
Abstract
Vilaprisan is a highly potent selective progesterone receptor modulator in development for the treatment of symptomatic uterine fibroids and endometriosis. Its pharmacokinetics are characterized by rapid absorption, almost complete bioavailability, and dose-proportional exposure. The intrinsic factors of age, bodyweight, and race have no clinically relevant effect on the pharmacokinetics and pharmacodynamics of vilaprisan and do not warrant a dose adjustment. Similarly, vilaprisan can be used in patients with mild or moderate renal or hepatic impairment without dose adjustment, but its use is not recommended in patients with severe organ impairment. Vilaprisan has no perpetrator potential on cytochrome P450 (CYP) enzymes or transporters and therefore restrictions in the concomitant use of their substrates are not required. Nonetheless, because it is a sensitive CYP3A4 substrate itself, concomitant use of vilaprisan with strong CYP3A inhibitors or inducers is not recommended. However, there is no risk for QTc prolongation when vilaprisan and a strong CYP3A inhibitor are administered concomitantly, as indicated by a vilaprisan concentration–QTc response analysis across all studies with triplicate electrocardiogram measurements. Furthermore, due to its mode of action, vilaprisan is also not recommended to be used together with progestin-containing oral contraceptives. Vilaprisan shows a steep exposure–response relationship for inducing amenorrhea in patients with uterine fibroids experiencing heavy menstrual bleeding. Based on simulations, a dose of 2 mg/day is expected to induce a maximum bleeding reduction and was thus selected for phase III.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Vilaprisan is a highly potent selective progesterone receptor modulator in development for the treatment of symptomatic uterine fibroids and endometriosis. |
Its clinical pharmacology was exhaustively investigated in a large number of phase I and II studies, the results of which are summarized this paper. |
Thus, the present paper offers a synopsis of the current state of knowledge of vilaprisan’s clinical pharmacology profile and provides guidance to clinicians. |
1 Introduction
Vilaprisan is a highly potent selective progesterone receptor modulator (SPRM) developed for the treatment of symptomatic uterine fibroids (UFs) and endometriosis.Footnote 1 It has potent antagonistic activity at the progesterone receptor (PR) but no agonistic activity [1].
UFs are the most common benign smooth muscle tumors of the myometrium [2] and can be found in up to 40% of women aged between 35 and 40 years [3]. Symptoms include heavy menstrual bleeding, which can lead to anemia and fatigue, painful periods, abdominal protuberance, painful intercourse, pelvic pressure, and bladder or bowel dysfunction. Furthermore, UFs can have adverse effects on fertility and pregnancy [4].
UF growth is estrogen- and, most importantly, progesterone-dependent. Therefore, current pharmacological treatment options aim at suppressing or modulating these ovarian sex hormones, e.g., by means of gonadotropin-releasing hormone (GnRH) agonists or antagonists. Other treatment options are aromatase inhibitors, oral contraceptives, and levonorgestrel-releasing intrauterine systems [5].
In a recent review of the current data on the efficacy and tolerability of vilaprisan, Ciebiera et al. [6] concluded that vilaprisan was effective in ameliorating UF-related clinical symptoms in phase I and II studies and that its tolerability tends to be more favorable than that of GnRH-agonists.
In this paper, we give an overview on the clinical pharmacology data obtained up to phase II of the clinical development of vilaprisan for the indication treatment of symptomatic UFs (electronic supplementary material [ESM] 1). Most studies in this program have already been published individually or have been accepted for publication; studies not yet published are summarized in ESM 2.
Focusing on the possible impact of intrinsic and extrinsic factors on the pharmacokinetics (PK) and pharmacodynamics (PD) of the drug and the analysis of the relationship between PK and PD in patients with UFs, this overview is intended to provide a practical guidance to clinicians.
2 Physicochemical Properties and Preclinical Pharmacology
2.1 Physicochemical Properties
Vilaprisan {(11β,17β)-17-hydroxy-11-[-4(methylsulfonyl)phenyl]-17-(pentafluoroethyl)estra 4,9-dien-3-one} (Fig. 1) is a highly lipophilic compound with a molecular weight of 544.6 g/mol. It is poorly soluble in aqueous solution under acidic and neutral conditions (< 0.1 mg/100 mL) and shows a high permeability in the Caco-2 cell assay. Therefore, it is a class II drug according to the Biopharmaceutics Classification System (low solubility, high permeability) and also according to the Biopharmaceutics Drug Disposition Classification System (low solubility, extensive metabolism) [7], as it is almost completely eliminated via metabolism.

Vilaprisan structural formula appearing as a steroid substructure
2.2 Mode of Action and Preclinical Pharmacology
In vitro, vilaprisan exhibits highly selective binding affinity to the human PR [8]. It exhibits no agonistic activity in the PR isoform-specific transactivation assays, but inhibits both PR B and PR A, with half maximal inhibitory concentration (IC50) values of 0.09 nM and 0.095 nM, respectively. Vilaprisan shows no glucocorticoid, mineralocorticoid, anti-mineralocorticoid, estrogenic, or anti-estrogenic activity. The anti-glucocorticoid activity in vitro is significantly lower compared with the standard antagonist mifepristone (Table 1). Following multiple-dose administrations of vilaprisan 0.1, 0.5, 1.0, 2.0 and 5.0 mg to premenopausal women over 84 days, no change from baseline in cortisol values was observed up to the maximum tested dose of 5 mg, suggesting no counter-regulation of the hypothalamic-pituitary-adrenal axis as a consequence of clinically relevant glucocorticoid receptor antagonism (data on file, Bayer AG). Adrenocorticotropic hormone concentrations were not measured in clinical pharmacology studies with vilaprisan.
The high antagonistic activity at the PR could also be demonstrated in several in vivo models, including an inhibitory activity on the progesterone-induced differentiation of the endometrium in the rabbit model as well as complete termination of pregnancy in the rat at a dose of 0.5 mg/kg/day subcutaneously and orally, showing approximately a tenfold higher potency than mifepristone.
Exploratory analysis of the messenger RNA (mRNA) expression profile in endometrial tissue of healthy women treated with different doses of vilaprisan showed that the majority of genes were highly downregulated. These genes were involved in processes directly related to cell-cycle regulation/progression or control of cell growth (partly overexpressed in endometrial carcinoma and other malignant tumors), including genes such AURKA, CCNB1, DLGAP5, EIF4EBP1, FOXM1, HOXA10, MELK, MKI67, MYC. Almost all PD effects were reversible or reduced after the first bleeding after treatment [9].
