
Abstract
Background and Objective
Pexidartinib is a novel oral small-molecule inhibitor that selectively targets colony-stimulating factor 1 receptor, KIT proto-oncogene receptor tyrosine kinase, and FMS-like tyrosine kinase 3 harboring an internal tandem duplication mutation. It is approved in the United States for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) associated with severe morbidity or functional limitations and not amenable to improvement with surgery. Pexidartinib in vitro data indicate the potential for absorption- and metabolism-related drug–drug interactions (DDIs). The objective was to present a comprehensive DDI risk assessment of agents that can impact pexidartinib exposure by altering its absorption and metabolism potentially affecting efficacy and safety of pexidartinib.
Methods
Four open-label crossover studies were performed to assess the effects of a pH modifier (esomeprazole), a strong cytochrome P450 (CYP) 3A4 inhibitor (itraconazole), a strong CYP3A/5′-diphospho-glucuronosyltransferase (UGT) inducer (rifampin), and a UGT inhibitor (probenecid) on the single-dose pharmacokinetics of pexidartinib. In addition, a physiologically based pharmacokinetic model was developed to predict the effect of a moderate CYP3A4 inhibitor (fluconazole) and a moderate CYP3A inducer (efavirenz) on the pharmacokinetics of pexidartinib.
Results
Co-administration of pexidartinib with esomeprazole modestly decreased pexidartinib exposure (maximum plasma concentration [Cmax], ng/mL: geometric mean ratio [90% confidence interval (CI)], 45.4% [36.8–55.9]; area under the drug plasma concentration–time curve from time 0 to infinity [AUC∞], ng•h/mL: geometric mean ratio [90% CI], 53.1% [47.4–59.3]), likely related to decreased solubility of pexidartinib at increased pH levels. As expected, the strong CYP3A4 inhibitor itraconazole increased pexidartinib exposure (Cmax, ng/mL: geometric mean ratio [90% CI], 148.3% [127.8–172.0]; AUC∞, ng•h/mL: geometric mean ratio [90% CI], 173.0% [160.7–186.3]) while the strong CYP3A/UGT inducer rifampin decreased exposure (Cmax, ng/mL: geometric mean ratio [90% CI], 67.1% [53.1–84.8]; AUC∞, ng•h/mL: geometric mean ratio [90% CI], 37.0% [30.6–44.8]). In addition, UGT inhibition increased pexidartinib exposure (Cmax, ng/mL: geometric mean ratio [90% CI], 105.8% [92.4–121.0]; AUC∞, ng•h/mL: geometric mean ratio [90% CI], 159.8% [143.4–178.0]), consistent with the fact that pexidartinib is a substrate of the UGT1A4 enzyme, which is responsible for the generation of the major metabolite, ZAAD-1006a.
Conclusions
The physiologically based pharmacokinetic model predicted that a moderate CYP3A4 inhibitor and a moderate CYP3A inducer would produce modest increases and decreases, respectively, in pexidartinib exposure. These results provide a basis for pexidartinib dosing recommendations when administered concomitantly with drugs with drug–drug interaction potential, including dose adjustments when concomitant administration cannot be avoided.
Clinical Trial Registration
Probenecid: phase I trial, NCT03138759, 3 May, 2017; esomeprazole, itraconazole, rifampin: phase I trials, not registered with ClinicalTrials.gov.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cytochrome P450 (CYP) 3A4 inhibitors, CYP3A inducers, 5′-diphospho-glucuronosyltransferase (UGT) inhibitors, and pH modifiers significantly affect and alter pexidartinib pharmacokinetics. |
These findings offer valuable insight for treating clinicians on how to safely administer certain agents (CYP3A4 inhibitors, CYP3A inducers, UGT inhibitors, and pH modifiers) in combination with pexidartinib. |
1 Background
Pexidartinib is an orally active, small-molecule kinase inhibitor that selectively targets colony-stimulating factor 1 receptor, KIT proto-oncogene receptor tyrosine kinase, and FMS-like tyrosine kinase 3 harboring an internal tandem duplication mutation [1, 2]. Based on the ENLIVEN trial, pexidartinib is approved for the treatment of adults with symptomatic tenosynovial giant cell tumor associated with severe morbidity or functional limitations and not amenable to surgery [1, 3]. In vitro data suggest that pexidartinib has the potential for absorption-related and metabolism-related drug–drug interactions (DDIs).
Pexidartinib solubility decreases as pH increases, suggesting that alterations in gastrointestinal pH may influence the absorption of pexidartinib prior to metabolism, resulting in altered bioavailability. Following oral administration, pexidartinib is extensively metabolized [1]. In vitro studies indicate cytochrome P450 (CYP) 3A4 as the main enzyme responsible for the metabolism of pexidartinib. In addition, the major metabolite of pexidartinib in plasma (ZAAD-1006a) is a N-glucuronide conjugate with an exposure similar to the parent compound. In vitro data indicate that ZAAD-1006a is generated by 5′-diphospho-glucuronosyltransferase (UGT). Therefore, altered enzymatic activity of any of these metabolizing enzymes due to induction or inhibition could potentially modify pexidartinib exposure. Overall, pexidartinib has DDI potential when administered with agents that influence pH or modify CYP3A4 and/or UGT activity and that have the potential to influence the pharmacokinetic (PK) profile of pexidartinib, resulting in a potential increase or decrease in pexidartinib exposure.
