
Abstract
Background
Site-of-action concentrations for bedaquiline and pretomanid from tuberculosis patients are unavailable. The objective of this work was to predict bedaquiline and pretomanid site-of-action exposures using a translational minimal physiologically based pharmacokinetic (mPBPK) approach to understand the probability of target attainment (PTA).
Methods
A general translational mPBPK framework for the prediction of lung and lung lesion exposure was developed and validated using pyrazinamide site-of-action data from mice and humans. We then implemented the framework for bedaquiline and pretomanid. Simulations were conducted to predict site-of-action exposures following standard bedaquiline and pretomanid, and bedaquiline once-daily dosing. Probabilities of average concentrations within lesions and lungs greater than the minimum bactericidal concentration for non-replicating (MBCNR) and replicating (MBCR) bacteria were calculated. Effects of patient-specific differences on target attainment were evaluated.
Results
The translational modeling approach was successful in predicting pyrazinamide lung concentrations from mice to patients. We predicted that 94% and 53% of patients would attain bedaquiline average daily PK exposure within lesions (Cavg-lesion) > MBCNR during the extensive phase of bedaquiline standard (2 weeks) and once-daily (8 weeks) dosing, respectively. Less than 5% of patients were predicted to achieve Cavg-lesion > MBCNR during the continuation phase of bedaquiline or pretomanid treatment, and more than 80% of patients were predicted to achieve Cavg-lung >MBCR for all simulated dosing regimens of bedaquiline and pretomanid.
Conclusions
The translational mPBPK model predicted that the standard bedaquiline continuation phase and standard pretomanid dosing may not achieve optimal exposures to eradicate non-replicating bacteria in most patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
It is important to understand the site-of-action tissue distribution of antituberculosis to optimize treatment strategies. We developed and validated a translational minimal physiologically based pharmacokinetic approach that allowed predictions for bedaquiline and pretomanid lung and cavitary lesion exposures. |
The model predicted that although current standard dosing of bedaquiline and pretomanid may achieve site-of-action exposures to exhibit bactericidal activity against replicating bacteria, additional treatment optimization may be required for eradication of non-replicating bacteria. |
1 Introduction
Tuberculosis (TB) continues to be a global health challenge. Rifampin and isoniazid are two key first-line antibiotics against Mycobacterium tuberculosis (Mtb). Increasing levels and frequency of resistance against rifampin and isoniazid over time necessitated the discovery of new anti-TB antibiotics [1]. Within the last decade, two promising second-line antibiotics, bedaquiline and pretomanid, were developed as a result of increased efforts to tackle drug-resistant TB [2, 3]. A combination regimen containing bedaquiline, pretomanid, and linezolid (BPaL) showed Mtb clearance (CL) as measured by negative culture status in 90% of patients with drug-resistant TB after 6 months of therapy [4]. BPaL is now recommended for the treatment of rifampin-resistant TB and multidrug-resistant TB patients [1].
Drug exposures at the site of action, within lungs and especially within cavitary lesions, are correlated with bactericidal and sterilizing activities against Mtb. Anaerobic conditions within the necrotic region of the lesion microenvironment, caseum, provide a survival niche for non-replicating but persistent Mtb [5]. Inadequate exposures within cavitary lesions can lead to resistance development. Measuring drug pharmacokinetics (PK) within the lungs and lesions of TB patients usually requires invasive methods to obtain samples and therefore is not readily possible. Preclinical animal models have traditionally been used to evaluate drug penetration at sites of action. These results require accounting for interspecies differences in physiological, pathological, and drug-specific aspects to translate to the patients [6]. Newer methods, such as positron emission tomography (PET) imaging methods, are increasingly being implemented to measure drug penetration at the site of action following intravenous administration of radiolabeled drugs to measure the relative fraction of drug penetration at the site of action [7]. A key benefit of this method is that it can be performed in the patients and as such can allow evaluations of tissue-specific PK with patient-specific covariates.
In silico approaches allow the incorporation of available data for the development of predictive models for optimal decision making. Population PK (PopPK) modeling approaches have been previously applied to quantify the relationship between plasma and lesion PK for several first- and second-line TB antibiotics; however, this approach can only be applied when sites of action measurements are available from patients [8, 9]. Minimal physiologically based PK (mPBPK) models are well suited for translational predictions of the relationship between drug PK in blood plasma and at sites of action. Such models can account for known species-related differences and can incorporate patient covariates and expected interindividual variability and uncertainty in the parameter estimate to allow population simulations. The probability of target attainment (PTA) can then be predicted by defining target attainment to the desired breakpoint based on experimental data [5]. In general, several experimental methods exist to understand drug exposures at the site of action. These methods should be used in combination with in silico approaches to rationally predict overall treatment response in TB patients.
Bedaquiline and its primary metabolite M2 exposures at the current standard dosing regimen (400 mg once daily for 14 days followed by 200 mg three times weekly) have the potential for safety concerns related to QTc interval prolongation and hepatic adverse events [10,11,12]. Alternative once-daily dosing (200 mg once daily for 8 weeks followed by 100 mg once daily) of bedaquiline is of interest to increase patient adherence to obtain optimal efficacy [12]. Pretomanid has overall acceptable clinical safety profiles; however, pretomanid combined with bedaquiline and linezolid can have additive effects, leading to an increased potential of certain adverse effects [13]. As such, understanding drug exposures within the systemic circulation and at sites of action is crucial to evaluate combination treatment strategies to ensure the attainment of adequate exposures to maximize efficacy, avoid resistance development, and minimize safety issues.