3 Pharmacokinetic (PK) Properties
3.1 General PK in Humans
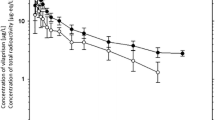
All data presented in this section were collected in populations of healthy postmenopausal women, mainly Caucasians, unless otherwise stated. Vilaprisan was generally administered in the form of 2 or 5 mg immediate-release tablets, and the validated liquid chromatography-tandem mass spectrometry method described by Liu et al. [10] was used to determine vilaprisan in plasma. A condensed overview of the absorption, distribution, and elimination characteristics of vilaprisan in humans is given in Fig. 2. Vilaprisan plasma concentration–time curves obtained after single and multiple administration of different doses of vilaprisan are shown in Fig. 3.

Creator of the central graph: istock.com/Pikovit44 (graph adapted)
Summary of vilaprisan absorption, distribution, and elimination in humans. Light-blue text boxes summarize the results of a mass-balance and metabolite-profiling study in six healthy women; dark-blue boxes summarize the results of a microtracer substudy in seven other healthy women [11]. All numbers are approximate. Total recovery of radiolabel in the mass-balance study amounted to 86.6 ± 2.81%. Geometric mean, geometric coefficient of variation [%], if applicable, and range are provided for absolute bioavailability, volume of distribution, and clearance. VPR vilaprisan.

Source: Schultze-Mosgau et al. [12]. Reproduced with permission of Dustri-Verlag Dr. Karl Feistle GmbH & Co. KG, Germany
Geometric mean plasma concentration versus time curves of vilaprisan obtained after single and multiple doses of vilaprisan 1–30 mg. Error bars = geometric standard deviations.
3.1.1 Absorption
After oral administration in the form of immediate-release tablets, vilaprisan is rapidly and almost completely absorbed, with times to maximum observed plasma concentrations (Cmax) between 1 and 2 h after single-dose administration [11, 12]. Food intake, as shown below, has a negligible impact on the PK of vilaprisan [13].
The systemic exposure of vilaprisan, expressed as area under the plasma concentration–time curve from time zero to infinity (AUC) after single-dose administration, or to 24 h after multiple-dose administration (AUC24md), increases approximately dose-proportionally in the dose range 1–30 mg. It accumulates after multiple dosing, with a factor in the range of 1.85–3.19 as expected based on the observed half-lives (31–38 h), indicating linear PK of vilaprisan in the expected therapeutic dose range up to the maximum tested dose, which exceeds the therapeutic dose by a factor of 15 [12]. Vilaprisan exposure shows low interindividual variability in healthy individuals (geometric mean coefficient of variation [CV]) after multiple dosing of approximately 20–40% for AUC24md and 17–30% for Cmax,md.
The absolute oral bioavailability was determined to be approximately 60% [11], primaily reflecting the contribution of presystemic metabolic first-pass during oral absorption. The overall systemic bioavailability of the immediate-release tablet was similar to that of an oral solution (estimated AUC ratio, 89.0%), whereas the rate of absorption was clearly decreased (estimated Cmax ratio, 62.1%) [data on file, Study 14720, Bayer AG; ESM 3].
3.1.2 Distribution
Vilaprisan is mainly distributed into plasma, with a blood:plasma partition coefficient of between 0.71 and 0.76 [11]. It is moderately and fully reversibly bound to human plasma proteins, showing a fraction unbound of 5.29% in vitro, which is not concentration-dependent; the main binding components are serum albumin and α-1 acid glycoprotein (data on file, Bayer AG). Vilaprisan shows a relevant affinity to tissues, as indicated by a geometric mean steady-state distribution volume (Vss) of approximately 360 L [11].
Based on studies in rats, it can be assumed that there is no relevant penetration of vilaprisan across the blood–brain barrier and the placental barrier, and that excretion in milk is low. Following administration of radiolabeled vilaprisan to lactating rats, an estimated 1% of the dose was excreted in milk (data on file, Bayer AG).
3.1.3 Metabolism and Elimination
Vilaprisan is a low to moderate clearance drug with an average systemic clearance of approximately 7 L/h in healthy subjects [11]. The mean terminal half-lives determined in different PK studies after single or multiple oral administration of vilaprisan doses between 1 and 30 mg were in the range of approximately 31–38 h [11, 12]. Vilaprisan is almost exclusively eliminated via metabolic processes (see Fig. 4 for the proposed main metabolic pathways of vilaprisan in humans. Following oral administration of radiolabeled vilaprisan to healthy subjects, vilaprisan was the dominant compound in plasma. No major plasma metabolites exceeding 10% of drug-related AUC were detected [11]. The main biotransformation pathways are oxidations at the steroid skeleton, as well as reductions in the 3-keto moiety and combinations of both. The formation of oxidation products is mainly catalyzed by CYP3A4; the reduction at the 3-keto moiety is catalyzed by aldoketoreductases [11].

Source: Schultze-Mosgau et al. [11] (no changes were made to the original graph). This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/)
Proposed main metabolic pathways of vilaprisan in humans. AKR aldoketoreductase, CYP cytochrome P450, VPR vilaprisan.
The pivotal role of CYP3A4 responsible for the oxidation reactions in the elimination of vilaprisan was confirmed in drug–drug interaction (DDI) studies with the strong CYP3A4 inhibitor itraconazole and the strong CYP3A4 inducer rifampicin (see Sect. 3.2.3.2 and Sect. 5.1). Vilaprisan is excreted predominantly as metabolites via the biliary/fecal route (74%; 13% via the renal route [11]). Excretion of unchanged vilaprisan accounts for < 2%. Overall, 37% of the dose is eliminated by oxidative biotransformation and 24% by a combined oxidation/reduction reaction. Approximately 24% of the dose could not be allocated to specific metabolites and 13% of the radioactivity was not recovered.