This report describes the results of four open-label crossover studies evaluating the effects of a pH modifier (esomeprazole), a strong CYP3A4 inhibitor (itraconazole) and a strong CYP3A/UGT inducer (rifampin), and a UGT inhibitor (probenecid) on the single-dose pharmacokinetics of pexidartinib. Furthermore, the effect of a moderate CYP3A4 inhibitor and inducer (fluconazole and efavirenz, respectively) on the single-dose pharmacokinetics of pexidartinib was assessed through physiologically based PK (PBPK) modeling. The overall objective is to present a comprehensive DDI risk assessment of the class of agents that can influence exposure of pexidartinib by altering its absorption and metabolism, potentially affecting the efficacy and safety of pexidartinib. The results from clinical studies and PBPK modeling were utilized to develop a dosing recommendation for pexidartinib when co-administered with a class of agents with DDI potential. The derived dosing recommendations based on the results from these studies are included in the US package insert [1].
2 Materials and Methods
2.1 Study Designs
Four separate phase I studies with one or two treatment sequences were conducted in healthy subjects at single sites in the USA. The studies evaluated the effect of a (1) pH modifier (proton pump inhibitor, esomeprazole); (2) strong CYP3A4 inhibitor (itraconazole); (3) strong CYP3A/UGT inducer (rifampin); and (4) UGT inhibitor (probenecid) on the single-dose pharmacokinetics of pexidartinib. The studies were conducted in compliance with the ethical principles of the Declarations of Helsinki and the International Conference on Harmonization consolidated Guideline E6 for Good Clinical Practice and approved by the applicable institutional review boards. All subjects provided written informed consent prior to study participation.
In all studies, subjects were screened from day − 21 through day − 1, and eligible individuals were confined at the clinical pharmacology unit for the study duration. The high-level design of the studies is presented in Fig. 1a–d. All four studies were two-treatment crossover studies investigating the pharmacokinetics of pexidartinib when administered alone and co-administered with perpetrator drugs. There was a washout period between periods and treatment, 10 days for the esomeprazole, rifampin, and itraconazole studies and 17 days in the probenecid study (Fig. 1a–d). A pexidartinib dose of 600 mg (3 × 200-mg capsules) was utilized in all studies. Depending on the study, the following doses were utilized, esomeprazole 40 mg, itraconazole 200-mg oral solution (20 mL as 10 mg/mL), rifampin 600 mg (2 × 300 mg capsules), and probenecid 500 mg (1 × 500-mg tablet). In all four studies, when pexidartinib was administered alone and on the day of co-administration, study drugs were administered in the morning with 240 mL of water after an overnight fast of at least 8–10 hours, followed by a continued fast for an additional 4 hours.

Study designs: a, esomeprazole study: an open-label randomized, two-treatment, two-period crossover study; b, itraconazole study: a single-sequence, two-treatment, two-period crossover study; c, rifampin study: a single-sequence, two-treatment, two-period crossover study; d, probenecid study: an open-label randomized, two-treatment, two-period crossover study
2.2 Subjects
Inclusion and exclusion criteria were identical between the four studies with minor exceptions.
2.2.1 Inclusion Criteria
Eligible subjects were healthy subjects aged 18–60 years with a body mass index between 18 and 30 kg/m2 (18–32 kg/m2 in the UGT inhibitor study). Female subjects required a negative serum pregnancy test, to be non-lactating, and either surgically sterile or naturally postmenopausal for at least 12 consecutive months, while male subjects were either surgically sterile or agreed to use double-barrier methods of contraception. Subjects required a negative urine test for drugs of abuse, human immunodeficiency virus antibody, hepatitis surface antigen, and hepatitis C virus antibody, and willing to refrain from consuming caffeine/xanthine or alcohol from 48 h prior to check-in until the end of the study, and willing to refrain from consuming grapefruit or Seville oranges 7 days prior to and through the end of the study.
2.2.2 Exclusion Criteria
Subjects with a history or current evidence of any significant disorder potentially preventing the successful completion of the study, and a history of clinically significant neutropenia were excluded. Those with clinical laboratory results outside the normal range and considered clinically significant, a history of stomach or intestinal surgery or resection that may alter drug absorption, or a history of alcoholism or drug addiction with 1 year or use of tobacco-containing or nicotine-containing products within 6 months were also excluded. Subjects were prohibited from taking prescription or over-the-counter medications within 14 days prior to the study (except acetaminophen and topical hydrocortisone cream) or medications known to induce or inhibit CYP3A4, CYP2C9, or UGT (UGT inhibitor study only) within 28 days prior to the study.
2.3 Assessments
Serial plasma samples were collected at the following timepoints for the measurements of pexidartinib and ZAAD-1006a: 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 8, 10, 12, 24, 36, 48, 72, 96, and 120 hours. In the strong CYP3A4 inhibitor (itraconazole) and UGT inhibitor (probenecid) studies, additional samples were collected at 144, 168, 192, 216, 240, 264, 288, and 312 hours post-pexidartinib dose. Plasma samples were analyzed for quantification of pexidartinib using the validated liquid chromatography-tandem mass spectrometry method over a concentration range of pexidartinib of 30–3750 ng/mL. For estimation of precision and accuracy of the assay, quality control samples were prepared at four concentrations (10.0, 30.0, 220, and 3750 ng/mL). Precision (coefficient of variation) was 0.8–7.2% within the run, and 3.3–6.3% between runs; accuracy was − 7.7 to 7.0% within the run, and − 3.3 to 0.3% between runs. Dilution integrity was verified at a concentration up to 30,000 ng/mL when diluted 20-fold. Recovery of pexidartinib was between 91 and 97% with the internal standard being 94%. Short-term stability was similar (24–26 h cumulative stability; 51–55 h [total all thaw cycles]) at − 20 °C and −80 °C storage, and between 25,000 and 30,000 ng/mL concentrations. Freeze-thaw stability consisted of seven freeze-thaws at − 20 °C. ZAAD-1006a was analyzed using a qualified liquid chromatography-tandem mass spectrometry assay over a concentration range of 10–5000 ng/mL. Precision (coefficient of variation) was 1.5–11.4% within the run, and 7.2–11.8% between runs; accuracy was − 14.1 to 17.0% within the run, and − 2.3 to 5.0% between runs. Dilution integrity was verified at a concentration up to 25,000 ng/mL when diluted ten-fold, and up to 45,000 ng/mL when diluted 15-fold. Recovery of pexidartinib was between 88 and 92% with the internal standard being 100%. Short-term stability in polypropylene tubes was 6 hours when stored at − 20 °C and 24 hours when stored at − 80 °C. Long-term stability storage at − 20 °C was over 400 days compared with 253 days at − 80 °C storage. Freeze-thaw stability was seven freeze-thaws at − 20 °C compared with five freeze-thaws at − 80 °C. Analyses performed to assess the interference of esomeprazole, itraconazole, and rifampin on the liquid chromatography-tandem mass spectrometry assay for the measurement of pexidartinib plasma concentrations revealed no interferences on the assay.
