
Abstract
Background and Objective
Renal impairment is common in patients with cancer and can alter the PK and thus the safety and efficacy of drugs. We assessed the impact of renal impairment during treatment with ribociclib, a cyclin-dependent kinase 4/6 inhibitor, and determined dose recommendations for patients with advanced breast cancer with renal impairment.
Methods
A comprehensive assessment integrating pharmacokinetic, safety, and efficacy data from a phase I dedicated renal impairment study in non-cancer subjects and six phase I–III trials in patients with cancer was performed.
Results
Ribociclib showed higher pharmacokinetic exposure in subjects with renal impairment than those with normal renal function following a single 400-mg dose in the dedicated renal impairment study. However, in patient trials, both single-dose and steady‑state ribociclib exposure was comparable between patients with cancer with mild/moderate renal impairment and those with normal renal function following the recommended starting dose of 600 mg. Model-predicted steady‑state exposure in patients with advanced breast cancer was also similar across the renal function groups. Progression-free survival was similar and safety profiles were generally consistent across the renal cohorts (normal/mild/moderate) in patients with advanced breast cancer, with low-grade and manageable adverse events, demonstrating a positive benefit-risk profile.
Conclusions
From the collective evidence and considering a real-world clinical setting, no dose adjustment is recommended for patients with mild/moderate renal impairment, whereas a reduced dose is recommended for patients with severe renal impairment. This report presented a holistic and innovative strategy to determine dose in patients with renal impairment and demonstrated the effectiveness of integrating the data of both a clinical pharmacology study and patient trials to justify doses in patients with renal impairment.
Clinical Trial Registration
Clinicaltrials.gov identifiers: NCT02431481, NCT01958021, NCT02422615, NCT02278120, NCT01237236, NCT01898845, NCT01872260.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
No clinically relevant difference in ribociclib PK, safety, and efficacy was observed between patients with advanced breast cancer with mild/moderate renal impairment and those with normal renal function; therefore, no dose adjustment is required; a reduced dose is recommended for patients with severe renal impairment based on data in subjects with renal impairment but healthy otherwise. |
This report described an alternative and holistic strategy integrating pharmacokinetic data of a dedicated clinical pharmacology renal impairment study and pharmacokinetic, safety, and efficacy data of patient trials to assess the effect of renal impairment on drug treatment and drive dose recommendations. |
1 Introduction
Ribociclib (Kisqali®), an orally bioavailable and selective small-molecule inhibitor of cyclin-dependent kinases 4 and 6, is approved worldwide for the treatment of women with hormone receptor-positive, human epidermal growth factor receptor-2 negative advanced breast cancer (ABC) at a recommended dose of 600 mg once daily for 21 consecutive days, followed by 7 days off treatment, with an option for a dose reduction to 400 and 200 mg [1, 2]. Ribociclib has demonstrated efficacy and safety, with a long-term overall survival benefit when combined with different endocrine therapy partners across a broad patient population [3,4,5,6,7,8]. Previous pharmacokinetic (PK) analyses showed that ribociclib is rapidly absorbed, with a time to maximum observed plasma drug concentration (Cmax) of 1–5 h and a mean effective half-life of 33–54 h at 600 mg [9,10,11,12]. Ribociclib exhibited slightly over-proportional increases in exposure across the dose range of 50–1200 mg [9, 13], and has an absolute oral bioavailability of 66% in healthy volunteers [11]. It is excreted primarily via hepatic elimination, with around 7% and 8% of renal and intestinal excretion, respectively [14], and is extensively metabolized, with a major contribution by cytochrome P450 (CYP) 3A4 (63%) and a smaller contribution by flavin-containing monooxygenase 3 (16%) [14, 15]. LEQ803 is a major metabolite formed via CYP3A4; however, it has no clinically relevant contribution to the pharmacological activity in vivo [15].
In recent years, the incidence of renal impairment (RI) has increased globally [16], with renal insufficiency being common in patients with cancer [17] and with only 39% of patients with breast cancer presenting a glomerular filtration rate (GFR) in the normal range (GFR ≥ 90 mL/min/1.73 m2) [18]. Impaired renal function can alter the PK of drugs eliminated either by renal or nonrenal pathways, resulting in changes in systemic drug exposure that can lead to toxicity-induced dose modifications [19,20,21]. In addition, renal disease can affect other organs and alter physiology, and patients with renal disease can also present with comorbidities [22], resulting in patients having an increased incidence of adverse events (AEs), altered pharmacodynamics, or drug efficacy [22]. Therefore, careful consideration must be taken during dose adjustment in order to optimize efficacy and minimize toxicity in this vulnerable population. Regulatory bodies have recognized the importance of assessing the effect of RI and provided guidance to conduct clinical pharmacology studies to evaluate its influence on PK [22,23,24]. At a recent US Food and Drug Administration Pharmaceutical Science and Clinical Pharmacology Advisory Committee Meeting, use of dedicated clinical pharmacology RI studies alone to determine dose recommendations in patients with RI was challenged and the utility of patient data via a population PK (popPK) analysis was proposed [22]. Here, we present a comprehensive assessment integrating both a dedicated RI study and patient clinical trial data on the PK, efficacy, and safety to evaluate the impact of RI in ribociclib treatment effect and determine whether dose adjustment is required for patients with RI.