3.1.4 Digression: Hepatic Safety of Vilaprisan
In all subjects treated with vilaprisan in phase I and II studies, no relevant changes in liver enzymes (aspartate aminotransferase [AST], alanine aminotransferase [ALT], alkaline phosphatase [ALP], γ-glutamyltransferase [γ-GT], bilirubin) were observed, and serum liver enzyme level assessment indicated no safety concerns. A very low dose of vilaprisan 2–4 mg was found to be efficacious. Despite high lipophilicity, none of the known risk factors for drug-induced liver injury [14] were found to be applicable for vilaprisan (e.g., no inhibition of bile-acid transporters or bilirubin metabolism, no hint on structural alerts leading to formation of reactive intermediates or covalent binding, no hint on mechanism-based inhibition of CYP enzymes, no mitochondrial or other cell toxicity). In contrast, liver safety signals have been observed with other SPRMs during clinical development, including telapristone and ulipristal acetate, which resulted in either termination of clinical development (telapristone) or severe restrictions in clinical use (ulipristal acetate). Furthermore, mifepristone, currently in single-dose clinical use, showed hints of liver effects after longer-term treatment in a clinical trial [15], as well as in preclinical studies [16]. All these SPRMs have an identical chemical subgroup structure in place consisting of a dimethyl-amino phenyl group that is subject to metabolic oxidation reactions resulting in the formation of aniline derivatives (see Fig. 5) [17,18,19]. Aniline derivatives have been reported to represent a ‘structural alert’ and could cause undesired liver effects by the formation of reactive intermediates [14, 20, 21] in rare cases, if common detoxification mechanism, for example via glutathione adduct formation or other phase II metabolic reactions, fail due to individual constitution. Available data from clinical studies with SPRMs that lack this dimethyl-amino phenyl group, such as vilaprisan and asoprisnil, have provided no evidence for such clinically relevant drug-related change in liver enzyme activity. Therefore, there is a clear differentiation with regard to metabolic pathways and possible metabolic activation between N-dimethylamino phenyl-substituted SPRMs and vilaprisan [5, 22]. Currently, further detailed mechanistic in vitro studies have been initiated, including investigations with liver microsomes, hepatocytes, and cytotoxicity tests under different cell culture conditions to support this hypothesis, and the results from these analyses will be published elsewhere.

Biotransformation pathways of SPRMs with different substructures. Ulipristal: R1 = O-COMe, R2 = COMe, R3 = NMe2; telapristone: R1 = O-COMe, R2 = COCH2OMe, R3 = NMe2; mifepristone: R1 = CCMe, R2 = OH, R3 = NMe2; asoprisnil: R1 = CH2-OMe, R2 = OMe, R3 = CHNOH; vilaprisan: R1 = C2F5, R2 = H, R3 = SO2Me. SPRM selective progesterone receptor modulator
3.1.5 Population PK Model for Vilaprisan in Premenopausal Women
A population PK meta-analysis showed that the PK of vilaprisan can be described by a linear two-compartment model with first-order elimination from the central compartment and first-order absorption kinetics including a lag time [23]. This population PK analysis was based on the PK data obtained in premenopausal or perimenopausal women between 21 and 50 years of age in two phase I [24, 25] and two phase II studies (ASTEROID 1 and ASTEROID 2 trials) [26, 27] as described in detail by Sutter et al. [23]. The results from the population PK model were consistent with the dense PK data established initially in postmenopausal women. Furthermore, no difference in PK was observed between healthy participants and patients.
Among a number of potentially relevant demographic covariates, the covariates body weight and body mass index (BMI) were identified as having a statistically significant effect on the apparent volume of distribution of the central compartment (V2/F) and apparent clearance (CL/F), respectively. The increase in V2/F with body weight was allometrically scaled.
The CL/F estimate for non-obese subjects (BMI <30 kg/m2) was 13.4 L/h, which was 77.3% of the value for obese subjects (BMI ≥30 kg/m2), suggesting slightly higher systemic exposure in obese subjects. This would be consistent with the hypothesis that morbidly obese individuals are under chronic inflammation conditions, leading to the suppression of CYP3A4 activity [28]. Overall, both covariates, i.e., body weight and BMI, are not considered clinically relevant for vilaprisan as they explain < 5% of the observed variability in the PK of vilaprisan. Further evaluations are warranted that include data from phase III studies to substantiate the findings of the covariate analysis reported by Sutter et al. [23]. The current database (N = 414 subjects) was too small for an extensive covariate analysis, and also too homogenous as it included mainly Caucasians (70.8%; 13.8% Asians, 12.6% Blacks or African Americans).
3.2 Impact of Intrinsic and Extrinsic Factors on the PK of Vilaprisan
3.2.1 Overview
The impact of various intrinsic and extrinsic factors on the exposure of vilaprisan was tested in a number of dedicated phase I studies. In addition, a few preselected potential impact factors were tested as covariates during the development of the above-described population PK model for vilaprisan [23]. An overview of the results of these studies and analyses is given in Table 2. Details are discussed in the subsequent sections.
3.2.2 Intrinsic factors
3.2.2.1 Age and Menopausal State
No major impact of age and menopausal state (pre- or postmenopausal) on vilaprisan exposure was detected. Furthermore, neither was age identified as a significant covariate in the population PK meta-analysis of data obtained in 414 premenopausal women between 21 and 50 years of age [23], and nor were there any marked differences in vilaprisan exposure between healthy postmenopausal women, who were mostly > 50 years of age, and women below 50 years of age with UFs and heavy menstrual bleeding (ESM 3).
3.2.2.2 Sex
No major impact of sex on vilaprisan exposure was detected. Due to the currently targeted indications, i.e., UFs and endometriosis, almost all studies with vilaprisan were conducted in women. Men were included in only two studies—a PK study in patients with renal impairment [29] and a PK study in patients with hepatic impairment [30]. A subgroup analysis within the latter study did not reveal any obvious differences in the vilaprisan exposure levels of men and women with normal hepatic function.
3.2.2.3 Body Size
As mentioned above, two covariates were identified—body weight as a significant covariate for V2/F and BMI as a significant covariate for CL/F; they explained 1.2% and 4.4% of the V2/F and CL/F variability, respectively [23]. Thus, no dose adjustment based on body size is necessary.
The CL/F estimate for non-obese subjects (BMI < 30 kg/m2) was 13.4 L/h; for obese subjects (BMI ≥ 30 kg/m2), the CL/F estimate was only 77.3% of that value. This finding is not unexpected: lower clearance in obese individuals than in non-obese individuals has also been reported for other CYP3A4 substrates [31].
