
Abstract
Background and Objective
Previous pharmacokinetic (PK) studies of ciprofloxacin in intensive care (ICU) patients have shown large differences in estimated PK parameters, suggesting that further investigation is needed for this population. Hence, we performed a pooled population PK analysis of ciprofloxacin after intravenous administration using individual patient data from three studies. Additionally, we studied the PK differences between these studies through a post-hoc analysis.
Methods
Individual patient data from three studies (study 1, 2, and 3) were pooled. The pooled data set consisted of 1094 ciprofloxacin concentration–time data points from 140 ICU patients. Nonlinear mixed-effects modeling was used to develop a population PK model. Covariates were selected following a stepwise covariate modeling procedure. To analyze PK differences between the three original studies, random samples were drawn from the posterior distribution of individual PK parameters. These samples were used for a simulation study comparing PK exposure and the percentage of target attainment between patients of these studies.
Results
A two-compartment model with first-order elimination best described the data. Inter-individual variability was added to the clearance, central volume, and peripheral volume. Inter-occasion variability was added to clearance only. Body weight was added to all parameters allometrically. Estimated glomerular filtration rate on ciprofloxacin clearance was identified as the only covariate relationship resulting in a drop in inter-individual variability of clearance from 58.7 to 47.2%. In the post-hoc analysis, clearance showed the highest deviation between the three studies with a coefficient of variation of 14.3% for posterior mean and 24.1% for posterior inter-individual variability. The simulation study showed that following the same dose regimen of 400 mg three times daily, the area under the concentration–time curve of study 3 was the highest with a mean area under the concentration–time curve at 24 h of 58 mg·h/L compared with that of 47.7 mg·h/L for study 1 and 47.6 mg·h/L for study 2. Similar differences were also observed in the percentage of target attainment, defined as the ratio of area under the concentration–time curve at 24 h and the minimum inhibitory concentration. At the epidemiological cut-off minimum inhibitory concentration of Pseudomonas aeruginosa of 0.5 mg/L, percentage of target attainment was only 21%, 18%, and 38% for study 1, 2, and 3, respectively.
Conclusions
We developed a population PK model of ciprofloxacin in ICU patients using pooled data of individual patients from three studies. A simple ciprofloxacin dose recommendation for the entire ICU population remains challenging owing to the PK differences within ICU patients, hence dose individualization may be needed for the optimization of ciprofloxacin treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A population pharmacokinetic model of ciprofloxacin was developed based on pooled data from three intensive care unit data sets, containing in total 140 intensive care unit patients and 1094 concentration–time samples. |
Despite the large amount of data, only bodyweight and renal function were associated with pharmacokinetic parameters of ciprofloxacin still leaving much inter-individual variability unexplained by commonly deployed covariates. |
A simple dose strategy of ciprofloxacin suitable for all intensive care unit patients remains challenging and dose individualization may be needed. |
1 Introduction
Ciprofloxacin is a commonly used antibiotic for treating infections in intensive care unit (ICU) patients given its broad spectrum of action against pathogenic bacteria including most Gram-negative species [1,2,3]. However, adequate dosing of ciprofloxacin remains a major challenge in these patients. The ICU patient population exhibits high pharmacokinetic (PK) variability between patients and over the time course of disease and therapy owing to the large and variable physiological changes associated with the type and severity of illness as well as therapy [4]. For example, the clearance (CL) of antibiotics may be decreased by acute kidney injury whereas the volume of distribution may be increased by capillary leakage and administration of intravenous fluids [5]. Thus, prescribing appropriate antibiotic doses for ICU patients is challenging for clinicians. This needs to be addressed as underdosing may give rise to treatment failure in at least some ICU patients but also to long-term increased antibiotic resistance [6].
Population PK models are increasingly being used to optimize and clinically guide antibiotic dose regimens. This is also the case for ciprofloxacin. Several studies have been performed characterizing ciprofloxacin population pharmacokinetics in ICU patients [7,8,9,10,11,12,13]. In theory, given sufficient data that includes all relevant model covariates, a well-developed model should be capable of accurately describing the entire patient population of the training data, i.e., the data used for the model development. However, considerable deviation can be observed in the reported population PK parameter estimates [6, 8,9,10,11,12, 14, 15]. Such results do not only highlight the high PK variability of ciprofloxacin in the ICU patient population, but also suggest that an important part of the variability may still be unexplained in the ICU patient population. As population PK models are often used to recommend dosing regimens, the use of a model that is not able to adequately describe the majority of patients to be treated is likely to lead to poor PK predictions and thus selection of suboptimal dose regimens. Therefore, it is important to further study the pharmacokinetics of ciprofloxacin in a larger population of ICU patients. To this end, we aimed to combine individual patient data from three previous studies to develop a pooled population PK model of ciprofloxacin representative for a large ICU population and to investigate the PK differences between studies.