Safety endpoints included adverse events, physical examination, vital signs, a 12-lead electrocardiogram, and laboratory assessments. Adverse events were assessed by the investigator for severity and causality to study medication.
2.4 Data Analysis
The PK analysis set included all subjects who received a dose of pexidartinib and had sufficient plasma concentration data to characterize PK parameters, while the safety analysis set included all subjects who received at least one dose of pexidartinib. Pharmacokinetic endpoints included maximum plasma concentration (Cmax), time to reach Cmax (tmax), area under the drug plasma concentration–time curve from time 0 to infinity (AUC∞), and half-life for pexidartinib and ZAAD-1006a when pexidartinib was administered alone or in combination with the perpetrator drug. The metabolite-to-parent exposure ratios (MPRs) were reported for Cmax, and AUC∞. Pharmacokinetic parameters for pexidartinib and ZAAD-1006a were computed using WinNonlin Professional (Version 6.4). Plasma concentration values that were below the limit of quantification at the beginning and end of a PK profile were set to zero, provided that the below the limit of quantification values were not flanked by measurable concentrations. Below the limit of quantification values that were flanked at the adjacent time by measurable concentrations were set to missing in the PK calculations. The mean PK parameters of pexidartinib and ZAAD-1006a were summarized descriptively.
The primary statistical analysis compared Cmax and AUC∞ of pexidartinib with and without the co-administration of the perpetrator drug using an analysis of variance (ANOVA) model for the natural log-transformed values of Cmax and AUC∞. The model included factors accounting for the following sources of variation: treatment, period, sequence, and subjects nested in sequences. Within the framework of an ANOVA, two-sided 90% confidence intervals (CIs) for geometric least-square mean ratios of PK parameters were calculated. Safety outcomes were analyzed descriptively.
2.4.1 PBPK Model
Based on the available in vitro and clinical data, the pharmacokinetics of pexidartinib is affected by strong CYP3A4 inhibitors and inducers. Therefore, a mechanistic PBPK model was developed to predict the effect of a moderate CYP3A4 inhibitor and inducer on pexidartinib pharmacokinetics. A base minimal PBPK model, with a single adjusting compartment, which considers metabolism in the liver, intestine, and kidney, and compiles other tissues together, was utilized for a pexidartinib PBPK analysis and further simulations (Simcyp Simulator Version 15, Certara Simcyp™).
2.4.2 Optimization of the ka Value
A sensitivity analysis was completed comparing the impact of pexidartinib ka on simulated Cmax across a range of ka values (0.12–1.2 h−1). The ka value was set to 0.12 h−1 in the original model, which was optimized using in vivo data for the 1200-mg dose obtained in healthy subjects from a different study (PL3397-A-U121). In the final model, a ka value of 0.6 h−1 in conjunction with a tlag time of 0.5 h was used to recover the observed steady-state Cmax and tmax in the clinical study after administration of multiple 400-mg oral doses of pexidartinib in subjects with cancer in the fasted state. Because of the inter-study variability in the observed Cmax of pexidartinib, a ka value of 0.24 h−1 was employed to retrieve the observed steady-state Cmax and tmax of pexidartinib in subjects with cancer. A ka value of 0.12 h−1 (as used in the original model) was utilized to recover the observed Cmax and tmax of pexidartinib in healthy subjects.
2.4.3 Fugut Input
Initial simulations showed that fugut set to 1 predicted a low F value of 0.06, resulting in insignificant under-estimations of Cmax and area under the drug plasma concentration–time curve (AUC) of pexidartinib following a single-dose administration of 400 mg. The predicted low F value was due to significant gut metabolism mediated by CYP3A4, and the default fugut value of 1 allowed for 100% drug availability for gut metabolism. A subsequent simulation indicated that fugut set to a predicted value (0.0038) led to a predicted F value of 0.5, improving the prediction of Cmax and AUC of pexidartinib. The forecasting of fugut utilized physchem, blood binding, and tissue composition inputs on the basis of methods for tissue distribution [4].
2.4.4 Optimization of fmCYP3A4
The model estimated the contribution of CYP3A to the metabolism of pexidartinib (fmCYP3A4) was 0.45. The fmCYP3A4 of the model was verified against the observed DDI effects in the strong CYP3A4 inhibitor (itraconazole) and the strong CYP3A/UGT inducer (rifampin) DDI study. Thereafter, the model was used to estimate the impact of moderate CYP3A4 inhibitors (fluconazole) and moderate CYP3A4 inducers (efavirenz) on the pharmacokinetics of pexidartinib for drugs that have not been evaluated within in vivo DDI studies.
