
Abstract
Background
Allopurinol, an xanthine oxidase (XO) inhibitor, is a promising intervention that may provide neuroprotection for neonates with hypoxic-ischemic encephalopathy (HIE). Currently, a double-blind, placebo-controlled study (ALBINO, NCT03162653) is investigating the neuroprotective effect of allopurinol in HIE neonates.
Objective
The aim of the current study was to establish the pharmacokinetics (PK) of allopurinol and oxypurinol, and the pharmacodynamics (PD) of both compounds on hypoxanthine, xanthine, and uric acid in HIE neonates. The dosage used and the effect of allopurinol in this population, either or not undergoing therapeutic hypothermia (TH), were evaluated.
Methods
Forty-six neonates from the ALBINO study and two historical clinical studies were included. All doses were administered on the first day of life. In the ALBINO study (n = 20), neonates received a first dose of allopurinol 20 mg/kg, and, in the case of TH (n = 13), a second dose of allopurinol 10 mg/kg. In the historical cohorts (n = 26), neonates (all without TH) received two doses of allopurinol 20 mg/kg in total. Allopurinol and oxypurinol population PK, and their effects on inhibiting conversions of hypoxanthine and xanthine to uric acid, were assessed using nonlinear mixed-effects modelling.
Results
Allopurinol and oxypurinol PK were described by two sequential one-compartment models with an autoinhibition effect on allopurinol metabolism by oxypurinol. For allopurinol, clearance (CL) was 0.83 L/h (95% confidence interval [CI] 0.62–1.09) and volume of distribution (Vd) was 2.43 L (95% CI 2.25–2.63). For metabolite oxypurinol, CL and Vd relative to a formation fraction (fm) were 0.26 L/h (95% CI 0.23–0.3) and 11 L (95% CI 9.9–12.2), respectively. No difference in allopurinol and oxypurinol CL was found between TH and non-TH patients. The effect of allopurinol and oxypurinol on XO inhibition was described by a turnover model of hypoxanthine with sequential metabolites xanthine and uric acid. The combined allopurinol and oxypurinol concentration at the half-maximal XO inhibition was 0.36 mg/L (95% CI 0.31–0.42).
Conclusion
The PK and PD of allopurinol, oxypurinol, hypoxanthine, xanthine, and uric acid in neonates with HIE were described. The dosing regimen applied in the ALBINO trial leads to the targeted XO inhibition in neonates treated with or without TH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The dosing regimen applied in the ALBINO trial leads to the targeted xanthine oxidase inhibition in hypoxic-ischemic encephalopathy (HIE) neonates treated with or without therapeutic hypothermia (TH). |
The disease-related autoinhibition effect of allopurinol metabolism, as well as higher hypoxanthine, xanthine and uric acid initial levels, were identified in HIE neonates. |
Clearances of allopurinol and oxypurinol were not significantly different between TH and non-TH patients. |
1 Introduction
Hypoxic-ischemic encephalopathy (HIE) caused by birth asphyxia is a major cause of neonatal death and long-term disability worldwide, occurring in 1–2 per 1000 births in the developed world [1]. At present, therapeutic hypothermia (TH) is the only established neuroprotective therapy for moderate to severe HIE [2, 3]. Despite hypothermia and modern supportive neonatal intensive care, around 50% of children with moderate or severe HIE still experience long-term neurodevelopmental impairment [4]. Therefore, besides hypothermia, additional early neuroprotective interventions are required.
During an hypoxic-ischemic (HI) event, two destructive pathways are responsible for HIE-associated permanent brain damage. First, fetal hypoxia leads to immediate necrosis and apoptosis of cells, and, second, reperfusion and reoxygenation occurring after the HI event further increase apoptosis of neuronal cells, cause inflammation and inhibit regeneration [5, 6]. During hypoxia, a highly active form of xanthine oxidase (XO) is formed by proteolytic effects, and hypoxanthine is accumulated due to the depletion of ATP [7, 8]. Hypoxanthine is subsequently catabolized by XO during the reperfusion and reoxygenation stage, which leads to the formation of free radicals and superoxide that are toxic for the immature brain (Fig. 1) [7, 8].