The main objective of this work was to predict bedaquiline and pretomanid lung and lesion exposures in TB patients to understand the PTA and the impact of patient covariates, body weight, and cavity size on PTA. To this end, we developed and validated a translational mPBPK modeling approach that allowed to predict bedaquiline and pretomanid PK at standard and alternative once-daily dosing and calculation of PTA in lungs and lesions using preclinical data.
2 Methods
A general translational mPBPK framework including lungs and lesions was first developed and validated using available lung PK data in mice and humans for pyrazinamide. The framework was then implemented for the development and assessment of bedaquiline and pretomanid models. Simulations were conducted to predict site-of-action exposures at standard bedaquiline and pretomanid dosing and an alternative bedaquiline dosing. The simulations were then compared against critical concentration estimates to evaluate the effects of body weight and cavity size on lesion PTA.
2.1 General Modeling Approach
2.1.1 Minimal Physiologically Based Pharmacokinetic (mPBPK) Model Structure
The model development for all drugs was based on a general mPBPK structural model containing central and lung compartments [14]. The mPBPK models were parameterized using species-specific physiological parameters, such as body weight, cardiac output, lung and other tissue volumes, and volume of blood in systemic circulation [15]. Drug-specific parameters, such as blood-to-plasma ratio and plasma protein binding were obtained from the literature. Other drug-specific parameter estimates, such as absorption rates, partition coefficients, and apparent CL were estimated using the data from PK studies in mice (electronic supplementary material [ESM] S1). Additional structural components were evaluated in a stepwise manner as suggested by the available data and prior knowledge. For instance, plasma concentrations of the primary metabolite of bedaquiline, M2, are of importance due to its relevance to safety concerns [16, 17]. Therefore, mechanistic representation of the metabolism of bedaquiline to M2 was described using a liver compartment represented by the well-stirred liver model. Although in vitro and mice experimental studies have demonstrated lower anti-Mtb activity of M2 compared with bedaquiline, its site-of-action exposures can be useful to evaluate its overall impact on bacterial load in TB patients using PK/pharmacodynamic (PD) models; therefore, M2 lung and lesion compartments were included in the model and the relevant parameters were estimated using mice M2 lung and lesion data. Different absorption models, i.e., multiple-site absorption, transit compartment absorption model, and dose-dependent bioavailability, were evaluated.
Drug uptake in lung lesions and uninvolved lungs was described using the effect compartment structural model as described previously [18]. Rates of drug transport between systemic circulation and lesion and uninvolved lung compartments were described by blood flow rates to these compartments calculated based on the approximate lesion and total lung volumes. The volume of lesions and uninvolved lungs were calculated based on the mean total lesion volume reported in the literature for cavitary TB patients and species-specific total lung volumes [19].
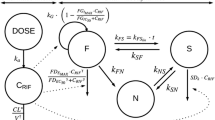
Here, i represents compartments, i.e., lesions or uninvolved lungs; Ci represents drug concentration in the respective compartment; Cbld is drug concentration in systemic circulation; ki is the transfer rate constant to the respective compartment; Ri is the penetration ratio for the respective compartment; Qc is cardiac output; Vlung is the total volume of lungs; VFi is the fractional volume of the respective compartment; and Vi is the volume of the respective compartment. Mean volume of lesions (VFle) was assumed to be 0.0216, calculated based on the mean total lesion volume, approximately 14 mL, reported in the literature for cavitary TB patients [19]. VFul was calculated as 1-VFle.
2.2 Translation of the mPBPK Models to Tuberculosis Patients
The mice mPBPK models developed using mice data were scaled to TB patients considering physiological differences between the species. Physiological parameters such as cardiac output and volumes of compartments for humans were obtained from the literature [15]. A drug-specific parameter, CL, was scaled from mice to humans using a previously known allometric exponent of 0.75 for CL [20]. For the absorption rate parameter (ka), plasma PK predictions using the model scaling with an allometric exponent of − 0.25 and models without any scaling were compared with the observed data for each drug prior to selecting whether or not to scale ka from mice to humans. The same estimated partition coefficients and penetration ratios for each drug and compartment from mice were assumed for TB patients. To simulate a standard TB population, body weights for 500 virtual patients were sampled from the body weight distribution from clinical trial data (ESM S1). Cavity presence or absence was also sampled from observed distribution from the clinical trials dataset. For virtual patients with a cavity, cavity size was simulated considering normal distribution using reported mean and standard deviation of total lesion volume in cavitary TB patients [19]. Log-normally distributed between-subject variability of 25–40% was added in the relevant parameters. To account for uncertainty in the parameter estimates, 50 trials with the estimated relative standard error (RSE) were simulated for each of the 500 virtual patients. All parameters for both mice and humans, including variability and RSE, are presented in Table 1.
2.3 Evaluation of the Modeling Approach Using Pyrazinamide Data
An external evaluation of the predictability of our overall approach was performed using pyrazinamide plasma, lungs, and lesions PK data from mice and TB patients [21, 22]. Therefore, preclinical mice data for pyrazinamide were digitized from the literature [21] and were used in pyrazinamide mPBPK model development. Next, pyrazinamide clinical studies data were compiled from the literature [22] and from the Platform for Aggregation of Clinical TB Studies (TB-PACTS; https://c-path.org/programs/tb-pacts/). Simulated lesion and uninvolved lung PK profiles of pyrazinamide were compared against available observed data in TB patients [22] (ESM S1).