2 Methods
2.1 Data
The data set used in the current study contained 1094 ciprofloxacin total plasma concentration–time data points, as well as associated patients’ demographics and biochemical data from 140 ICU patients (Table 1). The data were collected retrospectively from three prospective clinical trials conducted at four Dutch Hospitals. Henceforth, we refer to the study by Erasmus University Medical Center and Maasstad Hospital as study 1 [7], the study by Radboud University Medical Center as study 2 [8], and the study by Amsterdam University Medical Center (UMC) location Vrije Universiteit Medical Center as study 3. Overall, patient characteristics from these three studies were similar (Table 1). The primary diagnoses of the majority of the patients of study 1 were respiratory infection (47.6%) and sepsis (19%). Ciprofloxacin dosing was administered at the discretion of the clinician with most participants receiving ciprofloxacin intravenously 400 mg twice daily or three times daily (t.i.d.) with an infusion rate range from 30 to 60 min. For each patient, two trough samples and one peak sample were drawn. The patients of study 2 were mostly treated for pneumonia (72%). These patients received ciprofloxacin 400 mg twice daily intravenously. Within 24 h after the start of ciprofloxacin therapy, collection of two PK curves with eight timepoints each during a dosing interval were collected with a time interval of 12–24 h. For study 3, 55% of patients fulfilled the sepsis-3 criteria for septic shock and the cardiovascular system was the primary affected organ system upon admission [16]. The patients received ciprofloxacin dosing intravenously either 400 mg t.i.d. or individualized dosing following the advice of AutoKinetics, a bedside model-based, antibiotic precision dosing advice software [17, 18]. Plasma samples were drawn at peak concentration, mid-interval concentration, or trough concentration after the first dose and trough concentration for the following doses. The data of study 1 and study 2 were previously published and further details can be found in the respective publications [7, 8]. The use of patient data in these studies was approved by the local ethics committees and if applicable individual written informed consent was obtained. Data organization and visualization were carried out with R (version 4.0.3; R-project.org).
2.2 Population Pharmacokinetic Modeling
2.2.1 Base Model Development
A pooled population pharmacokinetic analysis was performed with the individual concentration–time data from the three studies using the nonlinear mixed-effects modeling approach. One-compartment, two-compartment, and three-compartment models with first-order elimination were evaluated to fit the PK data. Inter-individual variability (IIV) was tested exponentially on CL, volume(s) of distribution, as well as inter-compartmental CL when evaluating multi-compartment models. Assuming that dosing regimens may be evaluated and adjusted on a daily basis, we defined every 24 h as an occasion, which was taken into account for testing inter-occasion variability (IOV). Inter-occasion variability was only tested exponentially on ciprofloxacin CL, considering that CL is the most clinically relevant parameter for ciprofloxacin pharmacokinetics [19]. As the PK data were originally determined at different hospitals, we tested additive, proportional, or combined statistical error models separately for the data of each study. The analysis was performed using NONMEM software (version 7.5; ICON Development Solutions, Hanover, MD, USA). Parameters were estimated using the first-order conditional estimation method with interaction.
2.2.2 Covariate Selection
We tested demographic and biochemical data of the patients including age, sex, serum creatinine level, estimated glomerular filtration rate (eGFR), serum albumin level, Sequential Organ Failure Assessment score, and the use of continuous veno-venous hemofiltration support. However, despite efforts, we were not able to access a wider range of data items for all three studies. Thus, various clinical relevant data such as sepsis were not included for the covariate analysis. The eGFR (mL/min/1.73 m2) was calculated using the MDRD equation [20]. Additionally, creatinine level, eGFR, serum albumin level, and Sequential Organ Failure Assessment score were time-varying variables for which we also tested to separate the baseline effect and the time-varying effect [21].
In order to identify the covariate relationships, a stepwise covariate modeling procedure implemented in Perl-speaks-NONMEM was performed. Covariates were selected following a forward selection and a backward elimination procedure [22, 23]. Whether a covariate was retained or eliminated was determined based on the statistical significance using the likelihood ratio test. The significance level was set to 0.01 and 0.001 for the forward selection and the backward elimination, respectively. Both linear and power models were evaluated for covariate relationship selection. Missing covariates values (< 2%) were imputed using a carry forward method. Allometric scaling of body weight was added a priori to all PK parameters with a power of 0.75 on CL and/or intercompartmental CL parameters, and a power of 1 on volume of distribution parameters.
