
Abstract
Background and Introduction
The dispersible tablet formulation (DTF) of pretomanid has been developed to facilitate future use in children. This work aimed to assess the pharmacokinetics (PK) and relative bioavailability of the DTF compared to the marketed formulation (MF) and the potential influence of dose.
Methods
Pretomanid DTF was investigated in a single-dose, randomized, four-period, cross-over study, with 7 days of washout between doses. Forty-eight healthy volunteers were enrolled and randomized into one of two panels to receive doses either in the fasted state or after a high-fat meal. Each volunteer received doses of 10, 50, and 200 mg DTF, and 200 mg MF pretomanid. Blood samples for pharmacokinetic assessment were drawn following a rich schedule up to 96 h after each single dose. The study data from the panel receiving the high-fat meal were analyzed using a nonlinear mixed-effects modeling approach, and all data were characterized with noncompartmental methods.
Results
A one-compartment model with first-order elimination and absorption through a transit compartment captured the mean and variability of the observed pretomanid concentrations with acceptable precision. No significant difference in bioavailability was found between formulations. The mean absorption time for the DTF was typically 137% (86–171%) of that for the MF. The bioavailability was found to be dose dependent with a small positive and larger negative correlation under fed and fasted conditions, respectively.
Conclusion
Using data from a relative bioavailability study in healthy adult volunteers, a mathematical model has been developed to inform dose selection for the investigation of pretomanid in children using the new dispersible tablet formulation. Under fed conditions and at the currently marketed adult dose of 200 mg, the formulation type was found to influence the absorption rate, but not the bioavailability. The bioavailability of the DTF was slightly positively correlated with doses when administered with food.
Clinical Trial Registration
ClinicalTrials.gov Identifier: NCT04309656, first posted on 16 March 2020.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
We successfully characterized pretomanid pharmacokinetics across the child-friendly dispersible-tablet and marketed adult formulations from doses of 10–200 mg. |
Under fed conditions and at the currently marketed adult dose of 200 mg, the formulation type was found to influence the absorption rate, but not the bioavailability. |
The model developed here will support the design of planned pediatric studies of pretomanid where the dispersible-tablet formulation will be used. |
1 Introduction
Tuberculosis (TB) was the second most common cause of death due to infectious diseases globally in 2020, second only to COVID-19 [1]. The emergence of drug-resistant TB has created even more difficulties in the treatment of TB. Multidrug-resistant tuberculosis (MDR-TB) comprises TB strains that are resistant to at least isoniazid and rifampicin, the two most powerful first-line anti-TB drugs. There are higher orders of drug resistance defined according to which second-line drugs are no longer effective [2]. Hence, developing novel compounds and new regimens for TB infection is increasingly important.
Pretomanid is an antimycobacterial medicine in the nitroimidazooxazine class that kills active Mycobacterium TB by inhibiting mycolic acid biosynthesis and thereby preventing cell wall production [3]. Pretomanid 200 mg daily in combination with bedaquiline and linezolid (known as the BPaL regimen) for 6 months has been approved by the US Food and Drug Administration (FDA) in 2019 [4] and European Medicines Agency (EMA) in 2020 [5] for adults diagnosed with highly resistant TB.
Although drug-resistant TB is less common in children, the need for treatment of pediatric drug-resistant TB is highly unmet. According to Jenkins and Dodd et al. [6,7,8], an estimated 25,000–32,000 cases arise annually, and only 3–4% of them are likely to receive proper treatment. Therefore, it is important to develop treatment options that are also applicable to children. A pediatric investigation plan (PIP) for pretomanid has been agreed upon with the EMA. The purpose is to assess the appropriateness of the BPaL regimen in pediatric patients (< 18 years old). In the framework of the plan, a dispersible tablet formulation (DTF) of pretomanid with different dose strengths (10 and 50 mg) was developed to accommodate younger children who might experience difficulty swallowing whole tablets. However, the bioavailability could be altered by the formulation. It is therefore necessary to characterize the pharmacokinetics (PK) and safety of the new formulation of pretomanid and study potential differences from the marketed formulation (MF) before conducting studies in children.