3.2.2.4 Ethnicity and Race
No major impact of ethnicity or race on the PK of vilaprisan was detected. Furthermore, neither was ‘racial category’ identified as a significant covariate in the population PK analysis [23], and nor were there any major differences in vilaprisan exposure between the mainly Caucasian, Chinese, and Japanese populations studied in phase I studies of vilaprisan [10, 12] (ESM 2). However, as mentioned above, the number and racial and ethnic diversity of the patients included in the phase I and II studies was limited. Data from phase III studies with a larger sample size and focus on the exposure–response relationships are needed to conclusively assess the impact of race and ethnicity on the PK and, finally, the efficacy of vilaprisan. This question is of particular relevance because epidemiologic studies have shown that the prevalence of UFs is markedly higher in Black women than in White women [2, 3].
3.2.2.5 Renal Impairment
A study in patients with moderate or severe renal impairment (estimated glomerular filtration rate [eGFR] 30–59 mL/min/1.73 m2 and < 30 mL/min/1.73 m2, respectively) and matched healthy control subjects showed that the exposure of vilaprisan is very similar in individuals with impaired renal function and individuals with normal renal function [29]. The geometric mean AUC of vilaprisan was increased by a factor of 1.35 in participants with moderate renal impairment compared with healthy controls (90% confidence interval [CI] 0.918–1.973). The geometric mean CL/F was 9.3 L/h (CV 62.1%; N = 9) in moderately impaired subjects and 12.5 L/h (CV 31.9%; N = 9) in healthy controls. The CL/F observed in severely impaired subjects was very similar to the latter value (12.4 L/h; CV 63.5%; N = 3). There were no obvious correlations between eGFR and any of the PK parameters studied.
Overall, these findings are as expected for a drug that is eliminated from plasma almost completely by hepatic metabolism. Based on these data, no dose adjustment is required for patients with mild or moderate renal impairment, and the ‘standard’ 2 mg dose chosen for phase III studies [23] can also be used in this patient population. However, due to lack of sufficient data, it is not recommended to use vilaprisan in patients with severe renal impairment.
3.2.2.6 Hepatic Impairment
A study in patients with mild or moderate hepatic impairment (Child–Pugh A or B) and matched healthy control subjects showed that the exposure of vilaprisan is increased in patients with hepatic impairment [30], as expected for a compound that is mainly metabolized by the liver through oxidative and reductive metabolism.
Unbound vilaprisan exposure was 1.44‐fold higher in patients with mild hepatic impairment than in healthy controls (90% CI 0.91–2.26) and 1.74‐fold higher in patients with moderate impairment (90% CI 1.09–2.78). The maximum observed peak concentrations of unbound vilaprisan were similar in patients with hepatic impairment and healthy controls. There was a moderate positive correlation between AUCu and bilirubin level (r = 0.5593) and a moderate negative correlation between AUCu and albumin (r = −0.4568).
Overall, the observed impact of hepatic impairment on vilaprisan exposure was mild. Thus, adjustment of the vilaprisan dose is not considered necessary for patients with mild or moderate hepatic impairment. Due to missing data, vilaprisan is not recommended to be used in patients with severe hepatic impairment.
3.2.2.7 Uterine Fibroids
The vilaprisan exposure estimates for participants of the ASTEROID trials, i.e., premenopausal women with UFs and heavy menstrual bleeding, are similar to the values determined in healthy postmenopausal women (ESM 3).
3.2.3 Extrinsic Factors/Interaction Potential
3.2.3.1 Concomitant Food Intake
Food intake has a negligible impact on the PK of vilaprisan. In a food–drug interaction study [13], the extent of absorption (as reflected in the AUC) was increased by approximately 20% when vilaprisan was taken at the end of a high-fat/high-calorie or moderate-fat/moderate-calorie meal compared with fasted conditions. On the other hand, the rate of absorption was slightly decreased (Cmax reduced by approximately 10%) and the time to Cmax increased from 1.5 to 4 h. These differences are not clinically relevant and thus no specific recommendations on whether to take vilaprisan on an empty stomach or with a meal are required.
3.2.3.2 Concomitant Drug Intake/Drug–Drug Interactions with Vilaprisan as the Victim
CYP3A4 was identified as the predominant CYP isoform responsible for oxidative phase I biotransformation reactions of vilaprisan in in vitro studies. Aldoketoreductase (AKR) contributes to vilaprisan metabolism to a lower extent; therefore, the risk of clinically relevant DDIs with vilaprisan as a substrate of CYP3A4 has been confirmed in two clinical interaction studies. Concomitant administration of CYP3A4 inhibitors or inducers has a substantial impact on the PK of vilaprisan. In a PK DDI study with the CYP3A4 inhibitor itraconazole, pre- and coadministration of itraconazole (200 mg/day) increased the vilaprisan exposure approximately 6.2-fold and Cmax approximately 1.8-fold [11]. In another DDI study, pre- and coadministration of the CYP3A4 inducer rifampicin (600 mg/day) decreased vilaprisan exposure by approximately 96% and Cmax by about 86% [32]. Therefore, concomitant use of vilaprisan with strong CYP3A4 inhibitors or inducers is not recommended.
Vilaprisan is classified as a highly permeable compound. It was identified as a P-glycoprotein (P-gp) substrate but shows no substrate characteristics towards breast cancer resistance protein (BCRP) or the hepatic uptake transporters organic anion transporting polypeptides (OATP) 1B1, OATP1B3, and organic cation transporter (OCT) 1.
As vilaprisan is hardly eliminated as an unchanged compound via excretion into feces and/or urine, it is considered unlikely that coadministration of a potent P-gp inhibitor will affect the clearance pathways of vilaprisan. Vilaprisan was also identified as a highly permeable compound, and efflux transporters, such as P-gp, are believed to be less important for the absorption of substrates that are highly permeable. Subsequent clinical data from the DDI study with the strong CYP3A4 and P-gp inhibitor itraconazole supported this assumption, as the time to reach Cmax (tmax) of vilaprisan was not shortened in the presence of itraconazole, which would have been expected if P-gp is relevantly involved in the efflux of vilaprisan during the absorption phase.
Concomitant intake of a combined oral contraceptive with vilaprisan mildly increased vilaprisan exposure (estimated AUC ratio 112%; 90% CI 102–122%) [33].