2 Patients and Methods
2.1 Renal Function Categories
Renal function categories were based on absolute GFR (aGFR) for the RI study A2116 in non-cancer subjects (healthy subjects or subjects with RI but healthy otherwise) (NCT02431481) and for the subgroup PK analysis in patients with cancer, and based on estimated creatinine clearance by the Cockcroft–Gault equation for the pooled analysis for efficacy and safety across studies in patients with the following cut-off values: normal renal function (≥ 90 mL/min); mild RI (60–89 mL/min); moderate RI (30–59 mL/min); severe RI (15–29 mL/min); end-stage renal disease (ESRD; < 15 mL/min not on dialysis) [23, 24]. For the popPK analysis, eGFR was used with the following cut-off values: normal renal function (≥ 90 mL/min/1.73 m2); mild RI (60–89 mL/min/1.73 m2), moderate RI (30–59 mL/min/1.73 m2), and severe RI (15–29 mL/min/1.73 m2) [24]. Absolute glomerular filtration rate was derived from the eGFR through the conversion, aGFR = eGFR × body surface area/1.73 m2, where eGFR was determined by the Modification of Diet in Renal Disease equation [24].
2.2 Study A2116 in Non-Cancer Subjects
This was a phase I, open-label, multicenter, parallel-group, single-dose, two-staged study evaluating the PK and safety of a single 400-mg oral dose of ribociclib in subjects with varying degrees of RI compared with matched subjects with normal renal function. The study was conducted in two parts (Fig. S1 of the Electronic Supplementary Material [ESM]). Part I was conducted as a “reduced PK study design,” a worst-case scenario study that compared PK in subjects at the extremes of renal function: subjects with normal renal function, severe RI, or ESRD not yet on dialysis. Part II started only when the results from the interim analysis of Part I showed a potentially significant clinical effect (a > 50% increase in the area under the curve [AUC] from time zero extrapolated to infinity [AUCinf] of ribociclib) in subjects with severe RI compared with subjects with normal renal function and was then opened to subjects with mild/moderate RI. Pharmacokinetic parameters were derived by a non-compartmental analysis (NCA). The geometric mean ratio (GMR) and the 90% confidence intervals (CIs) of the PK parameters for the test versus reference (normal renal function) were calculated (see ESM for details).
2.3 Patients Studies X2101, X1101, X2107, MONALEESA-2, MONALEESA-3, and MONALEESA-7
The analysis of the effects of RI in patients with cancer was performed using pooled data from the following studies: a first-in-human phase I study of ribociclib as a single agent in patients with advanced solid tumors or lymphomas (X2101 [NCT01237236]) [9], a phase I study of ribociclib in Asian patients with advanced solid tumors (X1101 [NCT01898845]) [13], a phase Ib/II dose-escalation study of ribociclib and/or alpelisib with letrozole in postmenopausal adult patients with hormone receptor-positive ABC (X2107 [NCT01872260]) [25], and three pivotal phase III studies of ribociclib in combination with letrozole, anastrozole, or tamoxifen (MONALEESA-2 [NCT01958021] [3] and MONALEESA-7 [NCT02278120] (tamoxifen arm was excluded from the analysis) [4] or fulvestrant (MONALEESA-3 [NCT02422615]) [5] in patients with hormone receptor-positive, human epidermal growth factor receptor-2 negative ABC. In the three pivotal studies and Study X2107, the recommended dose modification scheme to manage study drug-related AEs is to reduce the dose from 600 mg to 400 or 200 mg. Pharmacokinetic data showed no drug–drug interactions between ribociclib and nonsteroidal aromatase inhibitors (NSAIs; letrozole and anastrozole) or fulvestrant [26, 27]. Patients with severe RI were not included in the analysis as they were excluded from the studies.
2.3.1 Patient Subgroup PK Analysis
The subgroup analysis evaluated PK parameters of ribociclib by renal function in patients with cancer (X2101) or ABC (X2107 and MONALEESA-7 [NSAI only]) following oral administration of 600 mg (once daily; 3 weeks on/1 week off) with no dose reduction. Pharmacokinetic parameters of ribociclib following single-dose administration or at steady state were derived by NCA and the GMR and the 90% CI of the PK parameters for the test (renal impaired) versus reference (normal renal function) was calculated (see ESM for details).