The PK characteristics of the MF of pretomanid in humans have previously been reported [5, 9]. The maximum concentration is reached 4–5 h after dose. Drug exposure increases 88% when administered with food. Approximately 86% of pretomanid is bound to plasma proteins, and albumin is responsible for most of the binding. Pretomanid is metabolized by multiple pathways, none of which is dominant alone. Half-life ranges from 14 to 26 h. A population PK model for pretomanid based on comprehensive data obtained from 14 studies covering phase 1 to phase 3 was previously published [10]. Multiple factors have been found to impact pretomanid PK, such as sex, food intake, types of resistance, and concomitant HIV infection. This report is the first to present data on the pretomanid DTF and pretomanid doses lower than 50 mg.
The objective of this modeling study was to evaluate the relative bioavailability of pretomanid DTF compared to pretomanid MF and investigate the potential dose dependency, using a population PK modelling approach. It uses data from a clinical relative-bioavailability and food-effect study. Here, only the data on pretomanid administered together with food were modeled given that pretomanid is labeled as being taken with food [11]. However, other results from all study participants are summarized.
2 Methods
2.1 Clinical Study Design
The PK of pretomanid DTF was investigated in a phase I single-dose, open-label, randomized, two-panel, four-period, cross-over study (CL-011, ClinicalTrials.gov Identifier: NCT04309656). The study was conducted in accordance with the International Council for Harmonization (ICH) Guideline for Good Clinical Practice (E6) by Worldwide Clinical Trials Early Phase Services, LLC., in San Antonio, Texas, USA, sponsored by Global Alliance for TB Drug Development.
Healthy adult participants were randomized 1:1 into two panels to receive doses with or without food. Participants in each panel were further randomized according to a four-sequence, four-period design, in which each subject received single-dose treatments of 10, 50, and 200 mg (4*50 mg) DTF, and 200 mg MF pretomanid after consuming the same FDA standard high-fat, high-calorie breakfast following an overnight fast of at least 10 h. Participants were administered DTF dispersed in 10 mL of ambient room temperature water, followed by an additional 10 mL of water that rinsed the same container, with an additional 25 mL to rinse their mouths. Each administration was separated by a 7-day washout period. One outpatient safety follow-up was done 7 days after the last PK sample in the study was collected.
2.2 Clinical Study Participants
Participants were eligible for the study if they were between 19 and 50 years old and weighed a minimum of 50 kg with body mass index (BMI) between 18.50 and 32 kg/m2. Exclusion criteria included any history or presence of the following: prolonged QT; positive test for hepatitis or human immunodeficiency virus (HIV); other medical conditions that were judged to jeopardize the safety of the subjects or affect study validity; using any other medications prior to the study that were considered to impact subject safety and affect study validity. Female volunteers were excluded if pregnant or breastfeeding.
2.3 Data Collection and Bioanalysis
During each treatment period, blood samples were drawn prior to each single-dose administration (pre-dose), and at 0.5, 1, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 72, and 96 h after dose. Hence, the pre-dose sample at dosing periods 2, 3, and 4 was taken 168 h after the preceding dose. Fifteen samples from each treatment period and a total of 60 samples were collected for each patient. Plasma samples were obtained by centrifuging blood samples and were collected into 2.0 mL Vacutainer tube(s) containing K2-EDTA, were placed in − 20 °C within 60 min of collection, and remained frozen until assayed. Concentrations were determined by a validated ultra-performance liquid chromatography/tandem mass spectrometric method. Inter-run accuracy ranged from 100.3 to 1003.8% over 10.0–10,000 ng/mL. Precision (%RSD, relative standard deviation) was ≤ 16.4% and ≤ 11.4% at the lower limit of quantification (LLOQ) intra- and inter-run, respectively; ≤ 11.1% and ≤ 7.1% above LLOQ.