2.4.5 Optimization of CYP Induction Data
The in vitro data showed that pexidartinib is a mechanism-based inactivator of CYP3A4 with a Ki (total) of 4.01 μM and a kinact of 1.356 h−1. In a human hepatocyte assay, pexidartinib was shown to induce CYP3A4. A review of the Emax and EC50 estimates determined in vitro revealed significant uncertainty associated with both parameters (i.e., wide 95% CI estimated), due to data variability. Hence, a sensitivity analysis was performed to determine the optimal combination of Emax (range 6–16) and EC50 (range 0.1–0.3 µM) estimates, enabling recovery of the observed DDI effect on midazolam (data on file). The sensitivity analysis was restricted to a narrow range, additional sensitivity analyses showed that a wider range of CYP3A4 induction parameters (i.e., more potent induction) led to a significant under-estimation of pexidartinib steady-state drug exposures. The Emax value was capped at 16 on the basis of (1) the Emax value of the most potent inducer, rifampicin, is 16 in the Simcyp model and (2) in vitro, the Emax value for pexidartinib is lower than those for rifampicin across three donors in the in vitro study. Based on the sensitivity analysis, Emax (= 16) and EC50 (= 0.16 µM) were identified.
2.4.6 CYP Enzyme Induction Mechanism
Prediction of drug interactions involving multiple inducers for the same enzyme is complex, as multiple inducers may affect the same or different transcription factors. The current PBPK analysis used the “default” option, which is the only option available in Simcyp simulator up to Version 15.1 (the version in this analysis). The “default” option assumes that when multiple inducers are co-administered, the highest induction factor of the compounds applied in the enzyme induction model is used to predict the net induction effect. Two additional options were introduced in Simcyp Version 16.1, additive and multiplicative options. These two additional options provide additional flexibility to investigate the net effect when multiple inducers are co-administered.
2.4.7 Final Model Parameters
All of the parameters utilized in the model are shown in Table 1 of the Electronic Supplementary Material (ESM).
2.4.8 Model Verification
2.4.8.1 Simulation of Plasma Concentration–Time Profiles of Pexidartinib in Patients After Multiple Daily Doses of 400–1000 mg for 15 Days (Fasted)
Verification of the final model for pexidartinib focused on daily doses between 400 mg and 1000 mg because (1) the target dosing regimen is 400 mg twice daily (BID) and (2) at lower dose levels there was a small number of patients (n = 3) per dose cohort. Simulated and observed plasma concentration–time profiles of pexidartinib in patients who received 400 mg, 600 mg, and 1000 mg (given as split doses of 400 mg/600 mg or 500 mg BID) of pexidartinib per day for 15 days are shown in Figs. 1, 2, 3a–d, and 4a–d of the ESM. A summary of predicted and observed PK parameters for pexidartinib are shown in Tables 2–5 of the ESM.


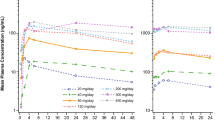
Mean (± standard deviation [SD]) plasma pexidartinib concentration–time profiles by treatment: a, esomeprazole study; b, itraconazole study; c, rifampin study; d, probenecid study. Note: Figures on the left are presented on a natural scale; figures on the right are presented on a logarithmic scale


Mean (± standard deviation [SD]) plasma ZAAD-1006a concentration–time profiles by treatment: a, esomeprazole study, b, itraconazole study, c, rifampin study, d, probenecid study

Forest plot (odds ratio) of the effect of a pH modifier (esomeprazole): a strong cytochrome P450 (CYP) 3A inhibitor (itraconazole) and strong CYP3A inducer (rifampin), and a 5′-diphospho-glucuronosyltransferase inhibitor (probenecid) and the simulated effects of a moderate CYP3A inhibitor (fluconazole) and a moderate CYP3A inducer (efavirenz) on the single dose: a, maximum plasma concentration (Cmax); and b, area under the drug plasma concentration–time curve from time 0 to infinity (AUC∞) of pexidartinib presented as 90% confidence intervals
For daily dose levels with a range from 400 to 1000 mg, the predicted mean day 15 Cmax and AUC from time 0 to 24 hours or AUC from time 0 to 6 hours values were within 0.66-fold to 1.39-fold of the observed data (Tables 2–5 of the ESM). The predicted mean day 1 Cmax and AUC from time 0 to 24 hours values were within 0.90-fold to 1.23-fold of the observed data at the dose level of 400 mg or 600 mg. However, there was a significant over-estimation of the day 1 Cmax and AUC from time 0 to 6 hours values at higher doses of 1000 mg.
A separate set of simulations were performed using the Sim-Healthy Subjects population. A comparison of the predicted PK parameters for pexidartinib using the two different populations showed that the predicted PK parameters are similar across all dose levels. This is expected because the key population characteristics considered in the cancer population include demographic data, reduced renal function, altered plasma binding proteins, and different transporter abundance (e.g., OATP1B1/3 and MATE1). Importantly, no change in the CYP3A4 activity was incorporated in this population.
2.4.8.2 Simulation of Plasma Concentration–Time Profiles of Pexidartinib in Healthy Subjects After a Single Dose of 600 mg With and Without Itraconazole Treatment (Fasted)
Simulations using the final model predicted geometric mean AUC∞ and Cmax ratios of 1.80 (95% CI 1.75–1.84) and 1.29 (95% CI 1.27–1.32), respectively, which are consistent with the reported ratios of 1.73 (95% CI 1.58–1.89) and 1.48 (95% CI 1.24–1.78), respectively, following concurrent itraconazole treatment (Table 6 and Fig. 5a–b of the ESM). These results are expected because the fmCYP3A4 value for pexidartinib was kept at 45% in the final model, and the effect of CYP3A4 induction is minimal after a single dose.