2.4 Bedaquiline and Pretomanid Model Development and Assessment
Bedaquiline and pretomanid model development was conducted using PK data from mice. PK data from plasma, liver, and lungs, including both lung lesions and uninvolved lungs of mice for bedaquiline and its primary metabolite M2, were extracted from the literature [21, 23]. Plasma PK data from mice treated with oral pretomanid were also digitized [24]. PK concentrations from PET imaging of plasma, lung lesions, and uninvolved lungs of mice that were administered a single intravenous dose of radioactive 18F-pretomanid were also digitized [25]. The PK concentrations obtained using PET imaging were represented as relative units (i.e., percentage of injected dose per millimeter [%ID/mL]); therefore, relevant doses were set to 100% in the analysis dataset. Model development was performed using the mPBPK generic structure as described above. Evaluations of additional drug-specific components were guided by data and the parameters were estimated using a model fitting to mice data. Model development and final model selection were guided by objective function value (OFV), the goodness-of-fit (predictions vs. observed) plots, physiological plausibility of parameter estimates, and precision (RSE) of the estimates.
Assessment of bedaquiline and pretomanid mice to human translated models was conducted using PK data from TB patients. Individual-level PK, body weight, and cavity presentation data from various clinical studies were retrieved from the TB-PACTS database. Data from relevant dose levels and clinical studies were compiled for bedaquiline and pretomanid. Simulated plasma PK profiles for bedaquiline and pretomanid in TB patients were compared against observed data to evaluate the translated mPBPK model performance in TB patients. No PK data on lesions or uninvolved lungs were available from human subjects for bedaquiline or pretomanid. Local sensitivity analysis was conducted to evaluate the impact of the parameters on steady-state plasma drug exposure and was examined by introducing 10% variation in the parameters one at a time and running the simulations for 50 times.
2.5 Pharmacokinetics and Target Attainment Predictions of Bedaquiline and Pretomanid at the Site of Action
Predictions of bedaquiline and pretomanid exposures in the lesions and uninvolved lungs were performed for 500 virtual humans using the body weight and cavity volume distributions from TB patients using the final translated mPBPK models (Table 1). Bedaquiline simulations included currently approved oral dosing of 400 mg once daily for 14 days followed by 200 mg three times weekly. The alternative bedaquiline dosing regimen included in the simulations was 200 mg once daily for 8 weeks followed by 100 mg once daily. Pretomanid simulations included oral dosing of 200 mg once daily. Plots of the predicted lesions and uninvolved lungs PK and drug exposure matrices were prepared. Next, predicted bedaquiline and pretomanid concentrations and exposures at the site of action following either standard or alternative bedaquiline or standard pretomanid dosing were compared against critical concentration estimates based on in vitro experiments obtained from the literature. The range for bedaquiline and pretomanid minimum inhibitory concentrations (MICs) for multidrug-resistant (MDR) TB strains, minimal bactericidal concentrations for replicating bacteria (MBCR), and minimal bactericidal concentrations for non-replicating bacteria (MBCNR) were obtained from the literature (Table 1) [5, 26]. Plots of predicted PK exposures within lesions and lungs were plotted and compared against the MDR-TB MIC range, MBCR, and MBCNR for both drugs. Attainment of target exposures in the lesions was defined as average daily PK exposure within lesions (Cavg-lesion) > MBCNR to ensure bactericidal activity against persisting, non-replicating bacteria. Body weight and cavity size from the virtual patient population were binned and PTA was calculated for each bin.
2.6 Software
All analyses were conducted in R (R for Windows, v4.1; https://www.r-project.org/) using RStudio (RStudio, v1-554; www.rstudio.com/). Data management and plotting were performed using the tidyverse package, and parameter optimization and model simulations were conducted using the nlmixr and RxODE packages. Final model codes are provided in ESM S2.
3 Results
3.1 Evaluation of the Modeling Approach Using Pyrazinamide Data
The pyrazinamide mPBPK model was first developed using mice PK data to evaluate the predictability of our translational mPBPK approach. Pyrazinamide plasma, lung lesions, and uninvolved lungs PK data from mice were best described by the mPBPK structural model with first-order absorption and first-order elimination (ESM S3). The addition of a tissue compartment and evaluations of other absorption models did not improve the model fit. The parameters describing plasma PK were estimated with good precision (RSE < 20%), and the parameters Rle and Rul were associated with an RSE of 52% and 102%, respectively (Table 1). The scaled mPBPK model appropriately described pyrazinamide plasma, lung lesion, and uninvolved lung concentrations at steady-state from TB patients using allometric exponents of − 0.25 for ka and 0.75 for CL (Table 1, Fig. 1). Overall, the pyrazinamide model predictions showed good agreement with the observed data from mice and TB patients, suggesting the reliability of our translational mPBPK approach for other TB antibiotics.

Evaluation of the modeling approach using pyrazinamide PK profiles in TB patients. Pyrazinamide model predictions showed good agreement with the observed data from TB patients, suggesting the reliability of our mPBPK framework for other TB antibiotics. Pyrazinamide 1500 mg oral dosing was simulated and compared against observed data from plasma, lungs, and lesions of TB patients [22, 28, 38] accessed through TB-PACTS. Both observed data and simulations for lungs and lesions represent one dosing cycle at steady state. Blue points represent observed data, solid grey line represents the median of the simulations, and shaded grey area represents the 95% PI of the simulations. PK pharmacokinetic, TB tuberculosis, mPBPK minimal physiologically based pharmacokinetic, TB-PACTS platform for aggregation of clinical TB studies, PI prediction interval
3.2 Model Development and Assessment for Bedaquiline and Pretomanid
Bedaquiline and M2 PK data from plasma, liver, lung lesions, and uninvolved lungs of Mtb-infected mice was best described by the mPBPK structural model containing two parallel first-order absorptions that are in alignment with prior bedaquiline PopPK models, parent-metabolite well-stirred liver compartment, tissue compartments for both bedaquiline and M2, first-order elimination of M2, and lung lesion and uninvolved lung compartments (Fig. 2a). Other components that were evaluated but did not provide improvements in the model fit included only one first-order absorption, transit compartment absorption, and saturable conversion of bedaquiline to M2. All parameters were estimated with reasonable precision (RSE < 40%) (Table 1), and the model predictions showed good agreement with the observed data in mice (Fig. 3a, ESM S4a).