4 Pharmacodynamic (PD) Properties and PK/PD Relationships
4.1 PD in Healthy Premenopausal Women
In healthy premenopausal women, multiple daily doses of vilaprisan (0.1–5 mg) resulted in a dose-dependent amenorrhea rate (spotting allowed) during the 3-month treatment period. The maximum amenorrhea rate was reached after vilaprisan 2 mg, with a point estimate of 93% (90% credible interval 83–98%) without a clear further increase after higher dosages [24]. After the end of treatment, menstrual bleeding returned within 20.3–25.8 days. In a further PD study in healthy premenopausal women, daily oral administration of vilaprisan at doses of 0.5, 1, 2 or 4 mg over 84 days was investigated. Vilaprisan effectively and dose-dependently inhibited ovulation in more than 80% of women receiving vilaprisan at dosages of ≥ 1 mg/day [25].Footnote 2 After the end of treatment, ovulatory cycles returned quickly, as evidenced by growing follicles and increased estradiol (E2) values followed by follicle rupture and rise of progesterone values.Footnote 3 Although vilaprisan effectively inhibited ovulation, the development of active follicle-like structures was not suppressed under treatment. Maximum follicle sizes were slightly increased during treatment across all dose groups (Fig. 6). At follow-up, values were similar to those measured in the pretreatment cycle. No persistent follicles/ovarian cysts of critical size were observed.

Maximum diameters of follicle-like structures before, during, and after 3-month administration of vilaprisan to healthy young women (box-and-whiskers plot). Box: 25% and 75% quartiles; horizontal line: median; cross: arithmetic mean; small square: outlying value. Whiskers extend as far as data, but to a distance of 1.5 times the interquartile range (data on file, Bayer AG). Details of the study and descriptive statistics for the depicted data were described by Schütt et al. [25]
E2 levels were moderately decreased during treatment with vilaprisan; however, mean average E2 concentrations remained above 80 pg/mL in line with the development of follicles. The decrease in E2 levels was fully reversible after the end of treatment.
As vilaprisan has an embryo-lethal effect in rats and rabbits, patients are required to use an acceptable non-hormonal method of contraception (e.g., condom with spermicide) during treatment with vilaprisan (see also Sect. 5.2).
4.2 PD in Premenopausal Women with Uterine Fibroids
4.2.1 Induction of Amenorrhea
The amenorrhea rates observed in premenopausal women with symptomatic UFs treated with different doses of vilaprisan [26, 27] increased in a similar dose-dependent manner as those described above for healthy premenopausal women. In the ASTEROID 1 trial, for example, amenorrhea was achieved in the majority of subjects after a few days of treatment with vilaprisan 1, 2, or 4 mg/day and maintained until the end of treatment in week 12 (cumulative amenorrhea rate 85%, 89%, and 83%, respectively) [26].
A recently published exposure–response analysis of the data from this trial shows a steep exposure–response relationship for induced amenorrhea (Fig. 7) [23]. The estimated maximum probability of inducing amenorrhea (maximum achievable response rate) was 59% (95% CI 49–68), with an AUC24 at steady state (AUC24ss) of 36.9 μg·h/L (95% CI 27.7–48.7). Simulations showed that an exposure resulting in 90% of the maximum probability of inducing amenorrhea will be reached by 98% of patients with once-daily administration of vilaprisan 2 mg, while this threshold will be reached by only 64% of patients at 1 mg/day. Therefore, the 2 mg dose was chosen for phase III studies in patients with UFs.

Source: Sutter et al. [23] (axis labels adapted to journal style). This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/)
Exposure–response relationship for vilaprisan-induced amenorrhea (ASTEROID 1 trial; N = 267). Red dashed line represents the probability of non-bleeding predicted by the final categorical pharmacokinetic/pharmacodynamic model; red area represents the 95% confidence interval of the predicted probability of non-bleeding; black circles represent the event rate = observed probability in distinct AUC24ss intervals; colored symbols with lines represent the median, minimum, and maximum of AUC24ss of the respective dose; colored symbols represent the individual AUC24ss values indicating occurrence/absence of a bleeding event. Of note: the highest AUC24ss values in the 4 mg dose group are not shown for scaling reasons. AUC24ss exposure at steady state (area under the concentration versus time curve from time zero to 24 h postdose), EAUC50 exposure for 50% of the maximum induced amenorrhea rate, EAUC90 exposure for 90% of the maximum induced amenorrhea rate, n number of binned individuals.
Graphical comparisons of simulated and observed probability for amenorrhea showed that the exposure–response relationship based on ASTEROID 1 was consistent with ASTEROID 2 data. A covariate analysis of the relationship between vilaprisan exposure and induction of amenorrhea did not reveal any statistically significant differences between different racial subgroups and the overall population. However, as mentioned before with regard to vilaprisan exposure, the sample used for this analysis was not large enough to allow qualified conclusions about factors potentially impacting the PD of vilaprisan.
4.2.2 Reduction of Estradiol Levels
As a safety parameter, exposure of unbound E2 during treatment with vilaprisan was investigated. In the ASTEROID 1 and 2 trials, the concentrations of unbound E2 were mostly within the physiologic range for the follicular phase during treatment with vilaprisan, although a moderate trend toward lower E2 levels with increasing vilaprisan exposure was observed [26]. An exposure–response analysis for E2 concentrations in serum confirmed this observation [23], as shown in Fig. 8. The clinical relevance of the decrease in E2 levels cannot be assessed based on the available vilaprisan short-term phase II data and should be further evaluated in long-term safety studies.

Source: Sutter et al. [23] (axis labels adapted to journal style). This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/)
Exposure–response relationship for vilaprisan-induced changes in estradiol concentrations (ASTEROID 1 and 2 trials; N = 354). Blue dots represent subjects from the ASTEROID 1 trial; cyan dots represent subjects from the ASTEROID 2 trial; solid line represents the moving median of the observations; blue band represents the 10th–90th percentile of the observations; and gray band represents the 10th–90th percentile of the baseline observations (individual observations not shown) from all subjects in ASTEROID 1 and ASTEROID 2. AUC24ss exposure at steady state (area under the concentration versus time curve from time zero to 24 h postdose).