The (1) During fetal hypoxia, hypoxanthine and xanthine oxidase levels rise because of primary energy failure and ATP degradation. (2) During reperfusion, hypoxanthine in combination with oxygen is converted by xanthine oxidase into xanthine, uric acid and toxic superoxide. (3) Allopurinol and oxypurinol inhibit the conversion of hypoxanthine to xanthine and xanthine to uric acid, thus preventing the conversion of oxygen to superoxide. (4) Accumulated hypoxanthine converts to inosine monophosphate (IMP) via the salvage pathway and increases the purine metabolite adenosine. IMP inosine monophosphate
There is evidence that allopurinol is a promising agent to prevent neuronal damage caused by fetal hypoxia and reperfusion, since both allopurinol and its active metabolite oxypurinol are XO inhibitors, which limit the production of toxic superoxide [8,9,10]. Furthermore, allopurinol at high concentrations acts as an iron-chelator and direct scavenger of free radicals [11, 12]. Moreover, as purine degradation is blocked, allopurinol intervention results in an accumulation of adenosine, a potent inhibitory neuromodulator, providing additional neuroprotection in HIE [13, 14]. To date, the positive neuroprotective effects of allopurinol in HIE have been proposed in several clinical studies [15]. Currently, a double-blind, placebo-controlled study (ALBINO) is in progress to investigate the neuroprotective effect of allopurinol in infants with HIE treated with and without TH [16].
Given the potential of allopurinol in the neuroprotective treatment of HIE, an optimal allopurinol dosing regimen is needed; however, this is hampered by the poor understanding of the pharmacokinetics (PK) and pharmacodynamics (PD) of allopurinol in this specific population. In adults, the population PK and PD of allopurinol have been fairly well studied [17, 18]. Allopurinol is rapidly metabolized to oxypurinol by enzymes such as XO and aldehyde oxidoreductase (AOR) [19]. Oxypurinol is cleared unchanged renally. The half-life of allopurinol is 1–2 h, whereas oxypurinol has a half-life of 25–30 h [20,21,22]. As a result, although allopurinol and oxypurinol showed similar ability in inhibiting XO, oxypurinol is assumed to be the major active moiety that provides pharmacological effects [23, 24].
The population PK of allopurinol and oxypurinol in asphyxiated neonates has been previously described by McGaurn et al. [25] and van Kesteren et al. [26]; however, the reported allopurinol half-life was distinctly different in the two studies, possibly due to the sparseness of available data. In these prior studies, oxypurinol PK were not quantified and all data were collected in the era before TH was implemented. Furthermore, neither physiological nor disease-related factors that might affect allopurinol and oxypurinol clearance (CL) in asphyxiated neonates were considered. Recently, Favié et al. [27] proposed an integrated mechanism-based population PK model for HIE neonates treated with TH based on seven drugs and five metabolites. In this integrated model, drugs and their metabolites were categorized into high, intermediate, and low CL compounds according to their PK characteristics. The effects of body size, maturation, recovery of organ function, and body temperature on drug CL were quantified within each group.
In order to optimize the allopurinol dosage used in current and future clinical studies, the first aim of this study was to describe the population PK of allopurinol and oxypurinol in HIE neonates under the framework reported by Favié et al. [27]. The second aim of this study was to describe the effects of allopurinol and oxypurinol by investigating the response of the three most relevant biomarkers: hypoxanthine, xanthine, and uric acid.
2 Methods
2.1 Patient Population and Data Collection
The final analyzed data originated from three clinical studies: the historical cohorts of van Bel et al. [28] and Benders et al. [29], with a pooled population PK analysis as described by van Kesteren et al. [26], and the PK substudy of the ongoing double-blind, placebo-controlled ALBINO study [16]. All studies investigated the effect of allopurinol on survival and neurodevelopmental outcome in (near-)term neonates with perinatal asphyxia and early signs of evolving encephalopathy.