Final model structures for a bedaquiline and b pretomanid. The mPBPK models for bedaquiline and pretomanid contain the relevant compartments, including lungs and lung lesions. Blood, liver, lungs, and lung lesions are represented by their volumes and the rest of the body is lumped into tissue compartment(s). Compartments are connected with each other by blood flows. CL plasma clearance, DoseF dose-dependent bioavailability, Eh hepatic extraction ratio, F1 bioavailability of depot1, ka1 absorption rate from depot1, ka2 absorption rate from depot2, Kp partition coefficient for tissue compartments, Qh blood flow to liver, Qle blood flow rate to lung lesions, Qt blood flow to tissues, QUL blood flow rate to unaffected lungs, Rle penetration ratio for lung lesion, RUL penetration ratio for unaffected lungs, Vblood volume of blood reservoir, Vliver volume of liver, Vtissues volume of the tissue compartment, mPBPK minimal physiologically based pharmacokinetic, IV intravenous

Predicted versus observed PK profiles in mice for a bedaquiline and b pretomanid. The model predictions for plasma, lungs, and lesions for bedaquiline and pretomanid agreed well with the observed data for mice. Bedaquiline 25 mg/kg was administered orally [21], and pretomanid was administered orally at varying doses between 18 and 486 mg/kg [24, 25]. Radioactive 18F-pretomanid was administered intravenously and the percentage of the injected dose was measured in plasma, lungs, and lesions of mice. Blue points represent observed data and black line represents model fit predictions. %ID percentage of injected dose, pretomanid concentrations for oral dosing units = ng/mL, 18F-pretomanid concentrations for IV dosing = %ID/mL, PK pharmacokinetic, IV intravenous
Slight underpredictions of bedaquiline and overpredictions of M2 plasma concentrations were noted when the mPBPK model was directly scaled from mice to humans using only allometric scaling. This was assumed to be due to the species difference between mice and humans in bedaquiline and M2 metabolism and has been previously noted [23, 27]. To account for this difference, calibration of two parameters, intrinsic CL (CLint) and CL of M2 (CLM2) was performed by fitting to median rich concentration-time profiles from TB patients for one dose group (400 mg on day 1, 300 mg on day 2, and 200 mg days 3–14) [28]. The updated model predicted both bedaquiline and M2 plasma concentration-time profiles well for all dose groups in the dataset (Table 2, Fig. 4). With a 10% or 50% variation in parameter estimates, the mean sensitivity index was low (between − 1 and 1) for both bedaquiline and M2, with the exception of absorption rate of depot2 (ka2), which showed high sensitivity for both bedaquiline and M2 plasma concentrations (ESM S6). Overall, this mPBPK model for bedaquiline and M2 was deemed reliable for predictions of exposures at the site of action, within lung lesions, in TB patients.

Predicted and observed plasma PK profiles in TB patients for bedaquiline, M2, and pretomanid. The model predictions for plasma bedaquiline, M2, and pretomanid agreed well with the observed data for TB patients from clinical trials at various doses [28, 39]. Blue points represent the observed data, solid grey line represents the median of the simulations, shaded grey area represents the 95% PI of the simulations. Panel titles represent bedaquiline or pretomanid doses in milligrams. Bedaquiline was administered as an increasing daily dose, i.e., panel 1 represents a group that received 200 mg on day 1 and 100 mg on day 2 onwards. PK pharmacokinetic, TB tuberculosis, PI prediction interval
Pretomanid oral absorption was best described by a first-order absorption model with dose-dependent bioavailability. Transit compartment absorption was evaluated but did not provide improvements in the model fits. Pretomanid plasma PK after oral dosing, and F18-pretomanid plasma, lung lesion, and uninvolved lung PK after F18-pretomanid intravenous dosing were best described by the mPBPK structural model with two tissue compartments and first-order elimination (Fig. 2b). All parameters were estimated with reasonable precision (RSE < 45%), except that Rle was estimated, with an RSE of 145% due to limited lesion PK data (Table 1). Overall, the model predictions showed good agreement with the observed data from mice following either F18-pretomanid intravenous dosing or pretomanid oral dosing (Fig. 3b). Reasonable agreement between observed data and scaled-up human model predictions was noticed for plasma PK data for pretomanid from TB patients, although some underpredictions were noted at 1000 mg and 1200 mg doses (Table 2, Fig. 4). With a 10% change in parameter estimates, the sensitivity for key pretomanid PK parameters was relatively low (between − 2.5 and 2.5), with the exception of partition coefficient for tissue compartment 1 (kpT1), which showed moderate sensitivity for pretomanid plasma concentrations. This further supported the reliability of the model (ESM S6). Overall, the mPBPK model for pretomanid was deemed reliable for prediction of exposures at the site of action, within lung lesions, following clinically relevant doses in TB patients.