2.2.3 Model Evaluation
During model development, models were evaluated primarily based on the likelihood ratio test, goodness of fit plots, and prediction- and variability-corrected visual predictive check plots [24]. Additionally, precision of the parameter’s estimate, i.e., relative standard error, was taken into account.
2.3 Post Hoc Analysis of Pharmacokinetic Variability between Studies
2.3.1 Posterior Distribution
Pooled random samples of individual PK parameters’ posterior distribution were previously proven to be able to reflect the distribution of these parameters among the entire population [25]. For each random-effect parameter, multiple samples can be drawn from its posterior distribution, whereas there exists only one empirical Bayes estimate sample. In addition, gathered random samples from posterior distributions do not suffer from shrinkage, whereas empirical Bayes estimate-based metrics tend to be less reliable when shrinkage is larger than 20% [25, 26]. Therefore, we drew 100 random samples from the random-effect posterior distribution of each PK parameter of each individual using a Markov-chain Monte Carlo method by tweaking the SAEM algorithm implemented in NONMEM: $EST METHOD=SAEM NBURN=0 NITER=0 MASSRESET=0 ETASAMPLES=1 ISAMPLE=100 EONLY=1. The posterior mean and posterior IIV were calculated using Eqs. 1 and 2 as below:
where \(P\) represents the posterior samples of a random-effect parameter, \(\overline{P }\) and \({P}_{i}\) denote posterior mean and \(i\)th posterior sample, respectively, \(n\) is equal to the number of drawn posterior samples of a random-effect parameter, i.e., 100 in this case, and \(\mathrm{VAR}\left(P\right)\) denotes the variance of posterior samples.
2.3.2 Differences in Pharmacokinetic Exposure
To study the potential differences in PK exposure due to the PK variability between the three studies, we simulated full concentration–time profiles based on the random samples drawn. As posterior samples can be traced back to the patient from whom they were drawn, no assumptions were made on the covariate distribution in the simulation. As the data set used in the current study contained 140 patients and we drew 100 posterior samples for each parameter of each patient, the number of subjects for the simulation was effectively 14,000. We performed the simulation of ciprofloxacin pharmacokinetics for the first 24 h of treatment assuming a commonly prescribed dose regimen of a 400-mg t,i.d. intravenous infusion. We specifically chose the first 24 h for this simulation because early appropriate antibiotic treatment is in general associated with a positive clinical outcome in ICU patients [27, 28]. The antimicrobial efficacy of ciprofloxacin is best described by the ratio of area under the concentration–time curve relative to the minimum inhibitory concentration (MIC), we thus calculated the area under the concentration–time curve at 24 h (AUC24) and the percentage of target attainment (PTA), defined as the percentage of patients of which the AUC24/MIC was equal to or greater than 125 [19, 29]. The PTA was calculated for a series of MICs from 0.0625 to 1 mg/L covering the epidemiological cut-off value for treating Pseudomonas aeruginosa with ciprofloxacin (0.5 mg/L) according to EUCAST [30]. This study aimed to only demonstrate the resulting differences in PK exposure between the three studies. The used PK target for this purpose was evaluated based on the steady-state concentrations [29]. Hence, it did not necessarily suggest any dosing strategies.
3 Results
3.1 Population Pharmacokinetic Model
A two-compartment model with IIV on CL, volume of distribution of the central compartment (V1), and volume of distribution of the peripheral compartment (V2), and IOV on CL best described the data. The parameters’ estimates of the final model were 14.7 (L/h) for CL, 61.2 (L) for V1, 44.9 (L/h) for inter-compartmental clearance (Q), and 71.6 (L) for V2 and all parameters were estimated with good precision with a relative standard error equal to or lower than 7.1% (Table 2). Apart from the included allometric scaling of body weight, only a linear covariate association between eGFR and ciprofloxacin CL was identified without separating the baseline effect and the time-varying effect. The estimated covariate coefficient was 0.008, which means that ciprofloxacin CL increases 0.008 L/h when eGFR increases 1 mL/min/1.73 m2 relative to the median eGFR value of 58.64 mL/min/1.73 m2. The IIVs were successfully estimated for CL, V1, and V2 with all estimates equal to or greater than 46.8%. While the inclusion of covariates resulted in an absolute decrease of 11.5% in the IIV of CL compared with the base model, the IIV of CL remained high (47.2%) compared with the original results (25.8%) of study 2 in which a model with the same parametrization and covariate as the current study was developed. Inter-occasion variability was estimated as 13.6% for CL in the final model. A combined residual error model for the data of study 1 and a proportional error model for the data of study 2 and study 3 were applied, respectively. The final model can be expressed as follows:
where \(\mathrm{WGT}\) denotes body weight and \(\mathrm{eGFR}\) denotes eGFR, \({\eta }_{\mathrm{CL}}\), \({\eta }_{V1}\), and \({\eta }_{V2}\) represent the random-effect parameters for IIVs, and \({\eta }_{\mathrm{IOV}}\) represents the random-effect parameters for IOV in ciprofloxacin CL.