2.3.2 Patient popPK Analysis
Using the popPK model of ribociclib developed based on a pooled dataset (X2101, X1101, X2107, MONALEESA-2, MONALEESA-7 [NSAI only], and MONALEESA-3) [28], individual estimates of steady-state AUC and steady-state Cmax at the 600-mg dose were computed for all patients with ABC included in the popPK dataset. The calculation of GMR and the 90% CI of the PK parameters for the test (renal impaired) versus reference (normal renal function) was repeated for these sets of parameters.
2.3.3 Patient Subgroup Safety and Efficacy Analysis
Safety was analyzed by renal function in patients with ABC from studies X2107 and three pivotal phase III studies (MONALEESA-2, MONALEESA-7 [NSAI only], and MONALEESA-3). Efficacy was analyzed by renal function from the three pivotal phase III studies. Median progression-free survival (PFS) was assessed using the Kaplan–Meier method; hazard ratios (HRs) were obtained from a Cox proportional-hazards model stratified by study.
3 Results
3.1 Effect of Renal Impairment on Ribociclib Pharmacokinetics and Safety in Non-Cancer Subjects
An increase of > 50% in ribociclib exposure in Part I (7, 3, and 7 with severe RI, ESRD and normal renal function, respectively) was observed in subjects with severe RI (1.96-fold and 1.51-fold increase in AUCinf and Cmax, respectively, Table S1 of the ESM) compared with subjects with normal renal function; therefore, Part II of the study was initiated including subjects with mild and moderate RI (total of 8, 6, 7, 3, and 14 with mild, moderate, severe RI, ESRD, and normal renal function, respectively in Part I and II) [Table S2 of the ESM].
Ribociclib exposure increased in subjects with mild, moderate, and severe RI compared with those with normal renal function for Cmax (by 80%, 79%, and 130%, respectively) and AUCinf (by 62%, 94%, and 167%, respectively), and apparent clearance reduced by 38.4%, 48.5%, and 62.5%, respectively (Table 1). Concurrently, the terminal-phase half-life increased correspondingly with the degree of RI as 36.0, 40.9, 50.2, and 56.2 h for normal, mild, moderate, and severe RI groups, respectively (Table S3 of the ESM). Time to Cmax values were similar across renal function groups. Consistent with increased systemic exposure, renal clearance was lower in subjects with RI and the reduction was related to the degree of RI (Table S3 of the ESM). However, it was only in the severe and ESRD groups that the extent of unchanged drug excreted into the urine from time 0 to 144 h was lower (11.4 and 8.11 mg) compared with the normal, mild, and moderate renal function groups (20.2, 19.9, and 19.3 mg) [Table S3 of the ESM]. Compared with subjects with normal renal function, exposures of the metabolite LEQ803 were 32%, 47%, and 80% higher in subjects with mild, moderate, and severe RI, respectively, and median metabolic ratio values were slightly lower in subjects with RI compared with subjects with normal renal function (Table S4 of the ESM).
Twelve subjects (31.6%) had at least one AE during the study; six (15.8%) had treatment-related AEs. All AEs reported were grade 1, except for two grade 2 AEs (headache in normal renal function cohort and back pain in the moderate RI cohort), which were promptly managed and resolved. No grade 3/4 AEs, deaths, or serious AEs were reported across the cohorts. No clinically meaningful changes from baseline in hematology, chemistry, vital signs, or electrocardiograms were noted in any of the cohorts.
3.2 Effect of RI on Ribociclib Pharmacokinetics, Efficacy, and Safety in Patients with Cancer
To rule out potential confounding factors, patient demographics, disease status, and hepatic functions in the pooled patient studies (Study MONALEESA-2, MONALEESA-7 [NSAI only], MONALEESA-3, X2107) were analyzed and did not reveal apparent differences across renal cohorts (data on file). The PK data from patients with cancer were analyzed using both an NCA and a popPK analysis as described in the following sections.
3.2.1 Subgroup PK Analysis
The intensive PK sampling implemented in the patients studies allowed for the calculation of exposure parameters using an NCA. The GMRs of the mild/moderate RI cohorts versus the normal renal function cohort for Cmax, AUC from 0 to 24 h, and trough concentration were close to 1 and/or the 90% CI included 1 at both single dose and steady state (Table 2), indicating no apparent difference in the PK between patients with cancer with mild/moderate RI and those with normal renal function.