3.3 Pharmacokinetics and Target Attainment Predictions of Bedaquiline and Pretomanid at the Site of Action
The simulations suggested good penetration for bedaquiline in both lung lesions and uninvolved lungs (Fig. 5a). The predicted bedaquiline median (95% prediction interval) lesion-to-plasma ratio was 11.0 (10.5–11.4) and uninvolved lungs-to-plasma ratio was 10.2 (9.8–10.5). Bedaquiline lesions and uninvolved lung concentrations were predicted to remain above the MIC range observed for MDR-TB isolates at standard dosing and alternative once-daily dosing [26]. Bedaquiline lesion and uninvolved lung concentrations were predicted to be slightly above MBCR for all virtual patients throughout the treatment period during the extensive phase of standard dosing and alternative once-daily dosing (Fig. 5a) [5, 26]. At standard bedaquiline dosing, 94% of virtual patients were predicted to have Cavg-lesion > MBCNR after extensive dosing (400 mg once daily) for the first 14 days of treatment, but < 5% of virtual patients were predicted to have Cavg-lesion > MBCNR throughout the continuation phase of treatment when dosing was reduced to 200 mg three times weekly. At alternative once-daily bedaquiline dosing, a total of 86% of virtual patients were predicted to have Cavg-lesion > MBCNR during the extensive phase (200 mg once daily) for the 8 weeks of treatment, but < 5% of virtual patients were predicted to have Cavg-lesion > MBCNR throughout the continuation phase of treatment when dosing was reduced to 100 mg once daily. The simulations also suggested relatively similar M2 exposures at the site of action as those of bedaquiline (Fig. 5). Simulated plasma M2 concentrations were below the reported M2 EC50 concentrations for QTcF prolongation for 83% of virtual patients during the extensive phase of standard bedaquiline treatment, and for all virtual patients during the continuation phase of standard bedaquiline and both phases of alternative once-daily dosing (ESM S4b). It should be noted that although the PTA is higher with bedaquiline standard dosing during the extensive phase, the period of extensive phase in standard dosing versus alternative once-daily dosing is shorter (2 vs. 8 weeks), which may have a large impact on overall efficacy. The probability of Cavg-lesion > MBCNR was predicted to increase with an increase in body weight for both dosing scenarios (Fig. 6). Cavity size was predicted to not affect the attainment of average lesion concentrations above MBCNR.

Simulated PK profiles at sites of action in TB patients. a Bedaquiline standard and once-daily dosing, and b pretomanid. Bedaquiline standard dosing included 400 mg once daily for 14 days followed by 200 mg 3 days a week. Bedaquiline once-daily dosing included 200 mg once daily for 8 weeks followed by 100 mg once daily. Pretomanid dosing included 200 mg once daily. The model predicted that lung and lesion concentrations of bedaquiline and pretomanid would remain above the MIC (for rifampin and/or isoniazid drug-resistant TB strains) and MBCR at current clinically relevant doses. Although it was predicted that 94% and 53% of patients would achieve target exposures at standard and alternative once-daily bedaquiline dosing, respectively, it should be noted that alternative once-daily dosing provides higher target attainment for a longer duration (2 vs. 8 weeks). M2 target attainment was predicted to be slightly higher than those of bedaquiline. It should be noted that M2 MBC and MICs were assumed to be the same as those of bedaquiline [40]. Blue or black lines represent the median of the simulations and the shaded blue or grey area represents the 95% PI of the simulations. MIC minimum inhibitory concentration, MDR multidrug-resistant tuberculosis, MBCR minimum bactericidal concentrations for replicating bacteria, MBCNR minimum bactericidal concentrations for non-replicating bacteria. PK pharmacokinetic, TB tuberculosis, PI prediction interval, QD once daily

Predicted probability of average bedaquiline concentrations above MBCNR in TB patients, by body weight. Body weight was predicted to have an impact on lesion PTA. PTA was defined as Cavg-lesion > MBCNR. PTAs were calculated at steady-state for each dosing group. MBCNR minimum bactericidal concentrations for non-replicating bacteria, TB tuberculosis, PTA probability of target attainment, Cavg-lesion average daily PK exposure within lesions, MBCNR minimum bactericidal concentration for non-replicating bacteria, QD once daily
The pretomanid predicted median (95% prediction interval) lesion-to-plasma ratio was 2.6 (2.5–2.8) and uninvolved lung-to-plasma ratio was 2.9 (2.7–3.1) [Fig. 5b]. Predicted lesion concentrations after pretomanid 200 mg daily dosing were predicted to remain above the pretomanid MIC range observed for MDR-TB isolates for 18% of virtual patients [13]. Pretomanid lesions and uninvolved lung concentrations were predicted to remain well above the MBCR for all patients. Less than 1% of patients were predicted to have pretomanid Cavg-lesion > MBCNR (Fig. 5b) [13].
4 Discussion
In this work, we developed translational mPBPK models for bedaquiline- and pretomanid-containing lungs, including cavitary lesion compartments, using physiological parameters describing blood flows and volumes of lesions and uninvolved lungs. The mPBPK models adequately described plasma, lungs, and lesions PK data from mice, while the translational mPBPK models adequately described dose-ranging plasma PK data from TB patients. The mPBPK models allowed predictions of bedaquiline and pretomanid exposures and target attainment in the lungs and lesions of TB patients.
One of the concerns against the newer antibiotics bedaquiline and pretomanid is the development of resistance [29]. A key mechanism of resistance development against antibiotics includes a subpopulation of Mtb, persisters, developing phenotypic tolerance against the drugs without genetic mutations. This process is reversible; however, suboptimal drug exposures within cavitary lesions where persisters dwell allow them to survive. This eventually allows Mtb strains to develop genotypic mutation and allow replication of genetically resistant strains against antibiotics [30]. As such, it is crucial to optimize therapeutic strategies to eliminate the non-replicating persistent Mtb population during the treatment phase. Our mPBPK models can be combined with various types of experimental in vitro and in vivo bacterial-kill dynamics and resistance development data to construct mechanistic PK/PD models that can be used to further evaluate optimal strategies against Mtb, especially against persisters within lesions [31].