2.4 Pharmacokinetic Analysis
The PK data from the panel of subjects receiving pretomanid in the fed state were analyzed using a nonlinear mixed-effects modeling (NLMEM) approach. A previously published population PK model for pretomanid was used as a starting point [10]. This was a one-compartment model with first-order elimination and absorption, parameterized by apparent oral clearance (\(\mathrm{CL}\)), apparent volume of distribution (\(\mathrm{Vd}\)), rate of absorption (\(\mathrm{KA}\)), and mean transit time (\(\mathrm{MTT}\)) describing the delay in absorption as due to passage of drug through three transit compartments between the depot (dosing) compartment and the central compartment. The number of the transit compartments and the MTT jointly absorption delay [12]. Absolute bioavailability (\(F\)) was fixed to 1 because only data obtained after oral administration were available, hence disposition parameters are relative to the true bioavailability. Theoretical values of 0.75 on \(\mathrm{CL}\) and 1 on \(\mathrm{Vd}\) for allometric scaling factors of body weight were included in this model [13]. Some other covariates that affected healthy subjects were sex, food intake, and dose (50–1500 mg).
The model was applied to the new data and updated to improve fit. The transit compartment model was retained to describe the delayed absorption phase, but the number of transit compartments was reevaluated. Allometric scaling factors for body weight on \(\mathrm{CL}\) and \(\mathrm{Vd}\) were retained and also fixed to their theoretical values. Effects of doses and formulations on relative bioavailability and other absorption parameters were step-wise introduced into the model as proportional change with the PK parameters (for formulation type) or using a power model (for dose levels) and were tested for statistical significance. Other components of the starting model were also revisited and evaluated for statistical significance. Random effects including the interindividual variability (IIV) and inter-occasion variability (IOV) were assumed to be log-normally distributed. For IOV, a new occasion was defined when a new dose was administered, hence four occasions in total. The magnitude of the variance was assumed to be the same for each occasion. Different types of transformation on IIV and IOV distributions were tested to explore the best fit, including log-modulus [14], Box–Cox [15], logit transformation [16], and uniform distribution. Different combinations of additive and/or proportional components were tested for residual unexplained variability (RUV).
The modeling work was conducted in NONMEM® 7.4, in which the first-order conditional estimation method with interaction was applied. Model development decisions were based on objective function values (OFVs), likelihood ratio tests (LRTs) at level p = 0.001, relative standard error of parameter estimates (RSE), and diagnostic checks as well as the quality assurance (QA) function available in PsN [17]. Statistics and graphic diagnostics listed here were aided by R, PsN, xpose4, and Pirana workbench [18,19,20].
To evaluate the updated PK model, parameter precision was calculated through the covariance step in NONMEM and visual predictive checks (VPCs) were produced based on 1000 simulations [21]. Because the model was to be used to guide the future pediatric study design, it was of importance to ensure the model’s ability to predict exposure metrics of interest. Therefore, posterior predictive checks for the area under plasma concentration curve (AUC) were generated using the ncappc tool [22] for non-compartmental analysis (NCA) posterior predictive checks. By performing NCA on 300 model-simulated datasets and the original dataset, the distribution of model predictions was evaluated compared to NCA characteristics derived from the original observations. The AUC was calculated using the “linear-up-log-down” trapezoidal method (linear interpolation in the increasing absorption phase; log interpolation in the decreasing elimination phase). Conventional statistical evaluations of relative bioavailability among formulations, doses, and food conditions using ANOVA following the regulatory guidelines of a typical bioequivalence study [23] were also performed, details of which are provided in the Electronic Supplementary Material (ESM) [23].
3 Results
3.1 Data Summary
Twenty-four participants including 16 males and eight females were enrolled in each of the fed and fasted panels of the study. Detailed demographics of the participants included are summarized in Table 1. All treatments were generally well tolerated. Adverse events (AEs) were infrequent and mild, and none led to study or treatment discontinuation.