3.2.2 PopPK Analysis
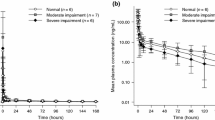
The popPK dataset pooled from studies in patients with cancer contains 57.8% of patients with mild (N = 488) or moderate (N = 113) RI and 42.2% of patients with normal renal function (N = 438). The model was parameterized as a two-compartmental model with first-order absorption and nonlinear clearance [28]. A covariate analysis showed that ribociclib PK in patients was not affected by RI using baseline eGFR as a continuous covariate [28]. In line with the covariate analysis, model-simulated steady-state AUC from 0 to 24 h and Cmax values in individual patients showed no apparent difference across the three RI cohorts (normal, mild/moderate) (Fig. 1). In addition, the ratios of geometric mean steady-state exposure of both 2.5th and 97.5th percentile eGFRs of the population versus normal renal function were close to 1 (Table S5 of the ESM). These results demonstrate the lack of effect of mild and moderate RI on ribociclib PK in patients, consistent with the results from the NCA-generated PK parameters.

Estimated steady-state exposure for maximum concentration [SS Cmax] (a), and area under the curve [AUC] (b) of ribociclib at 600 mg in patients with advanced breast cancer by renal function based on the population pharmacokinetic (popPK) simulation. Studies X2107, MONALEESA-2, MONALEESA-7, and MONALEESA-3 were included in the pooled analysis
3.2.3 Safety
In patients with ABC, the overall pattern, frequency, and severity of the majority of both hematological and non-hematological adverse events of special interest (AESIs) with ribociclib plus ET was consistent across normal, mild, and moderate RI cohorts (Table 3). As expected, AESIs classified as “renal toxicity” occurred more frequently with worsening renal function in both the ribociclib plus ET group and placebo plus ET groups.
Numerically higher increases in QT interval prolongation events were observed in the ribociclib group in patients with mild (10.3%) and moderate (10.0%) RI compared with patients with normal renal function (6.4%) (Table 3). However, numerical increases were also observed in the placebo group (4.5% in mild RI, 4.8% in moderate RI vs 1.8% in normal renal function), suggesting no additional impact of ribociclib on QT interval prolongation in patients with mild/moderate RI compared to patients with normal renal function. No AEs of torsade de pointes were reported. The vast majority of corrected QT prolongation events under the AESI category were asymptomatic electrocardiogram findings without clinical manifestations; these tended to occur early in treatment (median time to first occurrence of corrected QT interval according to Fridericia’s formula value > 480 ms and an increase from baseline of >60 ms was 15 days).
There was a higher incidence of thrombocytopenia in the ribociclib versus placebo groups in patients with mild (11.0%) or moderate (13.8%) RI versus those with normal renal function (7.0%) (Table 3). A numerical increase was also observed in the placebo group (2.4% in moderate RI vs 1.4% normal renal function). These events tended to be primarily grade 1 or 2 and did not result in bleeding events; no patients discontinued treatment because of thrombocytopenia in either the RI or the normal renal function cohorts.
An increase in on-treatment deaths was observed in patients in the ribociclib group with moderate RI (5.4%) compared with mild RI (1.3%) and normal renal function (1.4%); however, similar rates of on-treatment deaths were observed in the placebo group in moderate RI (3.6%) compared to the mild RI (2.1%) and normal renal function (1.1%) cohorts. Of the seven on-treatment deaths in patients with moderate RI who were treated with ribociclib, three were due to disease progression and neither of the others was suspected to be related to ribociclib.
Dose reductions occurred primarily within the first three cycles, regardless of renal function, and a similar median time (12.0–12.9 weeks) to the first dose reduction was observed across patients with dose reductions (Table 4). Although patients with mild RI (2.5%) and moderate RI (3.8%) in the ribociclib group had proportionally higher rates of dose modifications owing to renal toxicity compared with patients with normal renal function (0.7%), rates of discontinuation due to renal AEs were consistent across all three cohorts. Higher rates of AEs leading to discontinuations were observed in patients with mild/moderate RI compared with patients with normal function in the ribociclib group (16.0% and 25.4% vs 10.1%); similarly, higher rates were observed in the placebo group (5.9% and 6.0% vs 2.3%).
3.2.4 Efficacy
Analysis of the pooled data from the three pivotal phase III trials showed no differences in the estimated relative risk reduction in PFS in patients with mild (HR 0.57 [95% CI 0.45–0.72]) or moderate (HR 0.64 [95% CI 0.41–1.00]) RI versus patients with normal renal function (HR 0.58 [95% CI 0.48–0.70]) (Table 5). Median PFS per local investigator’s assessment was in favor of ribociclib regardless of renal function, with a median PFS of 23.8 (normal), 24.8 (mild), and 22.2 months (moderate) with ribociclib, and 14.6 (normal), 16.0 (mild), and 14.7 months (moderate) with placebo (Table 5). The Kaplan–Meier plots of PFS as per local investigator’s assessment were similar between normal and mild/moderate RI cohorts in both ribociclib and placebo groups (Fig. 2). These data demonstrated that patients with mild/moderate RI received a similar clinical benefit as patients with normal renal function.