Our predictions suggested that PTA decreases with an increase in patients’ body weight for both bedaquiline and pretomanid. The current dosing regimen for bedaquiline is associated with safety risks of QTcF prolongation and hepatic adverse events; therefore, an increase in bedaquiline dosing may not be a viable approach for all patients. Further efficacy and safety evaluations of bodyweight-based dose optimization for bedaquiline may be useful. The model also captured the PK of the bedaquiline metabolite M2 within plasma and at the site of action. To further evaluate the contribution of M2 on Mtb CL using the developed mPBPK framework, M2-specific PD parameters, such as derived in an in vitro experiment, would be required. Model-informed precision dosing approaches can be employed to ensure maximum risk-benefit balance for most patients considering totality of information [32, 33]. The current dosing regimen for pretomanid is considered to be well tolerated. The mechanistic PK/PD model can be used to predict the effects of alternative dosing of pretomanid in combination with bedaquiline and/or other antibiotics on CL of persisting Mtb from lesions.
Previously, standard compartmental models combined with effect compartment structural models or whole-body PBPK modeling combined with mechanistic lung models have been developed for mostly first-line TB antibiotics [8, 9, 34]. Middle-out approaches, such as mPBPK models, allow the balance between empirical compartmental models and rigorous whole-body PBPK models [35]. Our mPBPK model construct included standard physiological parameters, such as blood flow rates and volume for lungs. A key component of our structural model is the parameter describing flow rates to lesions, which were calculated based on total blood flow rates and volume fraction of cavitary lesions. As such, this approach suggests a simple technique to evaluate the impact of cavitary lesion size on target exposure attainment within lesions for anti-TB agents. Our simulations suggested no significant impact of total lesion volume on PTA within lesions for bedaquiline and pretomanid given other variability and uncertainty in the parameters that were estimated using limited preclinical data. Additional model-based analyses using our structural model and longitudinal PET imaging data from lesions of varying sizes from TB patients [7] may allow further insights into understating the impact of cavity size on bedaquiline and pretomanid target attainment within lesions to further optimize treatment strategies.
In vitro experimental evaluations of MIC and MBC values for antimicrobials are usually performed using free drug. As such, the role of plasma protein binding may be important for in vitro/in vivo response comparisons if plasma drug concentrations are used for predictions of response in vivo. Generally, only unbound drug from the systemic circulation is available for diffusion into the tissues. Both bound and unbound drug can penetrate the tissues via active transport; however, as distribution equilibrium is achieved, unbound plasma and unbound tissue concentrations reach equilibrium [36, 37]. The previously described empirical model structure for tissue penetration that was fitted to the observed lung and lesion concentration data allows estimation of lung and lesion distribution parameters relative to plasma drug concentrations, and may empirically account for the contributions of the plasma protein binding [8, 18]. Following drug penetration to the lung tissue, the fraction of drug available to exert the effect depends on the tissue content and drug physicochemical properties, such as lipophilicity, solubility, tissue protein binding, and acidity [36, 37]. Therefore, accounting for fraction unbound in tissue for predictions of the PK/PD relationship may be important [34]. In this work, we did not directly correct site-of-action exposures for protein or tissue binding processes as uncertainty exists in the overall impact of these parameters on the PK/PD relationship for bedaquiline and pretomanid. Evaluations of these relationships may be of interest in future studies.
Our mPBPK models were calibrated to mice PK data. The models were translated to TB patients using allometric scaling and were compared and qualified against dose-ranging plasma concentration data from clinical studies. Although our translational models provided reasonable agreement with the observed plasma concentration data for all three drugs, this work did not include evaluations of best structural model fit to the clinical data, or estimation of individual parameter estimation. The sensitivity analysis identified the most sensitive parameters. Future work may consider estimation of individual parameters using the mPBPK model structure, including further evaluations of the role of the sensitive parameters on exposure using clinical data. Mechanistic understanding included in the mPBPK construct combined with Bayesian estimation using our final parameter estimates as priors may provide a thorough understanding of individual variability and covariate/parameter relationships towards the goal of treatment optimization, especially against persisting Mtb.
Although the validation of our translational approach for predictions of lesion and lung concentrations using pyrazinamide give confidence in the applied approach, our model predictions for lesion and lung concentrations for bedaquiline and pretomanid cannot be compared with observed data from TB patients as such data are not available to date. Our models may be further validated or further developed in the future when such data are available. In general, the current construct of mPBPK models for bedaquiline and pretomanid are relevant for our primary objective, i.e., to predict exposures in the lungs and lesions of TB patients using preclinical data. Additionally, the good performance of our translational mPBPK approach for pyrazinamide gives confidence that similar translational performance may be expected for other drugs, such as bedaquiline and pretomanid.
5 Conclusion
We present the first translational mPBPK models for bedaquiline and pretomanid allowing comprehensive predictions of lung and lesion exposures in patients. Both extensive and continuation phases of current standard bedaquiline dosing were predicted to achieve target exposures within lungs and cavitary lesions to elicit bactericidal activity against replicating bacteria; however, only extensive phase treatment was predicted to achieve target exposures to elicit minimum bactericidal activity against non-replicating bacteria. Standard pretomanid dosing was predicted to achieve target exposures to elicit bactericidal activity against replicating bacteria but not against non-replicating bacteria for most patients. These models can also be further developed to be combined with PD, efficacy, or safety measures to optimize or individualize combination treatment strategies.
References
World Health Organization. Consolidated operational guidelines on handbook tuberculosis. Geneva: World Health Organization; 2020. p. 132.
Khoshnood S, Goudarzi M, Taki E, Darbandi A, Kouhsari E, Heidary M, et al. Bedaquiline: current status and future perspectives. J Glob Antimicrob Resist. 2021;25:48–59. https://doi.org/10.1016/j.jgar.2021.02.017.