2.4.8.3 Simulations to Impact of Rifampicin on the Pharmacokinetics of Single-Dose and Multiple-Dose Pexidartinib
Mean simulated plasma pexidartinib concentrations following a single oral dose of pexidartinib 400 mg in the absence of rifampicin, and on the 11th day of 20 days of dosing of rifampicin (600 mg once daily [QD]) to subjects with cancer are shown in Fig. 6 of the ESM. Mean simulated plasma pexidartinib concentrations following multiple oral doses of 400 mg BID in the absence and presence of rifampicin (600 mg QD) to subjects with cancer are shown in Fig. 7 of the ESM.
The predicted mean Cmax and AUC∞ or AUC from time 0 to 12 hours values and corresponding geometric mean ratios for pexidartinib in the presence and absence of rifampicin are shown in Table 7 of the ESM. The results indicate that all three induction models were able to predict the effect of rifampin on pexidartinib pharmacokinetics.
The simulated CYP3A4 activity profiles following single or multiple oral doses of pexidartinib (400 mg) in the absence and presence of rifampicin 600 mg QD in subjects with cancer are shown in Figs. 8a–b and 9a–b of the ESM. The hepatic CYP3A4 activity was induced up to 630% and 510%, respectively, following single and multiple oral doses of pexidartinib, respectively.
2.4.9 Simulations
Simulations were performed to estimate the effect of a moderate CYP3A4 inhibitor (fluconazole) and CYP3A4 inducer (efavirenz) on the PK parameters of pexidartinib. Predictions were based on ten virtual trials of ten subjects with cancer (50% female) aged 21–85 years for both fluconazole and efavirenz. In the fluconazole simulations, each virtual subject received a single 400-mg dose of pexidartinib orally on day 8, in the presence and absence of fluconazole treatment (400 mg on day 1 and 200 mg QD on days 2 through 20). Separate simulations were performed to assess the effect of fluconazole on multiple doses of pexidartinib 400 mg BID. In the efavirenz simulations, each virtual subject received a single 400-mg dose of pexidartinib orally alone and on the 11th day of the 20-day regimen with efavirenz (600 mg QD). In the multiple-dose efavirenz simulations, each virtual subject received multiple doses of pexidartinib 400 mg BID in the presence and absence of efavirenz 600 mg given QD for 15 days.
3 Results
3.1 Subjects
There were 16 subjects in each study. All 16 subjects completed the esomeprazole and rifampin DDI studies. Because of adverse events, three subjects discontinued the itraconazole DDI study after receiving pexidartinib alone and did not continue to receive pexidartinib plus itraconazole. One subject discontinued the probenecid DDI study because of an AE after receiving probenecid, 15 subjects received pexidartinib alone, and 13 subjects received pexidartinib with probenecid.
Subject characteristics are summarized in Table 1. The subjects were predominantly male in all four studies (56.3−93.8%), and the mean age ranged from 39.9 to 43.3 years. Most subjects were white in the esomeprazole and itraconazole studies, while the majority were African American in the rifampin and probenecid studies.
3.2 Pharmacokinetics
The mean plasma concentrations of pexidartinib and ZAAD-1006a administered alone and with the perpetrator drug in all four studies are summarized in Figs. 2 and 3, respectively, while Table 2 summarizes pexidartinib and ZAAD-1006a PK parameters and ANOVA models when administered alone and in combination with perpetrator agents. The mean tmax values ranged from 2.0 to 3.0 h for pexidartinib and from 4.3 to 7.0 h for ZAAD-1006a.
3.2.1 Esomeprazole (pH Modifier) DDI Study
Based on the ANOVA, Cmax and AUC∞ for pexidartinib were decreased by approximately 55% (2917 ng/mL vs 6431 ng/mL) and 47% (55,378 ng•h/mL vs 104,389 ng•h/mL), respectively, when pexidartinib was administered with esomeprazole (Table 3, Fig. 4a–b). Similarly, ZAAD-1006a Cmax and AUC∞ values were decreased by 53% (2472 ng/mL vs 5289 ng/mL) and 48% (67,646 ng•h/mL vs 130,366 ng•h/mL), respectively, when the drug was co-administered with esomeprazole (Table 4). However, the mean MPR for Cmax and AUC∞ were not significantly affected by co-administration with esomeprazole (Table 3, Fig. 4a, b), indicating in healthy subjects with normal hepatic and renal function and not taking any metabolism-altering drug, ZAAD-1006a pharmacokinetics is formation rate-limited pharmacokinetics.
3.2.2 Itraconazole (Strong CYP3A4 Inhibitor) DDI Study
Concomitant administration of itraconazole increased pexidartinib Cmax and AUC∞ by approximately 48% and 73%, respectively (Table 3, Fig. 4a, b). Similarly, Cmax and AUC∞ of ZAAD-1006a were approximately 50% (5858 ng/mL vs 3892 ng/mL) and 97% (238,561 ng•h/mL vs 121,049 ng•h/mL) higher when pexidartinib was co-administered with itraconazole (Table 4). The mean MPR for Cmax was similar for both treatments, while the mean MPR for AUC∞ was slightly increased (approximately 15%) when pexidartinib and itraconazole were co-administered (Table 2).