Kaplan–Meier plot of progression free survival as per local investigator’s assessment by treatment and renal function in patients with advanced breast cancer following administration of a starting dose of ribociclib (RIB) at 600 mg (3 weeks on/1 week off). The analysis includes MONALEESA-3 (first-line patients), MONALEESA-7 (nonsteroidal aromatase inhibitor [NSAI] only), and MONALEESA-2 studies (full analysis set). The Cox proportional hazards model was stratified by study
4 Discussion
In the dedicated clinical pharmacology RI study A2116, ribociclib exposure following a single-dose administration of 400 mg was higher in non-cancer subjects with RI compared with those with normal renal function. The associated exposure increase of ribociclib metabolite, LEQ803, was not expected to have clinical relevance because of a lack of clinically relevant activity [15, 29]. As ribociclib exhibits extensive hepatic elimination (84% of overall pathways) via CYP3A metabolism in humans [15, 29] with a minor contribution of renal clearance to ribociclib metabolism (7% of overall elimination routes in human absorption, distribution, metabolism, and excretion study [15], and around 5% of the apparent clearance in Study A2116), RI was not expected to have a substantial effect on ribociclib PK.
Growing evidence, however, indicates that RI may alter a variety of non-renal absorption, distribution, metabolism, and excretion pathways via effects attributed to elevated uremic toxins that may alter the expression and/or activities of plasma proteins, drug‑metabolizing enzymes, and transporters [19, 20]. Of note, ribociclib exposure was similar in subjects with mild hepatic impairment (HI) compared with subjects with normal hepatic function, consistent with the popPK analysis, and increased approximately 30% in subjects with moderate and severe HI [28, 29]. Therefore, the observed magnitude of 1.6-fold, 1.9-fold, and 2.7-fold increase of exposure in subjects with mild, moderate, and severe RI, respectively, is well above the 1.3-fold (30% increase) maximal increase in exposure observed in severe HI, ruling out that inhibition of drug-metabolizing enzymes (specifically CYP3A4) or hepatic transporters could account for this increase. Considering the moderate protein binding (70%) of ribociclib [15], changes in plasma protein binding are also not expected to cause major changes in the free drug concentration that would result in a clinically relevant impact. This finding, that a drug eliminated primarily via a hepatic pathway will have its oral clearance reduced more by RI than by HI, could not be explained using the principles of absorption, distribution, metabolism and excretion.
With the unexpected results from the dedicated RI study, it was pertinent to look at not only the PK data in non-cancer subjects, but also the PK as well as safety and efficacy data in the target patient population across renal functions. Interestingly, ribociclib PK exposure was found not to increase in patients with mild or moderate RI with cancer as compared to patients with normal renal function. Based on the NCA-generated PK parameters from three studies in patients with cancer, which had intensive PK sampling, as well as the PK parameters computed from the popPK analysis in patients with ABC from four studies, Cmax, AUC, or trough concentration in patients with mild/moderate RI were found to be comparable to those in patients with normal renal function. Of note, the PK parameters were grouped by the same RI criteria (aGFR) as A2116 to enable direct comparison of results. This result is consistent with previous popPK covariate analyses that eGFR had no clinically relevant effect on ribociclib PK [28]. The popPK analysis, along with the subgroup PK analysis for ribociclib in the target patient population using the same methodology (NCA) and the same renal function classification (aGFR) as the RI study, helped to strengthen our conclusion. Additionally, we performed a subgroup analysis for letrozole PK, which demonstrated comparable PK between normal renal function and mild RI (X2107 and MONALEESA-7, no moderate RI available, data on file), consistent with the prescribing information of letrozole that RI has no impact on its PK [30]. This further validates the conclusions on a lack of effect of mild and moderate RI for ribociclib using target patient data in real-world settings.
Furthermore, a positive benefit-risk profile was demonstrated in different (normal, mild, and moderate) renal function cohorts across all the four phase II and III clinical trials conducted in patients with ABC. Median PFS was in favor of ribociclib versus placebo across all renal cohorts. The overall pattern of AESIs in patients treated with ribociclib was consistent across renal function cohorts. Although there were numerical increases in the incidence of corrected QT prolongation and thrombocytopenia, relative increases in such AEs were also observed in the placebo group in patients with RI. Moreover, the majority of the events occurred early during treatment (within cycle 1 up to cycle 2 day 1), and thus would be adequately identified and effectively managed according to current safety monitoring and dose modification guidance in ribociclib prescribing information [1, 2].