4.3 QTc-Prolongation Potential
Critical evaluations of the electrocardiogram data obtained in clinical studies with vilaprisan did not suggest a risk for QT prolongation following administration of vilaprisan, and neither are any such class effects known for SPRMs. However, as it is possible that vilaprisan concentrations markedly above the expected therapeutic range are reached when the drug (against the recommended use) is taken together with a strong CYP3A4 inhibitor [11], an integrated vilaprisan concentration–QTc response analysis across all available studies with triplicate electrocardiogram measurements and corresponding PK data was conducted (ESM 4).Footnote 4 As illustrated in Fig. 9, there are no indications that the mean difference in QTc intervals from baseline and placebo (ΔΔQTc) exceeds 10 ms at systemic exposures up to 193 μg/L—a concentration that is approximately 17-fold above the maximum concentration observed after multiple administration of the intended therapeutic dose of 2 mg, i.e., even if a strong CYP3A4 inhibitor such as itraconazole is taken concomitantly, there appears to be no risk for QTc prolongation.

Relationship between vilaprisan plasma concentration and QTc interval. The numbers starting with ‘14…’ are study numbers. The symbols in the upper graph show the mean of the observed ∆∆QTcF for each concentration bin; the vertical lines show the 90% CIs of the mean; and the solid line is the predicted typical change in ∆∆QTcF with increasing vilaprisan plasma concentrations. The gray area shows the predicted 90% CI; the long dashed (blue) line shows the Cmax,ss for a 2 mg dose, and the short dashed (blue) line shows the Cmax,ss for a 2 mg dose when coadministered with itraconazole 200 mg once daily. The lower graph shows the distribution of the plasma concentrations in different subgroups of subjects as a boxplot. The upper whisker extends from the 75th percentile to the highest value that is within 1.5 × interquartile range, and the lower whisker extends from the 25th percentile to the lowest value within 1.5 × interquartile range. Data beyond the ends of the whiskers are outliers and are plotted as points. The data used for this concentration–response analysis covered a dose range from vilaprisan 0.25 mg administered as a single dose, up to vilaprisan 30 mg/day for 4 weeks. Altogether, 858 paired vilaprisan concentration/QTc values were analyzed using linear mixed-effect modeling. CI confidence interval, Cmax,ss maximum concentration at steady state, ΔΔQTc difference in QTc intervals from baseline and placebo
5 Interaction Potential
5.1 PK Interactions
As already described above, two dedicated DDI studies with vilaprisan as the victim of CYP3A inducers and inhibitors were conducted during the development of the drug. In contrast, interaction studies with vilaprisan as the perpetrator were not conducted.
Since no inhibition of CYP enzymes was observed up to 5 µM vilaprisan concentrations with CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4 in in vitro assays, the risk for clinically relevant DDIs of vilaprisan with substrates of these enzymes is considered to be low. Furthermore, there is no potential of induction of CYP3A4 activity when taking the maximum unbound plasma concentration at a dose of 4 mg/patient into account.
Uridine 5ʹ-diphospho-glucuronosyltransferase (UGT) 1A4 and UGT1A1 were moderately inhibited by vilaprisan, with IC50 values of 26.6 and 11.4 µM, respectively. No inhibition of UGT1A6, 1A9, 2B4, and 2B7 was observed. Maximum unbound concentration of an anticipated maximum therapeutic dose of 4 mg/day in the endometriosis indication is predicted to be about 2 nmol/L, which is >2000-fold lower than the maximum tested concentrations of 5 µM that showed no inhibitory effect. Therefore, the risk for clinically relevant DDIs with vilaprisan as the perpetrator and comedications that are substrates of these enzymes is considered to be low. Vilaprisan is no inhibitor for P-gp, BCRP, OATP1B1, OATP1B3, OCT1, or the renal uptake transporters OAT1, OAT3, and OCT2 as well as MATE1 and MATE2K and BSEP, investigated in appropriate in vitro assays (data on file, Bayer AG) [ESM 5].
5.2 PD Interactions
Since many women who suffer from symptomatic UFs are of reproductive age, hormonal contraception is widely used in this patient population. As the progestin component of hormonal contraceptives acts on the PR as an agonist, a PD interaction with SPRMs such as vilaprisan that act predominantly antagonistic on the PR in many tissues must be assumed. In a dedicated clinical PK and PD DDI study with vilaprisan and a combined oral contraceptive consisting of levonorgestrel 0.15 mg and ethinylestradiol 0.03 mg, the known contraceptive-driven suppression of ovarian activity, as assessed using Hoogland scores, was mildly affected by coadministration of vilaprisan [33, 34]. The effects of the contraceptive on cervical function, as assessed using the Insler score [35], was moderately affected when coadministered with vilaprisan, indicating that the efficacy of the contraceptive can no longer be taken for granted. Therefore, the combined used of vilaprisan with oral contraceptives is not recommended.
6 Conclusion
The PK and PD of vilaprisan, an SPRM indicated for the treatment of symptoms associated with UFs and endometriosis, have been extensively characterized in several phase I and II studies, including population PK/PD modeling and physiologically-based PK modeling analyses [36]. A comprehensive evaluation of all generated data covering preclinical in vitro and in vivo investigations up to clinical phase II data led to a thorough understanding of the relevant mechanistic PK and PD processes driving the clinical profile of vilaprisan, and provides a solid basis for practical guidance on the safe and effective use in patients.
Vilaprisan was well tolerated over a broad exposure range, and together with the favorable PK and PD profile, this supports the use of the same dose in all patients irrespective of body weight, race, and age, and also in patients with mild and moderate renally or hepatically impaired function.
Notes
Independently from developing vilaprisan for the indication ‘treatment of symptomatic uterine fibroids’, Bayer AG started a development program for the indication ‘treatment of endometriosis’.
Ovarian activity was classified according to Hoogland and Skouby (Contraception. 1993;47(6):583–590). Three parameters determining ovarian activity are combined to a 6-step scoring system: (a) the diameter of the maximum follicle-like structure; (b) the E2 serum concentration; and (c) the progesterone serum concentration. Ovulation inhibition was defined as Hoogland score < 6 in the complete treatment epoch.
Follicle size was measured by transvaginal ultrasound, and E2, progesterone, and follicle-stimulating hormone (FSH) and luteinizing hormone (LH) levels were determined repeatedly before, during, and after treatment [25].