Establishing population PK/PD data of allopurinol and oxypurinol was a predefined secondary aim of the ALBINO study. This substudy was conducted in four specific centres (University Medical Centre Utrecht and Amsterdam UMC location VUMC in The Netherlands, and University Hospital Tuebingen and Cnopf Children’s Hospital Nuremberg in Germany) involved in the ALBINO study and was approved by all relevant national and institutional Medical Ethics Committees (NL57237.041.16; EudraCT 2016-000222-19). Further details of the study protocol are published and the trial is registered at www.clinicaltrials.gov (NCT03162653) [16].
In brief, in the ALBINO study, all neonates in the verum group received an initial dose of allopurinol 20 mg/kg or placebo intravenously over a 10-min period within 45 min after birth. In neonates treated with TH, the second dose of allopurinol 10 mg/kg or placebo was administered 12 h after the initial dose. Blood samples were obtained to determine plasma concentrations of allopurinol, oxypurinol, hypoxanthine, xanthine and uric acid. Target drug exposure defined in the study protocol was area under the concentration curve (AUC) from time 0 to 12 h (AUC12) of 43.5 mg/L × h for allopurinol and 26.5 mg/L × h for oxypurinol [16]. These targets were based on the extrapolation of plasma levels reached in the previously reported observations in the prehypothermia era (i.e. van Bel et al. [28] and Benders et al. [29]) and animal observational studies [30, 31]. Both AUC targets should be reached in more than two-thirds (66%) of the analyzed patients, as a priori defined in the ALBINO study protocol, based on the Scientific Advice and Protocol Assistance communication with the European Medicines Agency [16].
In the historical cohorts of van Bel et al. [28] and Benders et al. [29], infants received two doses of allopurinol 20 mg/kg by slow intravenous infusion over 10–30 min. The first dose was administered within 4 h after birth, and the second dose was administered 12 h later. Blood samples were obtained to determine the plasma concentrations of allopurinol and oxypurinol.
An overview of the methods, sample intervals, and lower limit of quantification (LLOQ) of compounds for each clinical trial is summarized in Table 1. For other details on study design, sample collection, and bioanalysis, we refer to van Bel et al. [28], Benders et al. [29], van Kesteren et al. [26] and the ALBINO protocol [16].
2.2 Pharmacokinetic/Pharmacodynamic (PK/PD) Data Analysis
Population PK and PD analyses were performed using nonlinear mixed-effects modelling (NONMEM, version 7.3; Icon Development Solutions; Perl-speaks-NONMEM [PsN], version 4.9.0) with R (version 3.6.3) and Xpose (version 4) for data visualization, and with Piraña for run management [32]. Individual empirical Bayesian estimates for all parameters were obtained using the POSTHOC option of NONMEM. All models were fitted using the first-order conditional estimation with interaction (FOCE-I) method of NONMEM.
The development of the PK/PD model consisted of three steps. In the first step, a parent-metabolite model describing allopurinol and oxypurinol PK was established. The starting point for allopurinol PK model development was a one-compartment model based on the work of van Kesteren et al. [26]. Subsequent one- and two-compartment models were tested for describing oxypurinol data. The metabolism of allopurinol to oxypurinol and the elimination of oxypurinol were modelled as first-order processes. The unknown fraction of allopurinol converted to oxypurinol was denoted by a formation fraction (fm), which was further fixed to 1, assuming a complete conversion for model development. Therefore, oxypurinol CL and volume of distribution (Vd) were estimated relative to the fm (i.e. CL/fm and Vd/fm).