The goodness-of-fit plot shows that both the population predictions and the individual predictions of ciprofloxacin concentration data were well in accordance with the actual observations, suggesting that the structural model was adequately established (Fig. 1). The conditional weighted residual errors were evenly and randomly distributed around 0 across both time and the population predictions, indicating that the residual errors were properly specified. The prediction- and variability-corrected visual predictive check plot indicated that the model properly fits the data as the percentiles of observed data mostly fall within the prediction intervals except that the concentration data at low levels (2.5th percentile) were slightly overpredicted (Fig. 2).

Goodness of fit of the final model including observations vs individual predictions (a), observations vs population predictions (b), conditional weighted residuals (CWRES) vs population predictions (c), and CWRES vs time (d). The solid red line is the LOESS regression of the scatter plot; the black dashed line is the unity line; the dashed red line represents the 95%, 50%, and 5% percentile of the data

Prediction-corrected and variability-corrected visual predictive checks of the final model illustrating the pharmacokinetic profile over time (a) or over time after dose (b). The red dashed lines indicate the 97.5%, 50%, and 2.5% percentile of the observed data; the light gray area is the 95% prediction interval of the 97.5% or 2.5% percentile of the model predicted data, and the dark area is the 95% prediction interval of the 50% percentile of the model predicted data
3.2 Post Hoc Analysis of Pharmacokinetic Variability Between Studies
3.2.1 Posterior Distribution
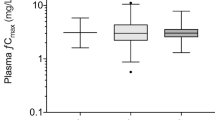
The results of the post hoc analysis showed that the PK parameters differed in both posterior mean and posterior IIV between the three studies (Table 3, Fig. 3). The CL showed the largest deviation between the three studies with a coefficient of variation of 14.3% for posterior mean and 24.1% for posterior IIV (Table 3). Overall, the results of study 1 and study 2 were relatively closer in terms of posterior means while the results of study 2 and study 3 were relatively closer in terms of posterior IIVs (Table 3).

Posterior distribution of pharmacokinetic parameters in the three studies, relative to the typical value of the final model including clearance (CL, upper panel), central volume of distribution (V1, middle panel) and peripheral volume of distribution (V2, lower panel). Dashed lines are the mean values of the three studies. The summary statistics are provided in Table 3
3.2.2 Differences in Pharmacokinetic Exposure
A clear distinction was observed in the calculated AUC24 between studies after a simulated intravenous ciprofloxacin dose of 400 mg t.i.d. (Fig. 4a). The AUC24 of study 3 was the largest among the three studies with a mean AUC24 of 58 mg·h/L compared with 47.7 mg·h/L for study 1 and 47.6 mg·h/L for study 2. Marginal differences were observed in the standard deviation of calculated AUC24 between studies, with a range from 22.8 to 25.8 mg·h/L for the three studies. Similar to the results of AUC24, the highest PTA was observed for the patients of study 3 (Fig. 4b). The PTA is good with an intravenous dose of 400 mg t.i.d. when MIC is not greater than 0.125 mg/L. However, at the EUCAST epidemiological cut-off value of 0.5 mg/L, only 21%, 18%, and 38% of the patients achieved the target for study 1, 2, and 3, respectively.

Box plot of the simulated AUC24 distributions of ciprofloxacin during the first 24 hours of treatment for patients of the three studies (a). Percentage of target attainment (PTA) of area under the concentration–time curve at 24 hours (AUC24)/minimum inhibitory concentration (MIC) equal or greater than 125 (b). The number of simulated patients is 4200, 5900, and 3900 for study 1, 2, and 3, respectively
4 Discussion
In this study, we performed a pooled population PK analysis of ciprofloxacin in a large cohort of ICU patients describing the data of three studies. As we developed the model based on a pooled data set consisting of individual patient data from multiple studies representing a larger sample of the ICU population, the estimated IIVs were overall higher compared with the original studies of study 1 and study 2 [7, 8]. The fact that substantial unexplained IIV and IOV were still present in the final model with only one covariate identified, despite the large size of the data set, indicates that ciprofloxacin pharmacokinetics is not yet comprehensively understood in the ICU patient population and remains difficult to predict on the individual level. The observed PK differences between the three studies in the post hoc analysis also illustrated that there are likely still factors to be identified that can explain the PK variability between studies and individuals.