In ESM 4, we provide an extended description of the new model-based concentration–QTc-effect modeling approach we have applied, as a successful example of how a comprehensive PK and electrocardiogram data set obtained in various clinical studies can be used to waive the otherwise obligatory thorough QT study.
References
Wagenfeld A, Saunders PTK, Whitaker L, Critchley HOD. Selective progesterone receptor modulators (SPRMs): progesterone receptor action, mode of action on the endometrium and treatment options in gynecological therapies. Expert Opin Ther Targets. 2016;20:1045–54. https://doi.org/10.1080/14728222.2016.1180368.
Stewart EA, Cookson CL, Gandolfo RA, Schulze-Rath R. Epidemiology of uterine fibroids: a systematic review. BCOG. 2017;124:1501–12. https://doi.org/10.1111/1471-0528.14640.
Baird DD, Dunson DB, Hill MC, Cousins D, Schectman JM. High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am J Obstet Gynecol. 2003;188:100–7. https://doi.org/10.1067/mob.2003.99.
Levy G, Hill MJ, Beall S, Zarek SM, Segars JH, Catherino WH. Leiomyoma: genetics, assisted reproduction, pregnancy and therapeutic advances. J Assist Reprod Genet. 2012;29:703–12. https://doi.org/10.1007/s10815-012-9784-0.
Ali M, Al-Hendy A. Uterine fibroid therapy: the pharmacokinetic considerations. Expert Opin Drug Metab Toxicol. 2018;14:887–9. https://doi.org/10.1080/17425255.2018.1506766.
Ciebiera M, Vitale SG, Ferrero S, Vilos GA, Barra F, Caruso S, et al. Vilaprisan, a new selective progesterone receptor modulator in uterine fibroid pharmacotherapy—will it really be a breakthrough? Curr Pharm Des. 2020;26:300–9. https://doi.org/10.2174/1381612826666200127092208.
Charalabidis A, Sfouni M, Bergström C, Macheras P. The Biopharmaceutics Classification System (BCS) and the Biopharmaceutics Drug Disposition Classification System (BDDCS): beyond guidelines. Int J Pharm. 2019;566:264–81. https://doi.org/10.1016/j.ijpharm.2019.05.041.
Wagenfeld A, Bone W, Schwede W, Fritsch M, Fischer OM, Moeller C. BAY 1002670: a novel, highly potent and selective progesterone receptor modulator for gynaecological therapies. Hum Reprod. 2013;28:2253–64. https://doi.org/10.1093/humrep/det247.
Wagenfeld A, Schulz A, Schultze-Mosgau M, Schuett B, Machens K, Rohde B. Exploratory analysis of molecular pharmacodynamic effects of vilaprisan in the endometrium. Fertil Steril. 2018;110:e134–5. https://doi.org/10.1016/j.fertnstert.2018.07.399.
Liu H, Jiang J, Chen Z, Zhang Y, Li J, Hoechel J, et al. Pharmacokinetics and safety of the selective progesterone receptor modulator vilaprisan in Chinese healthy postmenopausal women. Clin Pharmacol Drug Dev. https://doi.org/10.1002/cpdd.851(Epub 27 Jul 2020).
Schultze-Mosgau M-H, Höchel J, Prien O, Zimmermann T, Brooks A, Bush J, Rottmann A. Characterization of the pharmacokinetics of vilaprisan: bioavailability, excretion, biotransformation, and drug–drug interaction potential. Clin Pharmacokinet. 2018;57:1001–15. https://doi.org/10.1007/s40262-017-0607-4.
Schultze-Mosgau M-H, Schuett B, Hafner F-T, Zollmann F, Kaiser A, Hoechel J, Rohde B. Pharmacokinetics and safety of the selective progesterone receptor modulator vilaprisan in healthy postmenopausal women. Int J Clin Pharmacol Ther. 2017;55:16–24. https://doi.org/10.5414/CP202756.
Schultze-Mosgau M-H, Kaiser A, Zollmann FS. Effect of food intake on the pharmacokinetics of the selective progesterone receptor modulator vilaprisan: a randomized clinical study in healthy postmenopausal women. Clin Pharmacol Drug Dev. https://doi.org/10.1002/cpdd.876(Epub 5 Oct 2020).
Schadt S, Simon S, Kustermann S, Boess F, McGinnis C, Brink A, et al. Minimizing DILI risk in drug discovery—a screening tool for drug candidates. Toxicol In Vitro. 2015;30:429–37. https://doi.org/10.1016/j.tiv.2015.09.019.
Esteve JLC, Acosta R, Pérez Y, Campos R, Hernández AV, Texidó CS. Treatment of uterine myoma with 5 or 10mg mifepristone daily during 6 months, post-treatment evolution over 12 months: double-blind randomised clinical trial. Eur J Obstet Gynecol Reprod Biol. 2012;161:202–8. https://doi.org/10.1016/j.ejogrb.2011.12.018.
Xiao Y, Zhu Y, Yu S, Yan C, Ho RJY, Liu J, et al. Thirty-day rat toxicity study reveals reversible liver toxicity of mifepristone (RU486) and metapristone. Toxicol Mech Methods. 2016;26:36–45. https://doi.org/10.3109/15376516.2015.1118715.
Pohl O, Zobrist RH, Gotteland J-P. The clinical pharmacology and pharmacokinetics of ulipristal acetate for the treatment of uterine fibroids. Reprod Sci. 2015;22:476–83. https://doi.org/10.1177/1933719114549850.
Sitruk-Ware R, Spitz IM. Pharmacological properties of mifepristone: toxicology and safety in animal and human studies. Contraception. 2003;68:409–20. https://doi.org/10.1016/s0010-7824(03)00171-9.
Morris D, Podolski J, Kirsch A, Wiehle R, Fleckenstein L. Population pharmacokinetics of telapristone (CDB-4124) and its active monodemethylated metabolite CDB-4453, with a mixture model for total clearance. AAPS J. 2011;13:665–73. https://doi.org/10.1208/s12248-011-9304-7.
Liu R, Yu X, Wallqvist A. Data-driven identification of structural alerts for mitigating the risk of drug-induced human liver injuries. J Cheminform. 2015;7:4. https://doi.org/10.1186/s13321-015-0053-y.
Kalgutkar AS. Should the incorporation of structural alerts be restricted in drug design? An analysis of structure-toxicity trends with aniline-based drugs. Curr Med Chem. 2015;22:438–64. https://doi.org/10.2174/0929867321666141112122118.