The reason for the difference between non-cancer subjects and patients with cancer regarding the effect of renal function for the mild and moderate RI groups on ribociclib exposure is currently unclear. Differences in populations (non-cancer subjects vs patients with cancer), dosing and treatment duration (single vs multiple dosing), and comorbidities may account for the observed differences. Cytochrome P450 3A4 can be down-regulated in patients with cancer, which could result in 20–70% reductions in expression and thus cause lower clearance and higher exposure of its substrates [14, 31,32,33]. Ribociclib is a CYP3A4 substrate and showed a generally lower oral clearance in patients with cancer versus non-cancer subjects, although this difference was not considered clinically relevant based on the PK variability of ribociclib [14]. Ribociclib exposure was approximately 30% higher in subjects with moderate and severe HI; however, the increase was modest and therefore would not explain the approximate two-fold increase observed in subjects with moderate and severe RI. A substantial proportion of patients (~ 60%) assessed in our clinical trials had mild/moderate RI, which is consistent with reports that only 39% of patients with breast cancer have normal GFR [17], highlighting the clinical importance of this analysis.
The strengths of this analysis stem from the use of data close to real-world settings. It comprises both single-dose and steady-state PK of ribociclib administered at the recommended starting dose of 600 mg with the approved regimen of 3 weeks on/1 week off under long-term usage, in the target patient population. Furthermore, the PK analyses of patient data were based on much larger populations (N = 1104 in the pooled safety dataset) compared with that of the dedicated RI study in subjects with RI (N = 35) [Table S6 of the ESM]. More importantly, the PK-related findings were supplemented with the analysis of efficacy and safety data from four phase II and III clinical trials (including three pivotal phase III trials), which provides robust clinical evidence of the benefit-risk profile of ribociclib in patients with ABC and RI.
This work provides a more holistic approach on how to handle clinical pharmacology data when the results deviate from the real-world setting. The issue can be very pertinent in clinical development and life-cycle management of novel medicines, especially in oncology, where patients and treatment regimens are more heterogeneous because of the complexity in co-medications, comorbidities, disease status, and metastatic setting. Hence, based on the strengths of the patient PK, safety, and efficacy data, the same starting dose of ribociclib 600 mg as used for patients with normal renal function was recommended for patients with mild and moderate RI.
Finally, with respect to severe RI, the lack of data in patients receiving ribociclib during clinical development led us to a more conservative approach to rely upon the dedicated RI study results, wherein a 2.67-fold and 2.30-fold increase in AUCinf and Cmax were observed in subjects with severe RI versus those with normal renal function. Hence, a reduced starting dose of ribociclib 200 mg was recommended for this population. As previous analyses demonstrated that dose reductions of ribociclib from 600 mg to 400 and 200 mg do not compromise PFS or overall survival [34, 35], the reduced starting dose in this patient population is not expected to affect efficacy. The recommended doses of ribociclib for patients with mild, moderate, and severe RI were approved by various health authorities worldwide [1, 2].
5 Conclusions
This report provides a comprehensive analysis to determine dose in patients with RI. It went beyond a dedicated clinical pharmacology RI study in subjects by leveraging the PK, safety, and efficacy data of clinical trials from the target patient population, and integrating prior knowledge to arrive at the dose recommendation for patients with RI. The findings supported the current prescribing information for ribociclib in patients with hormone receptor-positive, human epidermal growth factor receptor-2 negative ABC, and RI. Such a holistic and innovative approach can represent an alternative and effective strategy to drive dose recommendations in the indicated patient populations with RI.
References
EMA. Summary of product characteristics. 2017. https://www.ema.europa.eu/en/documents/product-information/kisqali-epar-product-information_en.pdf. Accessed 17 Jun 2022.
US FDA. US Prescribing information. 2017. https://www.novartis.us/sites/www.novartis.us/files/kisqali.pdf. Accessed 17 Jun 2022.
Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375(18):1738–48.
Tripathy D, Im SA, Colleoni M, Franke F, Bardia A, Harbeck N, et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A randomised phase 3 trial. Lancet Oncol. 2018;19(7):904–15.
Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol. 2018;36(24):2465–72.
Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol. 2018;29(7):1541–7.
Im SA, Lu YS, Bardia A, Harbeck N, Colleoni M, Franke F, et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N Engl J Med. 2019;381(4):307–16.
Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Overall survival with ribociclib plus fulvestrant in advanced breast cancer. N Engl J Med. 2019;382(6):514–24.