Apart from the allometric scaling of body weight, the only covariate association identified was eGFR on CL. Expectedly, renal function-related variables should be strong predictors of CL, as ciprofloxacin is weakly bound to protein and the majority of the ciprofloxacin molecules in serum is eliminated through the kidneys [31, 32]. Notably, the original report of study 1 did not identify any covariate associations. Mathematically, the likelihood of a candidate covariate model is a composite of the likelihoods as contributed by the data of each study. In the covariate selection procedure, the data of study 1 might mask a weak or modest covariate effect that was more abundantly present in the data of study 2 and 3. It is worth mentioning that we purposely did not evaluate “study” as a covariate. This variable provides rather limited insight into how patients differ in the three studies, which should in principle be explained by differences in patients’ actual characteristics. Moreover, “study” being a covariate would have drastically diminished the model’s external applicability because such a model would be impossible to externally validate and use in any other clinical setting.
While the final model could describe the data of the three studies as a whole, we revealed that there was still some PK variability unexplained between studies (Table 3, Fig. 3). The largest variability was observed in CL for both the posterior mean and the posterior IIV. This may be attributable to the studied patient population. Study 3 included patients with severe sepsis and septic shock. Consequently, a high percentage of septic patients were included in that study, with 55% of patients fulfilling the criteria for septic shock. Patients with sepsis have an increased risk of elevated drug exposure owing to the derangement of renal and hepatic functions on which the elimination of ciprofloxacin mainly relies [33]. This may be indicated by the estimated posterior mean of CL, V1, and V2, which were all smaller for the patients of study 3 compared with the patients of study 1 and 2 (Table 3). In addition, as we previously mentioned, there is a deviation in the reported PK parameters in the published studies [6, 8,9,10,11,12, 14, 15]. The parameters’ estimates of our model differ from previously published models to varying extents as well. For instance, the most clinically relevant parameter CL is associated with a difference of up to 38.1% from the values of previously published two-compartment models, while for V1, Q, and V2 the differences may be as high as more than 200%. This could be due to the differences in the characteristics of the studied populations, for example, in underlying condition or disease severity. In comparison to a study in which a relatively similar population was studied, our results are in agreement with theirs, resulting in a difference in CL of below 5% [12]. However, we still observed large differences in PK parameters despite the studied patient population of our study being comparable to that of the published patient population [13]. Such a difference may be because of the differences that exist between the investigated populations that were not identified as a result of limitations in the collected data on patient characteristics and thus covariates analyzed in the respective studies, which we further discuss below. This is perhaps partially evidenced by the unreduced IIV of this study as well, despite a larger data set used for the analysis. It is also conceivable that the parameterization of a model can have an impact on the estimates of PK parameters. Nevertheless, the large remaining IIV suggests an insufficient knowledge on the ciprofloxacin pharmacokinetics in ICU patients because of the highly variable nature of this population. It is thus pivotal for clinical professionals to validate and perhaps calibrate an external model before the implementation in a clinical setting.
According to the simulation results, given the same dose regimen, the AUC24 and the PTA of ciprofloxacin differed between studies but mostly between study 3 and the other two (Fig. 4). When the MIC was equal to or greater than 0.125 mg/L, the PTA started to differentiate between studies. Such differences could be clinically relevant at an MIC of 0.25 mg/L. Because the PTA at a MIC of 0.5 mg/L was low for patients in the three studies, the need for a ciprofloxacin dose regimen higher than 400 mg t.i.d. is indicated for higher PTA if microorganisms are to be treated with such an expected MIC. This was in accordance with the findings of the original studies of both study 1 and 2 [7, 8]. However, a breakpoint of 0.5 mg/L is only applicable when treating P. aeruginosa caused infection for which a higher dose is required (≥ 1200 mg/day). Nevertheless, it may be difficult to determine a generally applicable dose regimen owing to the large unexplained existent PK variability between studies. Meanwhile, dosing strategies probably need to be tailored per treatment center likely because of the different subtypes of patients. This highlights that in order to adequately capture the PK variability between ICU patients, we may need better instead of larger datasets including a more diverse and detailed set of covariates. The primary reason for ICU admission may be associated with the pharmacokinetics of ciprofloxacin where, for example, trauma may be related to augmented renal CL while cardiosurgical patients might often have poor organ function. The underlying condition such as comorbidity scores and life expectancy reflecting the health status of a patient can probably also help to explain a fraction of PK variability. Local treatment policies may have an impact on the PK variability as well, for example, the intensity and duration of ventilation, types and doses of fluid resuscitation including total parenteral nutrition, and the doses and duration of the use of inotropes and their changes over time. A number of these factors have been previously identified as covariates for other antibiotics in ICU patients [34,35,36,37]. In addition, the immune response, for example, C-reactive protein, has also been shown to influence pharmacokinetics [38]. Such covariates are undoubtedly of interest to be collected and tested in future studies; however, these were unfortunately not available in the current pooled data analysis, which is an important limitation of this study.