In the second step, physiology (i.e. body size and maturation), disease (i.e. organ recovery) and treatment-related (i.e. TH) covariates, as identified by Favié et al. [27], were evaluated for allopurinol and oxypurinol PK. First, birth weight (BW), which is a descriptor of body size, was related to all parameters using allometric relationships [33]. The exponent defining the relationship of BW and CL was fixed to 0.75, and 1 for Vd. Gestational age (GA) was incorporated for describing maturation using a fixed sigmoidal Hill equation, in which the TM50 (the maturation half-time) was 54.2 weeks and the Hill coefficient was 3.92 [27, 34]. In addition to body size and maturation, the impact of organ recovery was validated by fixing the relative effect of postnatal age (PNA) on allopurinol and oxypurinol CL to the estimates of Favié et al. [27], which corresponds to an increase of 1.23% and 0.54% per hour of life for high and intermediate CL compounds, respectively. Based on the hypothesis of Favié et al. [27], the effect of PNA was expected to be negligible in allopurinol (a fully enzyme-cleared compound), and significant in oxypurinol (a renally cleared compound). To assess the impact of hypothermia, an infant treated with or without TH was incorporated as a categorical covariate on allopurinol and oxypurinol CL, respectively. Furthermore, considering the differences in behavioral and psychological responses and outcome, sex was tested as a covariate on drug CLs [35].
In the third step, a PK/PD model including interrelated biomarkers (hypoxanthine, xanthine, and uric acid) was developed. The starting point for development was a turnover model of hypoxanthine, which converts to xanthine and subsequently to uric acid, where the conversion of hypoxanthine to xanthine and xanthine to uric acid were inhibited by allopurinol and oxypurinol [17, 18]. The basis of this turnover model is the involvement of allopurinol and oxypurinol in XO inhibition, and the effect of XO inhibition (EFF) was tested by linear and saturable effect models. Under this framework, the biomarker time profiles of hypoxanthine, xanthine, and uric acid were described by the differential Eqs. 1–3, respectively:
in which A(n) describes the concentration in the nth compartment, kin3 represents the zero-order rate constant of the hypoxanthine production, k34 and k45 represent the first-order rate constant of the conversion of hypoxanthine to xanthine and xanthine to uric acid, and kout5 describes the first-order rate constant of uric acid elimination. This model assumed that kin3, k34, k45 and kout5 fully accounted for the production, conversion, and elimination of all biomarkers, therefore the baseline values for hypoxanthine, xanthine and uric acid were calculated by Eqs. 4–6, respectively:
Interindividual variability (IIV) in different PK and PD parameters was estimated using an exponential variance model, as shown in Eq. 7:
in which Pi is the individual parameter estimate for individual i, Ppop is the typical population parameter estimate, and \(\eta_{i}\) was assumed to be normally distributed with a mean of 0 and variance of ω2.
Residual unexplained variability was estimated for all compounds and was evaluated using a proportional error model, additive error model, and combined proportional and additive error model as described in Eqs. 8–10:
where \(C_{{{\text{obs}}.ij}}\) represents the observed concentration for individual i and observation j, \(C_{{{\text{pred}},ij}}\) represents the individual predicted concentration, \(\varepsilon_{p,ij}\) is the proportional error and \(\varepsilon_{{\alpha ,{\text{ij}}}}\) is the additive error, with both errors normally distributed with a mean of 0 and variance of σ2.
In the final PK/PD model, allopurinol, oxypurinol, hypoxanthine, xanthine and uric acid data were fitted simultaneously using NONMEM subroutine ADVAN6. Discrimination between different models was guided by the changes in objective function value (OFV), which is equal to minus two the log-likelihood. A decrease in OFV of ≥ 6.63, representing a p-value of < 0.001, was considered statistically significant for hierarchical models (Chi-square distribution with 1 degree of freedom [df]). Model adequacy was evaluated using goodness-of-fit plots, normalized prediction distribution errors (NPDEs) and physiological plausibility [36]. The stability of the model was evaluated by preconditioning (precond) and stochastic simulation and re-estimation (sse) tools in PsN [37, 38]. Parameter precision and 95% confidence interval (CI) was assessed with sampling importance resampling (SIR) [39]. Once the final PK/PD model was established, the individual allopurinol and oxypurinol AUC12 values were estimated for evaluating target achievements in the ALBINO study.
