Haider S, Chandrasekar P, Helfgott D, Jimenez JL, et al. Phase 3 pharmacokinetics and safety study of a posaconazole tablet formulation in patients at risk for invasive fungal disease. J Antimicrob Chemother. 2016;71(3):718–26.
Radej J, Krouzecky A, Stehlik P, Sykora R, Chvojka J, Karvunidis T, et al. Pharmacokinetic evaluation of voriconazole treatment in critically ill patients undergoing continuous venovenous hemofiltration. Ther Drug Monit. 2011;33(4):393–7.
Luke DR, Tomaszewski K, Damle B, Schlamm HT. Review of the basic and clinical pharmacology of sulfobutylether-beta-cyclodextrin (SBECD). J Pharm Sci. 2010;99(8):3291–301.
Neofytos D, Lombardi LR, Shields RK, Ostrander D, Warren L, Nguyen MH, et al. Administration of voriconazole in patients with renal dysfunction. Clin Infect Dis. 2012;54(7):913–21.
Lilly CM, Welch VL, Mayer T, Ranauro P, Meisner J, Luke DR. Evaluation of intravenous voriconazole in patients with compromised renal function. BMC Infect Dis. 2013;13:14.
Abdul-Aziz MH, Alffenaar JWC, Bassetti M, Bracht H, Dimopoulos G, Marriott D, et al. Antimicrobial therapeutic drug monitoring in critically ill adult patients: a position paper#. Intensive Care Med. 1–27.
Cies JJ, Moore WS 2nd, Giliam N, Low T, Marino D, Deacon J, et al. Oxygenator impact on voriconazole in extracorporeal membrane oxygenation circuits. Perfusion. 2020;35(6):529–33.
Zhang Y, Zeng Z, Zhang Q, Ou Q, Chen Z. Effect of extracorporeal membrane oxygenation on pharmacokinetics of antimicrobial drugs: recent progress and recommendations. Nan Fang Yi Ke Da Xue Xue Bao. 2021;41(5):793–800.
Peterson EL, Chittick PJ, Richardson CL. Decreasing voriconazole requirement in a patient after extracorporeal membrane oxygenation discontinuation: a case report. Transpl Infect Dis. 2021;23(3): e13545.
Ruiz S, Papy E, Da Silva D, Nataf P, Massias L, Wolff M, et al. Potential voriconazole and caspofungin sequestration during extracorporeal membrane oxygenation. Intensive Care Med. 2009;35(1):183–4.
Mathieu A, Thiboutot Z, Ferreira V, Benoit P, Grandjean Lapierre S, Hétu PO, et al. Voriconazole sequestration during extracorporeal membrane oxygenation for invasive lung aspergillosis: a case report. Asaio J. 2021.
Winiszewski H, Rougny AC, Lagoutte-Renosi J, Millon L, Capellier G, Navellou JC, et al. The pharmacokinetic challenge of treating invasive aspergillosis complicating severe influenzae assisted by extracorporeal membrane oxygenation. Crit Care. 2018;22(1):355.
Vu T, Feih J, Juul J. Fluctuating voriconazole concentrations during extracorporeal membrane oxygenation. J Pharm Pract. 2022:8971900211060959.
Lin XB, Hu XG, Xia YZ, Liu XM, Liang T, Chen X, et al. Voriconazole pharmacokinetics in a critically ill patient during extracorporeal membrane oxygenation. J Chemother. 2022;34(4):272–6.
Liu P, Ruhnke M, Meersseman W, Paiva JA, Kantecki M, Damle B. Pharmacokinetics of anidulafungin in critically ill patients with candidemia/invasive candidiasis. Antimicrob Agents Chemother. 2013;57(4):1672–6.
Aguilar G, Ferriols R, Carbonell JA, Ezquer C, Alonso JM, Villena A, et al. Pharmacokinetics of anidulafungin during venovenous extracorporeal membrane oxygenation. Crit Care. 2016;20.
Wang Q, Zhang Z, Liu D, Chen W, Cui G, Li P, et al. Population pharmacokinetics of caspofungin among extracorporeal membrane oxygenation patients during the postoperative period of lung transplantation. Antimicrob Agents Chemother. 2020;64(11):e00687-20.
Koch BC, Wildschut ED, Goede AL, Hoog M, Bruggemann RJ. Insufficient serum caspofungin levels in a paediatric patient on ECMO. Med Mycol Case Rep. 2012;2:23–4.