3.2.3 Rifampin (Strong CYP3A Inducer) DDI Study
Concomitant administration of rifampin decreased pexidartinib Cmax and AUC∞ approximately 33% and 63%, respectively (Table 3, Fig. 4a, b). A 36% increase in the Cmax (5942 ng/mL vs 4384 ng/mL) and a 45% decrease in AUC∞ (67,655 ng•h/mL vs 122,014 ng•h/mL) of ZAAD-1006a (Table 4) were observed when pexidartinib was co-administered with rifampin. The MPRs for Cmax and AUC∞ were increased on co-administration with rifampin (Table 2).
3.2.4 Probenecid (UGT Inhibitor) DDI Study
Pexidartinib AUC∞ was increased on co-administration with probenecid by approximately 60%, although Cmax values were unchanged (Table 3, Fig. 4a, b). ZAAD-1006a AUC∞ was also increased by approximately 125% (203,649 ng•h/mL vs 90,364 ng•h/mL) with co-administration with no difference for Cmax (4138 ng/mL vs 4162 ng/mL) (Table 4). Concomitant administration also increased the MPR for AUC∞ by approximately 42% compared with pexidartinib alone (Table 2).
3.2.5 Predicted DDI Risk with Moderate CYP3A Inducers/Inhibitors Using a PBPK Model
The predicted impact of a moderate CYP3A4 inhibitor and inducer on the single-dose and multiple-dose pharmacokinetics of pexidartinib is summarized in Table 5 and Fig. 5. The moderate CYP3A4 inhibitor, fluconazole, was predicted to increase Cmax and AUC∞ by 15% (8180 ng/mL vs 7106 ng/mL) and 54% (83,692 ng•h/mL vs 54,378 ng•h/mL), respectively, after single doses, and increase by 38% (11,426 ng/mL vs 8288 ng/mL) and 65% (72,541 ng•h/mL vs 43,956 ng•h/mL), respectively, after multiple doses (Table 5). In contrast, the model predicted that the moderate CYP3A4 inducer, efavirenz, would decrease Cmax and AUC∞ by 25% (4558 ng/mL vs 6106 ng/mL) and 49% (25,794 ng•h/mL vs 50,308 ng•h/mL), respectively, after single doses, and decrease by 20% (6046 ng/mL vs 7521 ng/mL) and 28% (30,173 ng•h/mL vs 42,090 ng•h/mL), respectively, after multiple doses (Table 5, Fig. 5b).

Simulated mean plasma concentration–time profiles of pexidartinib: a, multiple oral doses of pexidartinib (400 mg twice daily) in the absence of (solid black line) and presence (dashed black line) of fluconazole 200 mg once daily (400 mg on day 1) in subjects with cancer using the original model; b, single oral dose of pexidartinib (400 mg) in the absence of (solid black line) efavirenz and on the eighth day of 20 days of dosing of efavirenz (dashed black line) 600 mg daily in subjects with cancer using the final model
3.3 Safety
Safety results were generally consistent with the previously defined safety profile of pexidartinib. All treatment-emergent adverse events (TEAEs) in the four studies were grade 1 or 2. In the esomeprazole study, three subjects experienced a TEAE, one each during pexidartinib alone, esomeprazole alone, and pexidartinib plus esomeprazole. All TEAEs were of grade 1 intensity and none of the events (abdominal pain, constipation, toothache, musculoskeletal chest pain, headache, oropharyngeal pain) was considered related to study drug administration. In the itraconazole study, nine subjects experienced a TEAE with seven assessed as related to the study drug (pexidartinib or itraconazole). The most reported TEAEs were contact dermatitis and pruritis (n = 3 each), abdominal pain, constipation, diarrhea, nausea, and ventricular tachycardia (n = 2 each). Two subjects receiving pexidartinib alone discontinued therapy because of ventricular tachycardia. In the rifampin study, a single subject experienced a grade 1 TEAE (diarrhea). Eight subjects in the probenecid study experienced a TEAE of which four were considered study drug related. All events were of mild severity except an increase in alanine aminotransferase (n = 1) and rash (n = 1), both grade 2, which were related to pexidartinib and probenecid, respectively. There was one discontinuation because of rash, determined to be related to probenecid.
4 Discussion
The current studies were designed to evaluate the in vivo drug interaction potential when pexidartinib is administered as a single dose (600 mg) in healthy subjects with a pH modifier, esomeprazole, and strong inhibitors/inducers of CYP3A4 and UGT (itraconazole, rifampin, probenecid). Additionally, a PBPK model was developed that allows the ability to predict the effect of other agents that have CYP3A4 inhibition/induction potential. The PBPK model was utilized to predict the effect of a moderate CYP3A4 inhibitor (fluconazole) and an inducer (efavirenz) on pexidartinib pharmacokinetics. The overall outcome of the DDI assessment is presented in Table 5. Pexidartinib dose reduction is recommended when co-administered with moderate and strong CYP3A and UGT inhibitors. Concurrent administration of pexidartinib with strong CYP3A inducers and proton pump inhibitors have been recommended to be avoided. The current recommendation of pexidartinib dosing with strong and moderate CYP3A4 inhibitors and inducers and UGT inhibitors and pH modifiers are based on the study results and PBPK modeling reported in the article.