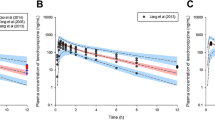
In total, 1377 concentration observations from the fed panel were included in the NLMEM analysis. The small number of observations below the quantification limit of 1 ng/mL (n = 39, i.e., 2.8%) were excluded. Mean concentration-time profiles are shown for the fed panel in Fig. 1 and for both panels in Figs. D1 and D2 in the ESM.

Mean plasma concentrations versus time after dose per dose and formulation of pretomanid at fed condition. Treatment A, 200 mg marketed formulation; B, 200 mg dispersible tablet formulation; C, 50 mg dispersible tablet formulation; D, 10 mg dispersible tablet formulation
3.2 Pharmacokinetic Analysis
The structural model mostly remained the same as the starting model, i.e., a one-compartment model with first-order elimination and first-order absorption. However, the number of transit compartments to describe the delayed absorption phase was reduced from three in the starting model to one in the final model. The mean absorption time (MAT, calculated as \(1/KA+MTT\)) for the DTF was typically 137% (95% confidence interval (CI): 86–171%) of that for the MF.
The bioavailability ratio (formulation effect) of DTF to MF was estimated by the model to be 1.00 with a 90% CI of 0.87–1.14, so formulation was not retained in the model as a covariate on bioavailability. Similar results were found by NCA, with geometric mean ratios for both Cmax and AUC in both fed and fasted panels at 200 mg having CIs within 80–125%, indicating bioequivalence (Tables D1 and D2 in the ESM).
A power model for dose dependence of the bioavailability parameter F yielded an estimated exponent of 0.082 (Table 2), consistent with an estimated slope of 1.086 in the standard noncompartmental assessment for ln(AUC0–inf) under fed conditions (Table D3 in the ESM), indicating nearly dose-proportional increase in exposure. The relative bioavailabilities of the 10 mg and 50 mg doses were 78% (95% CI 71–86) and 89% (95% CI 86–93) of a 200 mg dose. Under fasted conditions, however, the estimated slope in the noncompartmental assessment was 0.824, indicating a larger departure from dose proportionality and in the opposite direction relative to fed conditions (Table D4 in the ESM). At the 200 mg doses, food increased bioavailability by 66–88% per NCA; however, reflecting the opposite effects of dose, bioavailability under fed and fasted conditions were similar at 50 mg, and bioavailability was 18–31% less for fed versus fasted at 10 mg (Table D5 in the ESM).
IOV was included for absorption parameters, i.e., bioavailability, \(\mathrm{KA}\), and \(\mathrm{MTT}\); IIV for disposition parameters, \(\mathrm{CL}\) and \(\mathrm{Vd}\). The lognormal distribution was retained for all the IIVs and IOVs. The estimation of the IIV and IOV showed that the coefficients of variation (CV) of \(\mathrm{CL}\), \(\mathrm{Vd}\), and bioavailability were at most 25%, while the parameters determining the rate of absorption, \(\mathrm{KA}\), and \(\mathrm{MTT}\), were highly variable, with CV higher than 50%. The RUV was modeled by a combination of proportional and additive contributions. The residual error was found to be more than three times greater for observations from the absorption phase (< 10 h after dose) compared to the elimination phase.
The NONMEM control stream is provided in Section C of the ESM and parameter estimates are provided in Table 2. All parameters were estimated with acceptable precision. The VPC in Fig. 2 showed an acceptable fit of the model to the data (other forms of VPCs are available in Section B of the ESM). The results of the NCA posterior predictive checks showed reasonable predictions of the AUC, as shown in Fig. 3. The model predictions covered the observed means of AUC in all groups. Although the predicted variances were generally larger than the observed variance, they were within the acceptable prediction intervals.