3 Results
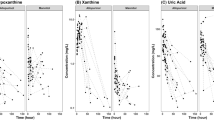
In total, 46 patients with 192 allopurinol observations, 164 oxypurinol observations, 97 hypoxanthine observations, 91 xanthine observations and 97 uric acid observations were analyzed. Patient characteristics are summarized in Table 2. All patients received a first dose of allopurinol 20 mg/kg, 52.2% (n = 24) of patients received a second dose of allopurinol 20 mg/kg, and 28.3% (n = 13) of patients received a second dose of allopurinol 10 mg/kg. In total, 28.3% (n = 13) of patients were treated with TH, all from the ALBINO study. The median number of samples per patient was 4 (range 2–6). In total, 14 allopurinol observations and 10 oxypurinol observations from the ALBINO study were below the LLOQ (7.3% and 6.1% of all available data, respectively). The first measurement below LLOQ in individuals was included and fixed to LLOQ/2. An additive residual error component of LLOQ/2, which was 0.05 mg/L for allopurinol and 0.0467 mg/L for oxypurinol, was further fixed [40]. The concentration-time curves of allopurinol, oxypurinol, hypoxanthine, xanthine, and uric acid in the study population are shown in Fig. 2. The final PK/PD model used is schematically depicted in Fig. 3, and the parameter estimates are provided in Table 3.

Concentration-time curves of a allopurinol, b oxypurinol, c hypoxanthine, d xanthine, and e uric acid

Schematic diagram of the PK/PD model. CL1 allopurinol clearance, V1 allopurinol volume of distribution, CL2 oxypurinol clearance, V2 oxypurinol volume of distribution, fm formation fraction, Kin3 hypoxanthine production rate, K34 hypoxanthine to xanthine metabolism rate, K45 xanthine to uric acid metabolism rate, Kout5 uric acid elimination rate, EBASE3 hypoxanthine baseline value, EBASE4 xanthine baseline value, EBASE5 uric acid baseline value, Init3D disease-related hypoxanthine initial value, Init4D disease-related xanthine initial value, Init5D disease-related uric acid initial value, EFF xanthine oxidase inhibition by allopurinol and oxypurinol, IV intravenous infusion, XO xanthine oxidase, PK pharmacokinetic, PD pharmacodynamic
3.1 PK Modelling
Initially, allopurinol and oxypurinol PK were described by two sequential one-compartment models. A one-compartment model adequately described allopurinol data; however, for oxypurinol, misspecification was observed, suggestive for distribution kinetics. Therefore, a two-compartment model was investigated, which improved the fit but led to highly implausible parameter estimates (i.e., a distribution half-life of only 0.07 h). Since excessive XO is produced from xanthine dehydrogenase during HI as well as the involvement of XO in allopurinol metabolism, autoinhibition of XO by allopurinol and/or oxypurinol was considered and the oxypurinol model was simplified to a one-compartment model [7, 8, 19]. The autoinhibition of XO was best described by a maximum effect (Emax) function driven by oxypurinol alone. This autoinhibition model showed superior goodness-of-fit compared with the previous two-compartment model and resulted in plausible parameter estimates
No systematic deviation was observed in the population conditional weighted residuals against BW and GA plots after the incorporation of BW and GA as covariates on CL and Vd for allopurinol and oxypurinol (electronic supplementary material [ESM] S1); however, including BW and GA as a covariate in the model did not result in OFV improvement. Considering the limitation of the collected dataset, which was only informative in neonates within 24 h of birth, the effect of body size and maturation was therefore excluded. For allopurinol, mainly enzymatically metabolized, the inclusion of PNA as covariates on CL did not improve the goodness-of-fit. For oxypurinol, mainly renally cleared, fixing the relative effect of PNA on CL to an increase of either 1.23%/h or 0.54%/h resulted in insignificant changes in OFV. No significant difference in allopurinol or oxypurinol CL was found between TH and non-TH patients as well as between males and females. Therefore, in the final PK model, no covariate was included.