The effect of esomeprazole on the pharmacokinetics of pexidartinib was evaluated because the solubility of pexidartinib decreases as pH increases, suggesting that alterations in gastrointestinal pH may influence drug absorption and, therefore, bioavailability of pexidartinib. The esomeprazole study showed that AUC∞ values of pexidartinib and ZAAD-1006a were decreased by approximately 47% each with no change in the MPR [1]. ZAAD-1006a has formation rate-limited pharmacokinetics in healthy subjects with normal hepatic and renal function, indicating its exposure is limited by the formation of ZAAD-1006 from pexidartinib and not by the elimination of ZAAD-1006a. Therefore, the decreased exposure of ZAAD-1006a with no change in the MPR when administered with esomeprazole indicates that the observed decrease in ZAAD-1006a exposure is due to the reduced bioavailability of pexidartinib and is not driven by increased elimination of ZAAD-1006a. Additionally, an exposure–response analysis showed that efficacy is related to exposure [5]. Therefore, as lower exposure may lead to lower efficacy, it is recommended to avoid the concomitant use of proton pump inhibitors while taking pexidartinib. If acid reduction is required, pexidartinib can be administered 2 h before or after locally acting antacids or 2 h prior or 10 h after histamine-2 receptor antagonists. This recommendation was developed to ensure that pexidartinib is administered in a time window during which there is no to minimum elevation of gastric pH following administration of the antacids and histamine-2 receptor antagonists [1].
Itraconazole, rifampin, and probenecid were selected in the metabolism-based DDI evaluation because these three drugs are the US Food and Drug Administration-recommended and commonly used probe drugs of a strong CYP3A inhibitor, CYP3A inducer, and UGT inhibitor, respectively, in a clinical DDI study setting [6,7,8,9,10]. Such strong index perpetrators are used to create worst-case scenarios where the results can be used to extrapolate the findings to concomitant medications sharing the same DDI potential. Additionally, a PBPK model was developed using the data from the clinical DDI studies along with data from other clinical PK studies. The PBPK model was then used to predict the effect of a moderate CYP3A4 inhibitor (fluconazole) and a moderate CYP3A4 inducer (efavirenz) on the pharmacokinetics of pexidartinib. Consistent with the knowledge that CYP3A4 is the main enzyme responsible for pexidartinib metabolism, the results of the current studies indicated that the exposure of pexidartinib (as measured by AUC∞) was increased 73% by a strong CYP3A4 inhibitor and decreased 63% by a strong CYP3A inducer. The inhibition of CYP3A4 by itraconazole also resulted in a greater production of the glucuronide conjugate (i.e., ZAAD-1006a) with AUC∞ values increased by 97% compared with when pexidartinib is administered alone. This reflects a greater role of UGT in the metabolism when a CYP3A4 inhibitor is present. As expected, co-administration of pexidartinib with the strong CYP3A/UGT inducer rifampin resulted in decreased exposure to pexidartinib (i.e., AUC∞ decreased 63%) [8]. The enzyme induction also resulted in an increase in ZAAD-1006a exposure and MPR. This is consistent with increased metabolism of pexidartinib via the UGT pathway in the presence of an inducer, rifampin. Results from the PBPK model predicting the effect of a moderate CYP3A4 inhibitor and a moderate inducer suggest that pexidartinib exposure was impacted to a lesser degree compared with when pexidartinib was co-administered with strong CYP3A inducer/inhibitors. The moderate CYP3A4 inhibitor fluconazole is predicted to modestly increase single-dose and multiple-dose exposure of pexidartinib (54% and 65%, respectively), while the moderate CYP3A4 inducer efavirenz modestly decreased single-dose and multiple-dose exposure of pexidartinib (49% and 28%, respectively).
Probenecid, a general inhibitor of a variety of UGT isozymes, which is responsible for glucuronidation reactions [7], also inhibits organic anion transporter (OAT) 1 and OAT3 [6]. While pexidartinib undergoes glucuronidation, in vitro data suggest that it is not a substrate for OAT1. ZAAD-1006a is generated by UGT, and it is a substrate of OAT [11]. Therefore, it is possible that OAT inhibition by probenecid may lead to a decrease in renal elimination of ZAAD-1006a and therefore, possibly increase ZAAD-1006a exposure. In this study, the observed 60% increase in pexidartinib exposure on co-administration with probenecid is consistent with the fact that pexidartinib is a substrate of UGT. In addition, the extent of increase in ZAAD-1006a exposure (more than two-fold) was higher than the observed increase in pexidartinib exposure (60%), resulting in a 42% higher MPR (based on AUC) when pexidartinib was co-administered with probenecid. This relatively higher extent of increase in ZAAD-1006a exposure could possibly be due to decreased renal elimination of ZAAD-1006a via OATs in the kidneys in the presence of probenecid. Previously, it was reported that co-administration with probenecid in healthy subjects resulted in an approximately 50% increase in olmesartan AUC [12].
Based on the findings of these CYP3A inhibitor/inducer and UGT inhibitor studies, it is recommended to avoid concomitant use of pexidartinib with strong CYP3A inducers as the reduced exposure may lead to suboptimal clinical benefit [1]. Mild and moderate CYP3A inducers may be co-administered with pexidartinib because a predicted decrease in pexidartinib exposure when administered with moderate CYP3A/UGT inducers is less than the variability in exposure following multiple doses of pexidartinib. To avoid potential increased adverse events associated with higher exposure, a reduced dose is recommended when pexidartinib is administered with moderate or strong CYP3A4 inhibitors, or UGT inhibitors. Pexidartinib pharmacokinetics is dose proportional, and currently pexidartinib is available only at a dose strength of 200 mg/capsule. Therefore, for the planned total daily pexidartinib doses of 800 mg and 400 mg, a reduced dose of 400 mg and 200 mg, respectively, has been recommended when co-administered with strong and moderate CYP3A4 inhibitors. For those with a planned daily dose of 600 mg, the dose should be reduced to 400 mg/day [1]. If concomitant use of a moderate or strong CYP3A inhibitor or UGT inhibitor is discontinued, the pexidartinib dose should be increased to the dose that was used before starting the inhibitor after three half-lives of the inhibitor.