Visual predictive checks of the final model on the CL-011 study data (fed conditions only) by formulations and doses from time after dose to 96 h. The concentrations are plotted on log scale. The open circles represent the observed samples. The solid and dashed black lines represent the 50th and 10th/90th percentiles of the observations in the data set. The red- and blue-colored areas represent the 95% model-predicted confidence intervals for the corresponding percentiles

Histogram of the simulated population of the AUC0–last from 300 simulations, compared with the observed AUC (red vertical lines), under fed state in the CL-011 study. The distribution of simulated AUC is represented by the 2.5th, 50th, and 97.5th percentiles in grey lines
4 Discussion
The current NLMEM analysis successfully described the PK of both pretomanid MF and DTF administered with food at different dose levels in healthy subjects. Our findings indicated comparable bioavailability between pretomanid MF and DTF at 200 mg. The developed model has also proved to adequately predict the pretomanid PK concentrations and AUC, with different formulations at different doses.
We found that the MAT was typically 37% longer for the DTF (3.7 h) than for the MF (2.7 h). The difference in the MAT between formulations is expected to be not clinically relevant. Together with the fact that the difference in bioavailability between formulations was within the limit for bioequivalence at 200 mg, the pediatric formulation could also be used for adults depending on the preference of the patient and for other populations with swallowing problems.
The bioavailability was found to be dose dependent with a small positive and a larger negative correlation under fed and fasted conditions, respectively, based on three dose levels from 10 to 200 mg. Such a difference in dose dependence between fed and fasted conditions has been noted previously for pretomanid, and may be due to increased solubility of pretomanid with food [11, 25].
NLMEM was the focus of this analysis, although results from NCA were also summarized. NLMEM identified which parameters exhibited significant between-subject (IIV) variability (\(\mathrm{CL}\) and \(\mathrm{Vd}\)) and which between-occasion (IOV) variability (bioavailability and absorption). The lesser regularity of absorption may be related to the solubility of pretomanid, noted above, and was also reflected in the larger within-occasion (RUV) variability during the absorption phase compared to the elimination phase [26].
As presented in Section B of the ESM, peak concentrations were slightly underpredicted in the model at 10 mg and 200 mg MF, which might influence the finding concluded by the model regarding the difference in the MAT between the formulations. However, mean residence times (MRTs) derived from the NCA method could be an option to evaluate such potential influence. Given that the dispositions (\(\mathrm{CL}\) and \(\mathrm{Vd}\)) are similar for both formulations, the difference in the MRT could be considered attributable to the difference in the MAT; the model-estimated typical difference of 1 h in MAT (3.7 vs. 2.7 h) could then be compared with the difference in MRT derived from NCA, which was about 1.6 h. Such similar results of MAT difference in both methods (1 h using the model and 1.6 h using NCA) suggest that the model-estimated difference is not an artefact of model misspecification. Regardless, it was not considered a major concern given that the primary application of the model was to assist pediatric study design; per the PIP, AUC is the key metric to determine pediatric doses for achieving clinical exposure and effect as in adults, rather than peak concentration, which was reported to have no clear correlation with drug effect [27]. Although the underpredicted peak affected model-predicted mean AUC more evidently at low dose of 10 mg (Fig. 3), the observed mean value was still covered by the model 95% CI.
A model-predicted formulation effect (ratio) on bioavailability of 1 with CI was compared to the standard bioequivalence criterion as a reference regarding the magnitude and clinical relevance of the estimated uncertainty. The sampling schedule in the PK study was dense, hence an inflated type I error (often seen in a sparse design using the model-based method) is not expected in this analysis [28].
5 Limitations
Some limitations of this analysis may be noted. This PK analysis was conducted only on healthy adult volunteers. When the model is used to extrapolate or predict the PK for children with TB, a larger variability may be expected due to rapid growth and development and other more uncontrolled factors in children. Properties such as dose dependency of bioavailability may also be different in children, for exaple, due to their premature metabolic system [29]. The fasted condition was not modeled because it is planned to administer pretomanid to children with food, as is the practice for adults [11]. Participants in this study were dosed after a high-fat meal, which is a meal-type not realistic to expect long term in practice. The magnitude of the food-effect may be different in children with other meal-types. The PIP includes a single-dose PK study, where the meal will not be standardized, to test the model’s predictions before a multiple-dose safety and efficacy study.