Published models of ciprofloxacin are to a large extent similar in terms of identified covariates [8, 11,12,13,14,15]. The most commonly found are body weight and renal function. We also observed this in other commonly used antibiotics such as vancomycin and meropenem [39, 40]. Undoubtedly, the body weight and renal function reflect a large portion of the PK variability. From a clinical perspective, however, there are likely additional factors that are explanatory for the PK variability between ICU patients, such as those mentioned earlier. We showed in this study that despite a larger dataset, the resulting model is not necessarily more elucidative, which may also explain the differences in the reported PK parameters of published studies and this study, as there may be differences in the distribution of yet unknown but potentially influential covariates between studies. This raises our concern on the covariates identifiability through conventional PK modeling approaches. As the traditional compartmental models represent the biological system with a high level of abstraction at the cost of omitting much detail, we may not be able to identify the covariates that are carried in granular clinical data. Dosing may alternatively be optimized through model-based therapeutic drug monitoring where individual dose advice can be produced using the individual PK parameters.
5 Conclusions
We developed a pooled population PK model of ciprofloxacin representative for a large ICU population on the basis of data from three studies. The model was able to describe the population but there was still a large amount of IIV unexplainable by commonly deployed covariates. In addition, a modest PK variability between studies was also revealed. Therefore, a simple dose strategy of ciprofloxacin suitable for all ICU patients remains challenging and improved models or feedback from therapeutic drug monitoring may be needed.
References
Barrett JS, Fossler MJ, Cadieu KD, Gastonguay MR. Pharmacometrics: a multidisciplinary field to facilitate critical thinking in drug development and translational research settings. J Clin Pharmacol. 2008;48:632–49.
de Velde F, Mouton JW, de Winter BCM, van Gelder T, Koch BCP. Clinical applications of population pharmacokinetic models of antibiotics: challenges and perspectives. Pharmacol Res. 2018;134:280–8.
Vance-Bryan K, Guay DR, Rotschafer JC. Clinical pharmacokinetics of ciprofloxacin. Clin Pharmacokinet. 1990;19:434–61.
Ridley S, Burchett K, Gunning K, Burns A, Kong A, Wright M, et al. Heterogeneity in intensive care units: fact or fiction? Anaesthesia. 1997;52:531–7.
Blot SI, Pea F, Lipman J. The effect of pathophysiology on pharmacokinetics in the critically ill patient: concepts appraised by the example of antimicrobial agents. Adv Drug Deliv Rev. 2014;77:3–11.
van Zanten ARH, Polderman KH, van Geijlswijk IM, van der Meer GYG, Schouten MA, Girbes ARJ. Ciprofloxacin pharmacokinetics in critically ill patients: a prospective cohort study. J Crit Care. 2008;23:422–30.
Abdulla A, Rogouti O, Hunfeld NGM, Endeman H, Dijkstra A, van Gelder T, et al. Population pharmacokinetics and target attainment of ciprofloxacin in critically ill patients. Eur J Clin Pharmacol. 2020;76:957–67.
Gieling EM, Wallenburg E, Frenzel T, de Lange DW, Schouten JA, Ten Oever J, et al. Higher dosage of ciprofloxacin necessary in critically ill patients: a new dosing algorithm based on renal function and pathogen susceptibility. Clin Pharmacol Ther. 2020;108:770–4.
Li X, Zoller M, Fuhr U, Huseyn-Zada M, Maier B, Vogeser M, et al. Ciprofloxacin in critically ill subjects: considering hepatic function, age and sex to choose the optimal dose. J Antimicrob Chemother. 2019;74:682–90.
Conil J-M, Georges B, de Lussy A, Khachman D, Seguin T, Ruiz S, et al. Ciprofloxacin use in critically ill patients: pharmacokinetic and pharmacodynamic approaches. Int J Antimicrob Agents. 2008;32:505–10.
Gai X, Shen N, He B, Zhou Q, Bo S, Li X, et al. Population pharmacokinetics of ciprofloxacin in Chinese elderly patients with lower respiratory tract infection. Zhonghua Yi Xue Za Zhi. 2015;95:1581–5.