IIV was estimated for the CL and Vd of allopurinol and oxypurinol. Residual variability in allopurinol was described using a combined proportional and additive error model, and in oxypurinol, residual variability was described using an additive error model. Higher residual errors were presented in the historical dataset, that is 1.51 times higher for allopurinol data and 4.3 times higher for oxypurinol data. In the ALBINO dataset, a dosing error in one patient occurred, as only a (unknown) fraction of the first dose was administered. To account for this, the fraction of the dose administered was modelled.
3.2 PK/PD Modelling
In the initial PK/PD model, hypoxanthine levels in the early period were considerably underpredicted. As fetal hypoxia results in the overproduction of hypoxanthine and XO, an additional disease-related initial level of hypoxanthine (Init3D) was included, which significantly improved the hypoxanthine model [7, 8]; however, the model still underestimated uric acid observations. Considering the catalyzation of hypoxanthine to xanthine, and, finally, to uric acid, was highly activated before the treatment, the high disease-related initial levels were further included for xanthine (Init4D) and uric acid (Init5D). In other words, the compartment initialization [A0(n)] of either biomarker was a combination of the baseline turnover estimate (EBASEn), defined by Eqs. 4–6, with a disease-related estimate (Initn) as described in Eq. 11.
Due to the sparse xanthine observations after 48 h of birth, the baseline xanthine value (EBASE4) was fixed to 0.1 mg/L, based on Fig. 2d, to improve model stability. Eventually, this biomarker turnover model with additional disease-related initial levels for hypoxanthine, xanthine and uric acid provided adequate description for the data.
After the structural PK/PD model was established, IIV in PD parameters was tested with a forward selection procedure. In the final PK/PD model, IIV in k34, k45, kout5, Init3D and Init5D were included, as any of them were identifiable, and improved the goodness-of-fit with a p-value < 0.001. A correlation between IIV in CL2, kout5, and k45 was found by visual examination and this correlation was described using a ω block structure. The result of the correlation is described in Table 3e. Residual variability in hypoxanthine, xanthine and uric acid was described using proportional error models.
The inhibition of allopurinol-oxypurinol conversion and hypoxanthine-xanthine/uric acid conversion could not be described by a single Emax equation. Therefore, two independent Emax inhibition models were used in the final PK/PD model. Model evaluation demonstrated that the final PK/PD model was stable and adequate in describing data and no identifiability problems could be detected. Goodness-of-fit plots, including NPDE plots, are provided in ESM S2–S3.
In the final PK/PD model, the combined allopurinol and oxypurinol concentration at the half maximal XO inhibition (IC50) was 0.36 mg/L (95% CI 0.31–0.42); 98% of the observed concentrations were above this dose. The mean ± standard deviation AUC12 value of all analyzed patients was 160.26 ± 62.57 mg/L × h for allopurinol and 37.40 ± 12.75 mg/L × h for oxypurinol. For subjects from the ALBINO study, 95% and 75% reached the predefined AUC12 target for allopurinol and oxypurinol, respectively. In the non-TH group, all patients reached the targets, while in the TH group, the allopurinol and oxypurinol exposure targets were reached by 92% and 61% of patients, correspondingly.
4 Discussion
A population PK/PD model for allopurinol, oxypurinol, hypoxanthine, xanthine and uric acid was developed for neonates with HIE that allowed evaluation of current allopurinol dosage used in clinical trials and quantification of drug effect on XO inhibition. We hereby confirmed the predefined target exposure in the ALBINO trial. Furthermore, the disease-related autoinhibition effect of allopurinol metabolism, as well as higher hypoxanthine, xanthine and uric acid initial levels were identified. The effects of body size, maturation and organ recovery on drug CL could not be identified. Between infants treated with and without TH, no significant differences in allopurinol and oxypurinol CL were found.