Autmizguine J, Hornik CP, Benjamin DK Jr, Brouwer KL, Hupp SR, Cohen-Wolkowiez M, et al. Pharmacokinetics and safety of micafungin in infants supported with extracorporeal membrane oxygenation. Pediatr Infect Dis J. 2016;35(11):1204–10.
López-Sánchez M, Moreno-Puigdollers I, Rubio-López MI, Zarragoikoetxea-Jauregui I, Vicente-Guillén R, Argente-Navarro MP. Pharmacokinetics of micafungin in patients treated with extracorporeal membrane oxygenation: an observational prospective study. Rev Bras Ter Intensiva. 2020;32(2):277–83.
Timsit JF, Azoulay E, Cornet M, Gangneux JP, Jullien V, Vesin A, et al. EMPIRICUS micafungin versus placebo during nosocomial sepsis in Candida multi-colonized ICU patients with multiple organ failures: study protocol for a randomized controlled trial. Trials. 2013;14:399.
Mourvillier B, Jullien V, Trouillet JL, Ruckly S, Capellier G, Wolff M, et al., editors. Increase micafungin dose for patients under ECMO. In: 27th ECCMID, Vienna, Austria, 2017.
Cabanilla MG, Villalobos N. A successful daptomycin and micafungin dosing strategy in veno-venous ECMO and continuous renal replacement. J Clin Pharm Ther. 2022;47(2):251–3.
Lepak AJ, Andes DR. Antifungal pharmacokinetics and pharmacodynamics. Cold Spring Harb Perspect Med. 2014;5(5): a019653.
Cornely OA, Maertens J, Bresnik M, Ebrahimi R, Ullmann AJ, Bouza E, et al. Liposomal amphotericin B as initial therapy for invasive mold infection: a randomized trial comparing a high-loading dose regimen with standard dosing (AmBiLoad trial). Clin Infect Dis. 2007;44(10):1289–97.
Lanternier F, Poiree S, Elie C, Garcia-Hermoso D, Bakouboula P, Sitbon K, et al. Prospective pilot study of high-dose (10 mg/kg/day) liposomal amphotericin B (L-AMB) for the initial treatment of mucormycosis. J Antimicrob Chemother. 2015;70(11):3116–23.
Love J, Kearns G, Wells T, Baker L, editors. Effect of ECMO on amphotericin disposition in vitro. In: Extracorporeal Life Support Organization Annual Meeting, 1993.
Watt K. Physiologically-based pharmacokinetics in critically ill children. Chapel Hill: University of North Carolina; 2016.
Hertzog JH, Brackett E, Sale M, Hauser GJ, Dalton HJ. Amphotericin B pharmacokinetics during extracorporeal membrane oxygenation: a case report. J Extra-Corpor Technol. 1996;28(2):94–8.
Foulquier JB, Berneau P, Frérou A, Verdier MC, Saint-Marcoux F, Petitcollin A, et al. Liposomal amphotericin B pharmacokinetics in a patient treated with extracorporeal membrane oxygenation. Med Mal Infect. 2019;49(1):69–71.
Branick K, Taylor MJ, Trump MW, Wall GC. Apparent interference with extracorporeal membrane oxygenation by liposomal amphotericin B in a patient with disseminated blastomycosis receiving continuous renal replacement therapy. Am J Health Syst Pharm. 2019;76(11):810–3.
Baracaldo-Santamaría D, Cala-Garcia JD, Medina-Rincón GJ, Rojas-Rodriguez LC, Calderon-Ospina CA. Therapeutic drug monitoring of antifungal agents in critically ill patients: is there a need for dose optimisation? Antibiotics (Basel). 2022;11(5):645.
Ha MA, Sieg AC. Evaluation of altered drug pharmacokinetics in critically ill adults receiving extracorporeal membrane oxygenation. Pharmacotherapy. 2017;37(2):221–35.
Acknowledgements
J. A. Roberts would like to acknowledge funding from the Australian National Health and Medical Research Council for a Centre of Research Excellence (APP2007007) and an Investigator Grant (APP2009736) as well as an Advancing Queensland Clinical Fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Conflict of interest
There are no conflicts of interest known for this manuscript.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
The paper was initiated by Roberts. The paper was initially drafted by Lyster and Abdul-Aziz. All authors provided critical review of the paper and approved the final version.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Lyster, H., Shekar, K., Watt, K. et al. Antifungal Dosing in Critically Ill Patients on Extracorporeal Membrane Oxygenation. Clin Pharmacokinet 62, 931–942 (2023). https://doi.org/10.1007/s40262-023-01264-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01264-0