The dosing recommendations when co-administered with moderate CYP3A inhibitors and inducers are based on the PBPK modeling, suggesting that an optimally defined PBPK model can be used to assess the CYP3A-based DDI potential of a molecule without performing specific clinical DDI studies. This model-based DDI risk assessment reduces the exposure of healthy subjects to the investigational agents and other drugs and reduces resource requirements, including the cost and time of drug development. In recent years, this methodology has been used to assess the DDI risk for multiple drugs [13].
5 Conclusions
The pharmacokinetics of pexidartinib is significantly altered by pH modifiers, CYP3A4 inhibitors, CYP3A inducers, and UGT inhibitors. The results of these studies offer guidance for clinicians on how to safely administer such agents in combination with pexidartinib.
References
Turalio (pexidartinib) capsules, for oral use [prescribing information]. Basking Ridge (NJ): Daiichi Sankyo, Inc.; 2022.
Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med. 2015;373(5):428–37.
Tap WD, Gelderblom H, Palmerini E, Desai J, Bauer S, Blay JY, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised phase 3 trial. Lancet. 2019;394(10197):478–87.
Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95(6):1238–57.
Yin O, Wagner AJ, Kang J, Knebel W, Zahir H, van de Sande M, et al. Population pharmacokinetic analysis of pexidartinib in healthy subjects and patients with tenosynovial giant cell tumor or other solid tumors. J Clin Pharmacol. 2021;61(4):480–92.
US Food and Drug Administration. Guidance for industry. Clinical drug interaction studies: Cytochrome P450 enzyme- and transporter-mediated drug interactions. 2020. Available at: https://www.fda.gov/media/134581/download. Accessed 9 Sept 2022.
Aceves Baldó P, Anzures-Cabrera J, Bentley D. In vivo evaluation of drug-drug interactions linked to UGT inhibition: the effect of probenecid on dalcetrapib pharmacokinetics. Int J Clin Pharmacol Ther. 2013;51(3):215–8.
Chen J, Raymond K. Roles of rifampicin in drug-drug interactions: underlying molecular mechanisms involving the nuclear pregnane X receptor. Ann Clin Microbiol Antimicrob. 2006;15(5):3.
Zhou S, Yung Chan S, Cher Goh B, Chan E, Duan W, Huang M, et al. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin Pharmacokinet. 2005;44(3):279–304.
Zhou SF. Potential strategies for minimizing mechanism-based inhibition of cytochrome P450 3A4. Curr Pharm Des. 2008;14(10):990–1000.
FDA Briefing Document. Oncologic Drugs Advisory Committee meeting. NDA 211810 Pexidartinib 2019.
Li KY, Qiu Y, Jiang Y, Luo CH, Lin XP, Wang J, et al. Effect of probenecid on pharmacokinetics and tolerability of olmesartan in healthy chinese volunteers. Curr Ther Res Clin Exp. 2014;76:7–10.
Zahir H, Kobayashi F, Zamora C, Gajee R, Gordon MS, Babiker HM, et al. Evaluation of potential drug-drug interaction risk of pexidartinib with substrates of cytochrome P450 and P-glycoprotein. J Clin Pharmacol. 2021;61(3):298–306.
Acknowledgements
Medical writing assistance was provided by Bret Fulton and Phillip Giannopoulos of SciStrategy Communications and funded by Daiichi Sankyo, Inc.
Author information
Authors and Affiliations
Contributions
HZ: conceptualization, formal analysis, investigation, methodology, project administration, supervision, writing (original draft), writing (review and editing). JG: conceptualization, methodology, project administration, supervision, validation, visualization, writing (review and editing). DS: conceptualization, funding acquisition, methodology, project administration, supervision, writing (original draft), writing (review and editing). CH: formal analysis, writing (review and editing). KW: conceptualization, data curation, formal analysis, methodology, resources, validation, writing (original draft), writing (review and editing). FL: conceptualization, funding acquisition, resources, supervision, writing (review and editing).
Corresponding author
Ethics declarations
Funding
The work was supported by Daiichi Sankyo, Inc.
Conflicts of interest/competing interests
Hamim Zahir reports employment with Daiichi Sankyo, Inc. during the time of the study. Jonathan Greenberg reports employment with and stock/stock options in Daiichi Sankyo, Inc. Dale Shuster reports employment, stock/stock options in, and patents with Daiichi Sankyo, Inc. Ching Hsu reports employment and stock/stock options in Daiichi Sankyo, Inc. Kengo Watanabe reports employment with Daiichi Sankyo, Co., Ltd. Frank LaCreta reports employment with and stock/stock options in Daiichi Sankyo, Inc.
Ethics approval
The studies were conducted in compliance with the ethical principles of the Declarations of Helsinki and the International Conference on Harmonization consolidated Guideline E6 for Good Clinical Practice and approved by the applicable institutional review boards.
Consent to participate
All subjects provided written informed consent prior to study participation.
Consent for publication
Not applicable.
Availability of data and material
De-identified individual participant data and applicable supporting clinical study documents may be available upon request at https://www.clinicalstudydatarequest.com//. In cases where clinical study data and supporting documents are provided pursuant to the sponsor’s policies and procedures, the sponsor will continue to protect the privacy of the clinical study participants. Details on data sharing criteria and the procedure for requesting access can be found at this web address: https://www.clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-DS.aspx.
Code availability
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Zahir, H., Greenberg, J., Shuster, D. et al. Evaluation of Absorption and Metabolism-Based DDI Potential of Pexidartinib in Healthy Subjects. Clin Pharmacokinet 61, 1623–1639 (2022). https://doi.org/10.1007/s40262-022-01172-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-022-01172-9