Stancil SL, Mirzayev F, Abdel-Rahman SM. Profiling pretomanid as a therapeutic option for tb infection: evidence to date. Drug Des Devel Ther. 2021;15:2815–30.
Conradie F, Diacon AH, Ngubane N, Howell P, Everitt D, Crook AM, et al. Treatment of highly drug-resistant pulmonary tuberculosis. N Engl J Med. 2020;382(10):893–902.
Sarathy JP, Dartois V. Caseum: a Niche for Mycobacterium tuberculosis drug-tolerant persisters. Clin Microbiol Rev. 2020;33(3):e00159-19.
Ernest JP, Strydom N, Wang Q, Zhang N, Nuermberger E, Dartois V, et al. Development of new tuberculosis drugs: translation to regimen composition for drug-sensitive and multidrug-resistant tuberculosis. Annu Rev Pharmacol Toxicol. 2021;61:495–516.
Ordonez AA, Wang H, Magombedze G, Ruiz- CA, Srivastava S, Chen A, et al. Heterogeneous drug exposures in pulmonary lesions. HHS Public Access. 2020;26(4):529–34.
Kjellsson MC, Via LE, Goh A, Weiner D, Low KM, Kern S, et al. Pharmacokinetic evaluation of the penetration of antituberculosis agents in rabbit pulmonary lesions. Antimicrob Agents Chemother. 2012;56(1):446–57.
Strydom N, Gupta SV, Fox WS, Via LE, Bang H, Lee M, et al. Tuberculosis drugs’ distribution and emergence of resistance in patient’s lung lesions: a mechanistic model and tool for regimen and dose optimization. PLoS Med. 2019;16(4):e1002773.
Dooley KE, Rosenkranz SL, Conradie F, Moran L, Hafner R, von Groote-Bidlingmaier F, et al. QT effects of bedaquiline, delamanid, or both in patients with rifampicin-resistant tuberculosis: a phase 2, open-label, randomised, controlled trial. Lancet Infect Dis. 2021;21(7):975–83. https://doi.org/10.1016/S1473-3099(20)30770-2.
Cohen K, Maartens G. A safety evaluation of bedaquiline for the treatment of multi-drug resistant tuberculosis. Expert Opin Drug Saf. 2019;18(10):875–82. https://doi.org/10.1080/14740338.2019.1648429.
Tanneau L, Karlsson MO, Rosenkranz SL, Cramer YS, Shenje J, Upton CM, et al. Assessing prolongation of the corrected QT interval with bedaquiline and delamanid coadministration to predict the cardiac safety of simplified dosing regimens. Clin Pharmacol Ther. 2022;112(4):873–81.
US FDA, Center For Drug Evaluation and Research. Center for Drug Evaluation and Research Application Number: 211810Orig1s000: multi-discipline review. US FDA; 2016. pp. 1–264. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212862Orig1s000MultidisciplineR.pdf.
Jermain B, Hanafin PO, Cao Y, Lifschitz A, Lanusse C, Rao GG. Development of a minimal physiologically-based pharmacokinetic model to simulate lung exposure in humans following oral administration of Ivermectin for COVID-19 drug repurposing. J Pharm Sci. 2020;109(12):3574–8.
Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health. 1997;13(4):407–84.
Ngwalero P, Brust JCM, van Beek SW, Wasserman S, Maartens G, Meintjes G, et al. Relationship between plasma and intracellular concentrations of bedaquiline and its m2 metabolite in South African patients with rifampin-resistant tuberculosis. Antimicrob Agents Chemother. 2021;65:11.
Tanneau L, Svensson EM, Rossenu S, Karlsson MO. Exposure–safety analysis of QTc interval and transaminase levels following bedaquiline administration in patients with drug-resistant tuberculosis. CPT Pharmacometrics Syst Pharmacol. 2021;10(12):1538–49.
Gobburu JVS, Tammara V, Lesko L, Jhee SS, Sramek JJ, Cutler NR, et al. Pharmacokinetic-pharmacodynamic modeling of rivastigmine, a cholinesterase inhibitor, in patients with Alzheimer’s disease. J Clin Pharmacol. 2001;41(10):1082–90.
Chen RY, Yu X, Smith B, Liu X, Gao J, Diacon AH, et al. Radiological and functional evidence of the bronchial spread of tuberculosis: an observational analysis. Lancet Microbe. 2021;2(10):e518–26. https://doi.org/10.1016/S2666-5247(21)00058-6.
Mahmood I, Balian JD. The pharmacokinetic principles behind scaling from preclinical results to phase I protocols. Clin Pharmacokinet. 1999;36(1):1–11.
Irwin SM, Prideaux B, Lyon ER, Zimmerman MD, Brooks EJ, Schrupp CA, et al. Bedaquiline and pyrazinamide treatment responses are affected by pulmonary lesion heterogeneity in mycobacterium tuberculosis infected C3HeB/FeJ mice. ACS Infect Dis. 2016;2(4):251–67.
Prideaux B, Via LE, Zimmerman MD, Eum S, Sarathy J, O’Brien P, et al. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat Med. 2015;21(10):1223–7.
Rouan MC, Lounis N, Gevers T, Dillen L, Gilissen R, Raoof A, et al. Pharmacokinetics and pharmacodynamics of TMC207 and its N-desmethyl metabolite in a murine model of tuberculosis. Antimicrob Agents Chemother. 2012;56(3):1444–51.
Ahmad Z, Peloquin CA, Singh RP, Derendorf H, Tyagi S, Ginsberg A, et al. PA-824 exhibits time-dependent activity in a murine model of tuberculosis. Antimicrob Agents Chemother. 2011;55(1):239–45.