Infante JR, Cassier PA, Gerecitano JF, Witteveen PO, Chugh R, Ribrag V, et al. A phase I study of the cyclin-dependent kinase 4/6 inhibitor rRibociclib (LEE011) in patients with advanced solid tumors and lymphomas. Clin Cancer Res. 2016;22(23):5696–705.
Yamada Y, Ishikawa N, Kakizume T, Tajima T, Hewes B, Doi T. Abstract B31: A phase I study of single-agent ribociclib in Japanese patients with advanced solid tumors. Mol Cancer Ther. 2015;14(122):B31-B.
Ji Y, Abdelhady AM, Samant TS, Yang S, Rodriguez LK. Evaluation of absolute oral bioavailability and bioequivalence of ribociclib, a cyclin-dependent kinase 4/6 inhibitor, in healthy subjects. Clin Pharmacol Drug Dev. 2020;9(7):855–66.
Laisney M, Heimbach T, Mueller-Zsigmondy M, Blumenstein L, Costa R, Ji Y. Physiologically based biopharmaceutics modeling to demonstrate virtual bioequivalence and bioequivalence safe-space for ribociclib which has permeation rate-controlled absorption. J Pharm Sci. 2022;111(1):274–84.
Doi T, Hewes B, Kakizume T, Tajima T, Ishikawa N, Yamada Y. Phase I study of single-agent ribociclib in Japanese patients with advanced solid tumors. Cancer Sci. 2018;109(1):193–8.
Samant TS, Huth F, Umehara K, Schiller H, Dhuria SV, Elmeliegy M, et al. Ribociclib drug-drug interactions: clinical evaluations and PBPK modeling to guide drug labeling. Clin Pharmacol Ther. 2020;108(3):575–85.
James AD, Schiller H, Marvalin C, Jin Y, Borell H, Roffel AF, et al. An integrated assessment of the ADME properties of the CDK4/6 inhibitor ribociclib utilizing preclinical in vitro, in vivo, and human ADME data. Pharmacol Res Perspect. 2020;8(3): e00599.
GBD. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390(10100):1211–59. https://www.sciencedirect.com/science/article/pii/S0140673617321542?via%3Dihub
Launay-Vacher V, Janus N, Deray G. Renal insufficiency and cancer treatments. ESMO Open. 2016;1(4): e000091.
Launay-Vacher V, Gligorov J, Le Tourneau C, Janus N, Spano JP, Ray-Coquard I, et al. Prevalence of renal insufficiency in breast cancer patients and related pharmacological issues. Breast Cancer Res Treat. 2010;124(3):745–53.
Xiao JJ, Chen JS, Lum BL, Graham RA. A survey of renal impairment pharmacokinetic studies for new oncology drug approvals in the USA from 2010 to early 2015: a focus on development strategies and future directions. Anticancer Drugs. 2017;28(7):677–701.
Nolin TD, Naud J, Leblond FA, Pichette V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin Pharmacol Ther. 2008;83(6):898–903.
Abdel-Kahaar E, Zolk O. Prescribing of anticancer drugs in renal impairment: why can’t we do better? Naunyn Schmiedebergs Arch Pharmacol. 2018;391(2):107–9.
US FDA. May 7, 2019: Meeting of the pharmaceutical science and clinical pharmacology advisory committee meeting announcement. 2019. https://www.fda.gov/advisory-committees/advisory-committee-calendar/may-7-2019-meeting-pharmaceutical-science-and-clinical-pharmacology-advisory-committee-meeting. Accessed 17 Jun 2022.
EMA. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. 2016. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-decreased-renal-function_en.pdf. Accessed 17 Jun 2022.
FDA. Pharmacokinetics in patients with impaired renal function: study design, data analysis, and impact on dosing and labeling. 2010. https://www.fda.gov/media/78573/download. Accessed 17 Jun 2022.
Munster PN, Hamilton EP, Estevez LG, De Boer RH, Mayer IA, Campone M, et al. Ph IB study of LEE011 and BYL719 in combination with letrozole in ER+, HER2- breast cancer. J Clin Oncol. 2014;32(26_Suppl.):143.
Lu YS, Im SA, Colleoni M, Franke F, Bardia A, Cardoso F, et al. Updated overall survival of ribociclib plus endocrine therapy versus endocrine therapy alone in pre- and perimenopausal patients with HR+/HER2− advanced breast cancer in MONALEESA-7: a phase III randomized clinical trial. Clin Cancer Res. 2022;28(5):851–9.
Slamon DJ, Neven P, Chia S, Jerusalem G, De Laurentiis M, Im S, et al. Ribociclib plus fulvestrant for postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer in the phase III randomized MONALEESA-3 trial: updated overall survival. Ann Oncol. 2021;32(8):1015–24.