For the ALBINO study, the drug exposure target for allopurinol and oxypurinol were defined based on the lowest individual values observed during previous studies of allopurinol in neonates [28, 29]. The predefined target for allopurinol and oxypurinol were reached in 95% and 75% of analyzed patients, respectively. More importantly, the IC50 estimate of 0.36 mg/L for the combined allopurinol and oxypurinol concentration was below all available observed concentrations in the first 72 h. The relatively low IC50 value indicated that over 90% of XO was inhibited by allopurinol and oxypurinol during the first 24 h after administration. This would indicate that the dosing regimen as applied in the ALBINO trial leads to sufficient XO inhibition and would not require adjustment for both infants treated with and without TH. However, since the half-life of oxypurinol is 25–30 h, AUC12 only describes part of its PK profile, which might account for the lower target attainment. Therefore, additional justification for this predefined target exposure for oxypurinol might be required.
This study identified that the metabolic CL of allopurinol to oxypurinol was autoinhibited by oxypurinol. As allopurinol is widely recognized as a substrate of XO, the self-inhibitory effect of oxypurinol on allopurinol metabolism has been proposed [19, 41, 42]. Nevertheless, some recent studies suggested AOR is the major enzyme responsible for the conversion of allopurinol to oxypurinol, as they found the metabolism of allopurinol in adults is rapid and non-saturable [17]. In neonates with HIE, it could be reasonably hypothesized that XO takes a more crucial role in allopurinol metabolism, since excessive amounts of highly activated XO are formed during fetal hypoxia [7]. Therefore, the autoinhibition effect conceivably occurred specifically in this population.
Due to the impact of autoinhibition, the estimated CL of allopurinol in this study decreased by 70% in the first 24 h after birth, and slowly recovered until the end of the study period (PNA of 7 days). The dynamic allopurinol metabolism in asphyxiated neonates provides a possible elucidation for the marked difference in allopurinol half-life between the McGaurn et al. [25] and van Kesteren et al. [26] studies, which allopurinol half-life was approximately 2.5 h for neonates with a PNA of 3–21 days, and 7 h for neonates within 0.5–24 h after birth [25, 26]. Although the elimination of excessive XO over time could also result in variable allopurinol CL, as allopurinol is a substrate of XO, the likelihood of autoinhibition in this population was undoubtable.
Although the autoinhibition model suggested XO is primarily responsible for allopurinol metabolism in asphyxia neonates, the role of AOR in allopurinol conversion remains essential. Using a single inhibition model to describe allopurinol metabolism and hypoxanthine-xanthine/uric acid conversion simultaneously was found unfeasible in our study, which indicated that enzymes besides XO, most likely AOR, also account for allopurinol metabolism [19]. Although AOR enzyme activity soon after birth was not able to be identified, a previous study found minimal AOR activity at 4 months after birth, reaching a plateau around 2 years of age [43].
Additional initial high hypoxanthine, xanthine and uric acid levels were estimated in our model to capture the fetal hypoxia-related effects in this population. Unlike previous studies in adults, where the hypoxanthine levels increased after allopurinol administration, a decrease of hypoxanthine was found in our population [21, 44]. In theory, under the condition of XO inhibition, accumulation of hypoxanthine levels could occur. However, for HIE neonates whose initial hypoxanthine baseline was rather high, the effect of hypoxanthine elimination might supersede its accumulation, which explained the declining hypoxanthine level. In addition, compared with hypoxia neonates with a PNA of 1.5–21 days, observed initial hypoxanthine levels in our population were 3.5- to 10-fold higher, which suggested a large impact of fetal hypoxia after birth [25, 44]. Consequently, the quantified extra initial hypoxanthine, xanthine and uric acid levels were likely due to the fetal hypoxia. However, verifying these assumptions in the present study is challenging due to the absence of such data in asphyxia cases not exposed to allopurinol (‘natural course’).
Serum uric acid has been regarded as the most significant biomarker for the drug response of allopurinol and oxypurinol, as it directly reflects the effect of XO inhibition [17]. The present model provided satisfactory description of the uric acid observations. For the descriptions of hypoxanthine and xanthine, misspecification in the early time period remained. It is known that the drug effects on hypoxanthine and xanthine metabolism could lead to greater salvage of adenosine, as well as the feedback inhibition of de novo purine synthesis [13, 14]. However, these complicated mechanisms could not be captured in the established model, therefore this would be a limitation when applying the present model for predictions outside the study observation window.