Mota F, Ruiz-Bedoya C, Tucker E, De Jesus P, Flavahan K, Turner M, et al. Noninvasive assessment of intralesional antimicrobial concentration-time profiles in pulmonary and central nervous system tuberculosis using dynamic 18F-pretomanid positron emission tomography. Open Forum Infect Dis. 2021;8(Suppl 1):S789–90. https://doi.org/10.1093/ofid/ofab466.1603.
Ismail NA, Omar SV, Joseph L, Govender N, Blows L, Ismail F, et al. Defining bedaquiline susceptibility, resistance, cross-resistance and associated genetic determinants: a retrospective cohort study. EBioMedicine. 2018;28:136–42. https://doi.org/10.1016/j.ebiom.2018.01.005.
van Heeswijk RPG, Dannemann B, Hoetelmans RMW. Bedaquiline: a review of human pharmacokinetics and drug-drug interactions. J Antimicrob Chemother. 2014;69(9):2310–8.
Diacon AH, Dawson R, Von Groote-Bidlingmaier F, Symons G, Venter A, Donald PR, et al. Bactericidal activity of pyrazinamide and clofazimine alone and in combinations with pretomanid and bedaquiline. Am J Respir Crit Care Med. 2015;191(8):943–53.
Chesov E, Chesov D, Maurer FP, Andres S, Utpatel C, Barilar I, et al. Emergence of bedaquiline resistance in a high tuberculosis burden country. Eur Respir J. 2022;59(3):1–10. https://doi.org/10.1183/13993003.00621-2021.
Allué-Guardia A, Garcia-Vilanova A, Olmo-Fontánez AM, Peters J, Maselli DJ, Wang Y, et al. Host- and age-dependent transcriptional changes in Mycobacterium tuberculosis cell envelope biosynthesis genes after exposure to human alveolar lining fluid. Int J Mol Sci. 2022;23(2). https://www.mdpi.com/1422-0067/23/2/983
Drusano GL, Kim S, Almoslem M, Schmidt S, D’Argenio DZ, Myrick J, et al. The funnel: a screening technique for identifying optimal two-drug combination chemotherapy regimens. Antimicrob Agents Chemother. 2021;65:2.
Jarugula P, Scott S, Ivaturi V, Noack A, Moffett BS, Bhutta A, et al. Understanding the role of pharmacometrics-based clinical decision support systems in pediatric patient management: a case study using Lyv software. J Clin Pharmacol. 2021;61(Suppl 1):S125–32.
Hughes JH, Tong DMH, Lucas SS, Faldasz JD, Goswami S, Keizer RJ. Continuous learning in model-informed precision dosing: a case study in pediatric dosing of vancomycin. Clin Pharmacol Ther. 2021;109(1):233–42.
Humphries H, Almond L, Berg A, Gardner I, Hatley O, Pan X, et al. Development of physiologically-based pharmacokinetic models for standard of care and newer tuberculosis drugs. CPT Pharmacometrics Syst Pharmacol. 2021;10(11):1382–95.
Cao Y, Jusko WJ. Applications of minimal physiologically-based pharmacokinetic models. J Pharmacokinet Pharmacodyn. 2012;39(6):711–23.
Rowland M, Tozer TN. Clinical pharmacokinetics and pharmacodynamics: concepts and applications; 1980.
Shargel L, Wu-Pong S, Yu ABC. Chapter 10. physiologic drug distribution and protein binding. In: Applied biopharmaceutics and pharmacokinetics, 6e. New York: The McGraw-Hill Companies; 2012. http://accesspharmacy.mhmedical.com/content.aspx?aid=56603200.
Diacon AH, Dawson R, von Groote-Bidlingmaier F, Symons G, Venter A, Donald PR, et al. 14-day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: a randomised trial. Lancet. 2012;380(9846):986–93.
The global alliance for tb drug development. Evaluation of Early Bactericidal Activity in Pulmonary Tuberculosis (TMC207-CL001). 2017. https://clinicaltrials.gov/show/NCT01215110.
Kim S, Yamada WM, Duncanson B, Nole J, Rogers S, Parker S, et al. Building optimal three-drug combination chemotherapy regimens to eradicate mycobacterium tuberculosis in its slow-growth acid phase. Antimicrob Agents Chemotherapy. 2021;65(10):e00693-e721. https://doi.org/10.1128/AAC.00693-21.
Lyons MA. Pretomanid dose selection for pulmonary tuberculosis: an application of multi-objective optimization to dosage regimen design. CPT Pharmacom Syst Pharmacol. 2021;10(3):211–9.
Muliaditan M, Della PO. Evaluation of pharmacokinetic–pharmacodynamic relationships and selection of drug combinations for tuberculosis. Br J Clin Pharmacol. 2021;87(1):140–51.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for this work.
Conflicts of interest
Piet H. van der Graaf is an employee of Certara, UK. Krina Mehta, Tingjie Guo, and J.G.C. van Hasselt declare no conflicts of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors provided consent for publication.
Availability of data and material
All data used in the analysis were obtained from either the literature (as cited) or from the TB-PACTS database (https://c-path.org/programs/tb-pacts/).
Code availability
Final model codes are provided in the ESM.
Author contributions
All authors contributed to the study conception and design. KM performed the analysis and wrote the first draft of the manuscript. All authors reviewed and edited the manuscript, and reviewed and approved the final version.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Mehta, K., Guo, T., van der Graaf, P.H. et al. Predictions of Bedaquiline and Pretomanid Target Attainment in Lung Lesions of Tuberculosis Patients using Translational Minimal Physiologically Based Pharmacokinetic Modeling. Clin Pharmacokinet 62, 519–532 (2023). https://doi.org/10.1007/s40262-023-01217-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01217-7