Roberts JA, Alobaid AS, Wallis SC, Perner A, Lipman J, Sjövall F. Defining optimal dosing of ciprofloxacin in patients with septic shock. J Antimicrob Chemother. 2019;74:1662–9.
Khachman D, Conil J-M, Georges B, Saivin S, Houin G, Toutain P-L, et al. Optimizing ciprofloxacin dosing in intensive care unit patients through the use of population pharmacokinetic-pharmacodynamic analysis and Monte Carlo simulations. J Antimicrob Chemother. 2011;66:1798–809.
Zahr N, Urien S, Aubry A, Chauvin C, Comets E, Llopis B, et al. Ciprofloxacin population pharmacokinetics during long-term treatment of osteoarticular infections. J Antimicrob Chemother. 2021;76:2906–13.
Šíma M, Michaličková D, Ryšánek P, Cihlářová P, Kuchař M, Lžičařová D, et al. No time dependence of ciprofloxacin pharmacokinetics in critically ill adults: comparison of individual and population analyses. Pharmaceutics. 2021;13:1156. https://doi.org/10.3390/pharmaceutics13081156.
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315:801–10.
Roggeveen LF, Fleuren LM, Guo T, Thoral P, de Grooth HJ, Swart EL, et al. Right Dose Right Now: bedside data-driven personalized antibiotic dosing in severe sepsis and septic shock: rationale and design of a multicenter randomized controlled superiority trial. Trials. 2019;20:745.
Roggeveen LF, Guo T, Driessen RH, Fleuren LM, Thoral P, van der Voort PHJ, et al. Right Dose, Right Now: development of AutoKinetics for real time model informed precision antibiotic dosing decision support at the bedside of critically ill patients. Front Pharmacol. 2020;11:646.
Roberts JA, Abdul-Aziz MH, Lipman J, Mouton JW, Vinks AA, Felton TW, et al. Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. Lancet Infect Dis. 2014;14:498–509.
Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130:461–70.
Wählby U, Thomson AH, Milligan PA, Karlsson MO. Models for time-varying covariates in population pharmacokinetic-pharmacodynamic analysis. Br J Clin Pharmacol. 2004;58:367–77.
Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN): a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75:85–94.
Lindbom L, Pihlgren P, Jonsson EN, Jonsson N. PsN-Toolkit: a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79:241–57.
Nguyen THT, Mouksassi M-S, Holford N, Al-Huniti N, Freedman I, Hooker AC, et al. Model evaluation of continuous data pharmacometric models: metrics and graphics. CPT Pharmacometrics Syst Pharmacol. 2017;6:87–109.
Lavielle M, Ribba B. Enhanced method for diagnosing pharmacometric models: random sampling from conditional distributions. Pharm Res. 2016;33:2979–88.
Savic RM, Karlsson MO. Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11:558–69.
MacArthur RD, Miller M, Albertson T, Panacek E, Johnson D, Teoh L, et al. Adequacy of early empiric antibiotic treatment and survival in severe sepsis: experience from the MONARCS trial. Clin Infect Dis. 2004;38:284–8.
Pea F, Viale P. Bench-to-bedside review: appropriate antibiotic therapy in severe sepsis and septic shock: does the dose matter? Crit Care. 2009;13:214.
Forrest A, Nix DE, Ballow CH, Goss TF, Birmingham MC, Schentag JJ. Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob Agents Chemother. 1993;37:1073–81.
The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters, version 10.0. 2020. http://www.eucast.org/clinical_breakpoints/. Accessed 22 Feb 2022.
Drusano GL. An overview of the pharmacology of intravenously administered ciprofloxacin. Am J Med. 1987;82:339–45.
Dudley MN, Ericson J, Zinner SH. Effect of dose on serum pharmacokinetics of intravenous ciprofloxacin with identification and characterization of extravascular compartments using noncompartmental and compartmental pharmacokinetic models. Antimicrob Agents Chemother. 1987;31:1782–6.
Chai MG, Cotta MO, Abdul-Aziz MH, Roberts JA. What are the current approaches to optimising antimicrobial dosing in the intensive care unit? Pharmaceutics. 2020;12:638.
Pařízková RČ, Martínková J, Havel E, Šafránek P, Kaška M, Astapenko D, et al. Impact of cumulative fluid balance on the pharmacokinetics of extended infusion meropenem in critically ill patients with sepsis. Crit Care. 2021;25:251.
Winter SD, De Winter S, van Hest R, Dreesen E, Annaert P, Wauters J, et al. Quantification and explanation of the variability of first-dose amikacin concentrations in critically ill patients admitted to the emergency department: a population pharmacokinetic analysis. Eur J Drug Metab Pharmacokinet. 2021;46:653–63. https://doi.org/10.1007/s13318-021-00698-w.