6 Conclusion
In conclusion, the developed model adequately described pretomanid PK across the two formulations (DTF and MF) and doses from 10 to 200 mg. Under the fed conditions and the currently marketed adult dose of 200 mg, the formulation type was found to influence the absorption rate, but not the bioavailability. Characterizing the effects of formulation, dose, and body weight in the context of model as developed here will support the design of planned pediatric studies of pretomanid.
References
Global tuberculosis report 2021. Geneva: World Health Organization. Published online 2021. licence: CC BY-NC-SA 3.0 IGO.
WHO updated definitions of extensively drug-resistant tuberculosis. 2021. https://www.who.int/news/item/27-01-2021-who-announces-updated-definitions-of-extensively-drug-resistant-tuberculosis. Accessed 6 Jan 2022.
Manjunatha U, Boshoff HIM, Barry CE. The mechanism of action of PA-824: novel insights from transcriptional profiling. Commun Integr Biol. 2009;2(3):215. https://doi.org/10.4161/CIB.2.3.7926.
FDA Approves New Drug for Treatment-Resistant Forms of Tuberculosis That Affects the Lungs | FDA.; 2019. https://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-treatment-resistant-forms-tuberculosis-affects-lungs. Accessed 21 Nov 2021
Dovprela (previously Pretomanid FGK) | European Medicines Agency. Published online 2020. https://www.ema.europa.eu/en/medicines/human/EPAR/dovprela-previously-pretomanid-fgk. Accessed 21 Nov 2021
Jenkins HE, Tolman AW, Yuen CM, et al. Incidence of multidrug-resistant tuberculosis disease in children: systematic review and global estimates. Lancet. 2014;383(9928):1572–9. https://doi.org/10.1016/S0140-6736(14)60195-1.
Dodd PJ, Sismanidis C, Seddon JA. Global burden of drug-resistant tuberculosis in children: a mathematical modelling study. Lancet Infect Dis. 2016;16(10):1193–201. https://doi.org/10.1016/S1473-3099(16)30132-3.
Jenkins HE, Yuen CM. The burden of multidrug-resistant tuberculosis in children. Int J Tuberc Lung Dis. 2018;22(5):S3–6. https://doi.org/10.5588/ijtld.17.0357.
Keam SJ. Pretomanid: first approval. Drugs. 2019;79:1797–803. https://doi.org/10.1007/s40265-019-01207-9.
Salinger DH, Subramoney V, Everitt D, Nedelman JR. Population pharmacokinetics of the antituberculosis agent pretomanid. Antimicrob Agents Chemother. 2019. https://doi.org/10.1128/AAC.00907-19.
FDA. Pretomanid Tablets label. Published online 2019. www.fda.gov/medwatch. Accessed 28 Nov 2021
Savic RM, Jonker DM, et al. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn. 2007;34:711–26. https://doi.org/10.1007/s10928-007-9066-0.
Anderson BJ, Holford NHG. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet. 2009;24(1):25–36. https://doi.org/10.2133/dmpk.24.25.
John JA, Draper NR. An alternative family of transformations. Appl Stat. 1980;29(2):190. https://doi.org/10.2307/2986305.
Box GEP, Cox DR. An analysis of transformations. Source J R Stat Soc Ser B. 1964;26(2):211–52. https://www.jstor.org/stable/2984418. Accessed 6 Jan 2022.
Collins JJ, Fanciulli M, Hohlfeld RG, Finch DC, Sandri GVH, Shtatland ES. A random number generator based on the logit transform of the logistic variable. Comput Phys. 1998;6(6):630. https://doi.org/10.1063/1.168442.