Möller C, Bone W, Cleve A, Klar U, Rotgeri A, Rottmann A, et al. Discovery of vilaprisan (BAY 1002670): a highly potent and selective progesterone receptor modulator optimized for gynecologic therapies. ChemMedChem. 2018;13:2271–80. https://doi.org/10.1002/cmdc.201800487.
Sutter G, Frei M, Schultze-Mosgau M-H, Petersdorf K, Seitz C, Ploeger B. Assessment of the safe and efficacious dose of the selective progesterone receptor modulator vilaprisan for the treatment of patients with uterine fibroids by exposure-response modelling and simulation. Br J Clin Pharmacol. https://doi.org/10.1111/bcp.15014(Epub 30 Jul 2021).
Schütt B, Kaiser A, Schultze-Mosgau M-H, Seitz C, Bell D, Koch M, Rohde B. Pharmacodynamics and safety of the novel selective progesterone receptor modulator vilaprisan: a double-blind, randomized, placebo-controlled phase 1 trial in healthy women. Hum Reprod. 2016;31:1703–12. https://doi.org/10.1093/humrep/dew140.
Schütt B, Schultze-Mosgau M-H, Draeger C, Chang X, Löwen S, Kaiser A, Rohde B. Effect of the novel selective progesterone receptor modulator vilaprisan on ovarian activity in healthy women. J Clin Pharmacol. 2018;58:228–39. https://doi.org/10.1002/jcph.998.
Bradley LD, Singh SS, Simon J, Gemzell-Danielsson K, Petersdorf K, Groettrup-Wolfers E, et al. Vilaprisan in women with uterine fibroids: the randomized phase 2b ASTEROID 1 study. Fertil Steril. 2019;111:240–8. https://doi.org/10.1016/j.fertnstert.2018.10.012.
Gemzell-Danielsson K, Heikinheimo O, Zatik J, Poka R, Rechberger T, Hudecek R, et al. Efficacy and safety of vilaprisan in women with uterine fibroids: Data from the phase 2b randomized controlled trial ASTEROID 2. Eur J Obstet Gynecol Reprod Biol. 2020;252:7–14. https://doi.org/10.1016/j.ejogrb.2020.05.043.
Brill MJ, van Rongen A, van Dongen EP, van Ramshorst B, Hazebroek EJ, Darwich AS, et al. The pharmacokinetics of the CYP3A substrate midazolam in morbidly obese patients before and one year after bariatric surgery. Pharm Res. 2015;32:3927–36. https://doi.org/10.1007/s11095-015-1752-9.
Schultze-Mosgau M-H, Lasseter KC, Marbury T, Loewen S, Riecke K. Pharmacokinetics and safety of the novel selective progesterone receptor modulator vilaprisan in participants with renal impairment. J Clin Pharmacol. 2020;60:1030–8. https://doi.org/10.1002/jcph.1608.
Chattopadhyay N, Riecke K, Ligges S, Zimmermann T, Halabi A, Schultze-Mosgau M-H. Effect of hepatic impairment on the pharmacokinetics of vilaprisan: an open-label, single-dose, parallel-group study. Br J Clin Pharmacol. 2019;85:2011–21. https://doi.org/10.1111/bcp.13992.
Brill MJE, Diepstraten J, van Rongen A, van Kralingen S, van den Anker JN, Knibbe CAJ. Impact of obesity on drug metabolism and elimination in adults and children. Clin Pharmacokinet. 2012;51:277–304. https://doi.org/10.2165/11599410-000000000-00000.
Chattopadhyay N, Kanacher T, Casjens M, Frechen S, Ligges S, Zimmermann T, et al. CYP3A4-mediated effects of rifampicin on the pharmacokinetics of vilaprisan and its UGT1A1-mediated effects on bilirubin glucuronidation in humans. Br J Clin Pharmacol. 2018;84:2857–66. https://doi.org/10.1111/bcp.13750.
Hoogland HJ, Skouby SO. Ultrasound evaluation of ovarian activity under oral contraceptives. Contraception. 1993;47(6):583–590.
Schultze-Mosgau M-H, Schütt B, Draeger C, Casjens M, Loewen S, Zimmermann T, Rohde B. The effects of vilaprisan on the pharmacodynamics and pharmacokinetics of a combined oral contraceptive—a randomized controlled trial. Br J Clin Pharmacol. 2021;87:925–36. https://doi.org/10.1111/bcp.14443.
Insler V, Melmed H, Eichenbrenner I, Serr DM, Lunenfeld B. The Cervical Score. A simple semiquantitative method for monitoring of the menstrual cycle. Int J Gynecol Obstet. 1972;10:223–8.
Kanacher T, Frechen S, Wendl T, Block M, Rottmann A, Chattopadhyay N, et al. Towards a detailed understanding of the impact of drug drug interactions on vilaprisan exposure by PBPK modeling & simulation. Poster presented at the DDI Workshop at Marbach Castle, Öhningen, Germany; 2017.
Acknowledgements
The authors would like to thank Barbara Schütt, previously employed by Bayer AG, Berlin, who was involved in the planning and conducting of several phase I studies on vilapisan, for her contributions to the first draft of this paper, and C. Hilka Wauschkuhn, Bonn, Germany, for medical writing support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
All studies reported in, as well as the preparation of, this paper were sponsored by Bayer AG.
Conflict of interest
Marcus-Hillert Schultze-Mosgau, Bart A. Ploeger, Matthias Frei, Joachim Höchel, and Antje Rottmann are employees of Bayer AG.
Availability of data and material
Available upon reasonable request.
Code availability
Not applicable.
Ethics approval
All studies reported in this paper were approved by the relevant Ethics Committees.
Consent to participate
All study subjects gave their written informed consent.
Consent for publication
Not applicable.
Author contributions
All authors contributed to the conception and content of this paper. MSM wrote the first draft, which the other authors critically revised.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Schultze-Mosgau, MH., Ploeger, B.A., Frei, M. et al. Clinical Pharmacokinetics and Pharmacodynamics of the Selective Progesterone Receptor Modulator Vilaprisan: A Comprehensive Overview. Clin Pharmacokinet 61, 1–16 (2022). https://doi.org/10.1007/s40262-021-01073-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01073-3