Selig DJ, Akers KS, Chung KK, Pruskowski KA, Livezey JR, Por ED. Meropenem pharmacokinetics in critically ill patients with or without burn treated with or without continuous veno-venous haemofiltration. Br J Clin Pharmacol. 2021. https://doi.org/10.1111/bcp.15138.
Werumeus Buning A, Hodiamont CJ, Lechner NM, Schokkin M, Elbers PWG, Juffermans NP, et al. Population pharmacokinetics and probability of target attainment of different dosing regimens of ceftazidime in critically ill patients with a proven or suspected Pseudomonas aeruginosa infection. Antibiotics. 2021;10:612.
Niibe Y, Suzuki T, Yamazaki S, Uchida M, Suzuki T, Takahashi N, et al. Identification of factors affecting meropenem pharmacokinetics in critically ill patients: impact of inflammation on clearance. J Infect Chemother. 2021. https://doi.org/10.1016/j.jiac.2021.12.017.
Cunio CB, Uster DW, Carland JE, Buscher H, Liu Z, Brett J, et al. Towards precision dosing of vancomycin in critically ill patients: an evaluation of the predictive performance of pharmacometric models in ICU patients. Clin Microbiol Infect. 2020. https://doi.org/10.1016/j.cmi.2020.07.005.
Wong G, Farkas A, Sussman R, Daroczi G, Hope WW, Lipman J, et al. Comparison of the accuracy and precision of pharmacokinetic equations to predict free meropenem concentrations in critically ill patients. Antimicrob Agents Chemother. 2015;59:1411–7.
Acknowledgements
We thank Ronald Driessen for kindly helping us with data extraction.
The Dutch Antibiotic PK/PD Collaborators: Luca F. Roggeveen, Department of Intensive Care Medicine, Amsterdam UMC, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands. Lucas M. Fleuren, Department of Intensive Care Medicine, Amsterdam UMC, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands. Nicole G. M. Hunfeld, Department of Hospital Pharmacy, Erasmus University Medical Center, Rotterdam, The Netherlands; Department of Intensive Care, Erasmus University Medical Center, Rotterdam, The Netherlands. Tim M.J. Ewoldt, Department of Intensive Care, Erasmus University Medical Center, Rotterdam, The Netherlands. Anouk E. Muller, Department of Medical Microbiology and Infectious Diseases, Erasmus University Medical Center, Rotterdam, The Netherlands; Department of Medical Microbiology, Haaglanden Medical Center, The Hague, The Netherlands. Annemieke Dijkstra, Department of Intensive Care, Maasstad Hospital, Rotterdam, The Netherlands. Dylan W. de Lange, Department of Intensive Care and National Poisons Information Center, UMC Utrecht, Utrecht, The Netherlands. Emilie Gieling, Department of Pharmacy, Radboud UMC, Nijmegen, The Netherlands; Department of Pharmacy, UMC Utrecht, Utrecht, The Netherlands. Peter Pickkers, Department of Intensive Care, Radboud UMC, Nijmegen, The Netherlands. Jaap ten Oever, Radboud Center for Infectious Diseases, Radboud UMC, Nijmegen, The Netherlands.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Funding
This work is part of the Right Dose, Right Now project by Amsterdam UMC, location Vrije Universiteit Medical Center, and OLVG Oost and the DOLPHIN project by Erasmus University Medical Center, and was partially funded by the ZonMw Rational Pharmacotherapy program (Grant ID: 848017008 and 80-83600-98-40050).
Conflict of interest/competing interests
There are no competing interests to declare.
Ethics approval
The use of patient data in these studies was approved by the local ethics committees.
Consent to participate
Written informed consent forms were obtained from all participants prior to the initiation of the project.
Consent for publication
Not applicable.
Availability of data and material
The data sets used in this analysis are not available.
Code availability
The code is available upon reasonable request.
Authors’ contributions
RvH, RB, BK, and TG conceived the project; TG conducted the analysis; HE, JS, and PE collected the data; TG, RvH, AA, and CvH wrote the manuscript; and all authors revised the manuscript prior to the submission.
Additional information
The members of the Dutch Antibiotic PK/PD are listed in acknowledgements section.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Guo, T., Abdulla, A., Koch, B.C.P. et al. Pooled Population Pharmacokinetic Analysis for Exploring Ciprofloxacin Pharmacokinetic Variability in Intensive Care Patients. Clin Pharmacokinet 61, 869–879 (2022). https://doi.org/10.1007/s40262-022-01114-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-022-01114-5