Karlsson MO, Freiberga S, Yngman G, Nordgren R, Ueckert S. Extensive and automatic assumption assessment of pharmacometric models. PAGE 27 abstract 8754. Published 2018. https://www.page-meeting.org/default.asp?abstract=8754. Accessed 6 May 2021.
Lindbom L, Pihlgren P, Jonsson N. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–57. https://doi.org/10.1016/j.cmpb.2005.04.005.
Jonsson EN, Karlsson MO. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1998;58(1):51–64. https://doi.org/10.1016/S0169-2607(98)00067-4.
Keizer RJ, van Benten M, Beijnen JH, Schellens JHM, Huitema ADR. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101(1):72–9. https://doi.org/10.1016/j.cmpb.2010.04.018.
Karlsson MO, Holford N. A tutorial on visual predictive checks. https://www.page-meeting.org/?abstract=1434. Accessed 6 May 2021.
Acharya C, Hooker AC, Türkyılmaz GY, Jönsson S, Karlsson MO. A diagnostic tool for population models using non-compartmental analysis: the ncappc package for R. Comput Methods Programs Biomed. 2016;127:83–93. https://doi.org/10.1016/j.cmpb.2016.01.013.
FDA, Cder, Purdief. Guidance for Industry Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs-General Considerations; 2014. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm. Accessed 21 Nov 2019.
Winter H, Ginsberg A, Egizi E, et al. Effect of a high-calorie, high-fat meal on the bioavailability and pharmacokinetics of PA-824 in healthy adult subjects. Antimicrob Agents Chemother. 2013;57(11):5516.
Karlsson MO, Beal SL, Sheiner LB. Three new residual error models for population PK/PD analyses. J Pharmacokinet Biopharm. 1995;23(6):651–72. https://doi.org/10.1007/BF02353466.
Ahmad Z, Peloquin CA, Singh RP, et al. PA-824 exhibits time-dependent activity in a murine model of tuberculosis. Antimicrob Agents Chemother. 2011;55(1):239. https://doi.org/10.1128/AAC.00849-10.
Dubois A, Gsteiger S, Pigeolet E, Mentré F. Bioequivalence tests based on individual estimates using non-compartmental or model-based analyses: Evaluation of estimates of sample means and type i error for different designs. Pharm Res. 2010;27(1):92–104.
Holford N, Heo YA, Anderson B. A pharmacokinetic standard for babies and adults. J Pharm Sci. 2013;102(9):2941–52. https://doi.org/10.1002/jps.23574.
Acknowledgements
We thank all the volunteers participating in this study and all the staff who contributed to the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was a collaborative work between Uppsala University and TB Alliance, supported by TB Alliance (Global Alliance for TB Drug Development) with funding from Australia’s Department of Foreign Affairs and Trade, Bill and Melinda Gates Foundation [OPP1129600], the Foreign, Commonwealth and Development Office (United Kingdom), Germany’s Federal Ministry of Education and Research through KfW, Irish Aid, the Netherlands Ministry of Foreign Affairs, and the US Agency for International Development.
Conflicts of interest
The authors declare that they have no competing interests. JN, AL, and FP are employees of TB Alliance, the organization developing pretomanid. ES is involved in development of multiple novel anti-tuberculosis compounds through the UNITE4TB consortium.
Ethics approval
This study was performed in line with the principles of the Declaration of Helsinki and the International Council for Harmonisation (ICH) Guideline for Good Clinical Practice: Consolidated Guidance (E6).
Consent to participate
Written informed consent was obtained from all the participants in the study.
Consent for publication
Not applicable.
Availability of data and material
The data of this study are not openly available due to sensitivity issues.
Code availability
The NONMEM model code is available in the ESM.
Authors’ contributions
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Zou, Y., Nedelman, J., Lombardi, A. et al. Characterizing Absorption Properties of Dispersible Pretomanid Tablets Using Population Pharmacokinetic Modelling. Clin Pharmacokinet 61, 1585–1593 (2022). https://doi.org/10.1007/s40262-022-01163-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-022-01163-w