Lu Y, Yang S, Ho Y-Y, Ji Y. Ribociclib population pharmacokinetics and pharmacokinetic/pharmacodynamic analysis of neutrophils in cancer patients. J Clin Pharmacol. 2021;61(8):1054–68.
Samant TS, Yang S, Miller M, Ji Y. Pharmacokinetics of ribociclib in subjects with hepatic impairment. J Clin Pharmacol. 2021;61(8):1001–9.
US FDA. US Prescribing information. 2020. https://www.novartis.us/sites/www.novartis.us/files/Femara.pdf?TB_iframe=true. Accessed 17 Jun 2022.
Cheeti S, Budha NR, Rajan S, Dresser MJ, Jin JY. A physiologically based pharmacokinetic (PBPK) approach to evaluate pharmacokinetics in patients with cancer. Biopharm Drug Dispos. 2013;34(3):141–54.
Coutant DE, Kulanthaivel P, Turner PK, Bell RL, Baldwin J, Wijayawardana SR, et al. Understanding disease-drug interactions in cancer patients: implications for dosing within the therapeutic window. Clin Pharmacol Ther. 2015;98(1):76–86.
Schwenger E, Reddy VP, Moorthy G, Sharma P, Tomkinson H, Masson E, et al. Harnessing meta-analysis to refine an oncology patient population for physiology-based pharmacokinetic modeling of drugs. Clin Pharmacol Ther. 2018;103(2):271–80.
De Laurentiis M, Merino LdlC, Hart L, Bardia A, Im SA, Sohn J, et al. 331P Impact of ribociclib (RIB) dose reduction on overall survival (OS) in patients (pts) with HR+/HER2- advanced breast cancer (ABC) in MONALEESA (ML) -3 and -7. Ann Oncol. 2020;31:S378–9.
Beck JT, Neven P, Sohn J, Chan A, Sonke GS, Bachelot T, et al. Ribociclib treatment benefit in patients with advanced breast cancer with ≥1 dose reduction: data from the MONALEESA-2, -3, and -7 trials. Cancer Res. 2019;79(4D):P6-18–06.
Acknowledgments
We thank the subjects who took part in this trial and their families, as well as the staff members at each study site. Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals. We thank Serge Winter for his contribution to the pharmacokinetic sample analysis, Yasong Lu and Tanay Samant, formerly of Novartis Pharmaceuticals, for their contribution to the population pharmacokinetic analysis and Study A2116, respectively, and Sara Henriques, Healthcare Consultancy Group LLC, and Bhavana Muddana, Novartis Healthcare Pvt Ltd, for their medical editorial assistance with this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The studies were supported by Novartis Pharmaceuticals Corporation, who also funded medical writing assistance.
Conflict of Interest
Yan Ji, Vitaly Yartsev, Michelle Quinlan, Yingbo Wang, Paolo Serra, Abhijit Chakraborty, and Michelle Miller are employees of Novartis and hold stock in the company.
Ethical Approval
All studies were conducted according to the relevant ethical principles, and were approved by the local ethics committee.
Consent to Participate
Written informed consent was obtained from all patients participating in the study.
Consent for Publication
Not applicable (no personal data were reported).
Availability of Data and Material
Novartis will not provide access to patient-level data, if there is a reasonable likelihood that individual patients could be re-identified. Phase I studies, by their nature, present a high risk of patient re-identification; therefore, patient individual results for phase I studies cannot be shared. In addition, clinical data, in some cases, have been collected subject to contractual or consent provisions that prohibit transfer to third parties. Such restrictions may preclude granting access under these provisions. Where co-development agreements or other legal restrictions prevent companies from sharing particular data, companies will work with qualified requestors to provide summary information where possible.
Code Availability
SAS® version 9.4 (Cary, NC, USA); Phoenix WinNonlin™ version 6.4 (Pharsight, Mountain View, CA, USA).
Author Contributions
YJ, MM, VY, and AC designed the research. YJ and VY supported in the acquisition of data and data curation. YJ, MQ, PS, YW, and VY analyzed, interpreted, and validated the data. All authors wrote or reviewed the manuscript. YJ, MQ, YW, PS, and VY provided administrative, technical, or material support. YJ, AC, and MM supervised the development of the manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ji, Y., Yartsev, V., Quinlan, M. et al. Justifying Ribociclib Dose in Patients with Advanced Breast Cancer with Renal Impairment Based on PK, Safety, and Efficacy Data: An Innovative Approach Integrating Data from a Dedicated Renal Impairment Study and Oncology Clinical Trials. Clin Pharmacokinet 62, 493–504 (2023). https://doi.org/10.1007/s40262-022-01206-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-022-01206-2