Incorporating BW, GA, and PNA as covariates in the model failed to provide OFV improvement. According to the hypothesis of Favie et al., body size and maturation were related to the CL of all tested compounds, while recovery of organ function from asphyxia was suggested to affect compounds whose CL is closely linked to organ perfusion. Therefore, PNA was identified as a covariate on fully renally cleared compounds and hepatically cleared compounds with high and intermediate CL, but not a covariate for low-CL compounds such as phenobarbital [27]. The limitation of our small and constrained dataset, with most data collected within 24 h after birth, may hamper the characterization of the possible effects of BW, GA and PNA. Therefore, the present dataset and analysis was unable to provide adequate evaluation of the framework proposed by Favie et al. [27]. However, the negligible influence of PNA on allopurinol was in coherence with the hypothesis of Favie et al., showing that the CL of allopurinol, whose metabolism is mainly driven by enzymes such as XO and AOR, might be independent of renal and hepatic perfusion.
Despite the limitations of the short sampling period, the insignificant increase in oxypurinol CL with PNA might underline the importance of oxypurinol reabsorption by uric acid transporters [45]. It has been suggested that approximately 80% of glomerular filtered oxypurinol is actively reabsorbed, in which the reabsorptive transporter, urate transporter 1 (URAT1), plays the most important role [46, 47]. Our model supported that oxypurinol shares the same renal transport mechanism system with uric acid, since the elimination rate constants of oxypurinol (0.024 h−1) and uric acid (0.035 h−1) were comparable [22, 48]. Moreover, a strong correlation between IIV in oxypurinol CL and uric acid elimination rate constant was found. Previously, a very high fractional excretion of uric acid at birth, followed by a rapid decline, was reported, which was further related to the increasing expression of URAT1 from birth until childhood [49, 50]. As the excretion of oxypurinol might be comparable with that of uric acid, the recovery of renal perfusion might have a relatively small influence on the CL of oxypurinol, highlighting the possible substantial impact of the ontogeny of renal transporters.
In this study, the CLs of allopurinol and oxypurinol were not significantly different between TH and non-TH patients, while patients who failed to achieve the target oxypurinol AUC were all from the TH group. TH has been found to affect the CL of different drugs in previous studies since the dynamic body temperature can alter cardiac output and activity of enzymes and transporters [27]. Nevertheless, as only limited observations in our study covered the entire hypothermia process, the impact of alterations in body temperature on allopurinol and oxypurinol CL were impossible to evaluate in this study.
5 Conclusion
This study described the PK and PD of allopurinol, oxypurinol, hypoxanthine, xanthine and uric acid in HIE neonates. The PK of oxypurinol and the effect of allopurinol and oxypurinol on inhibiting XO were quantified for the first time in this population. Furthermore, the disease-related autoinhibition effect of allopurinol metabolism and increased initial hypoxanthine, xanthine and uric acid levels were identified. These findings may form the basis for further treatment optimization and dose individualization in HIE neonates.
References
Lee ACC, Kozuki N, Blencowe H, Vos T, Bahalim A, Darmstadt GL, et al. Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr Res. 2013;74:50–72.
Azzopardi DV, Strohm B, Edwards AD, Dyet L, Halliday HL, Juszczak E, et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med. 2009;361:1349–58.
Jacobs SE, Berg M, Hunt R, Tarnow-Mordi WO, Inder TE, Davis PG. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev. 2013;1:CD003311.
Azzopardi D, Strohm B, Marlow N, Brocklehurst P, Deierl A, Eddama O, et al. Effects of hypothermia for perinatal asphyxia on childhood outcomes. N Engl J Med. 2014;371:140–9.
Webster D. Neonatal brain injury. Neonatal Intensive Care Nurs. 2019;351:351–74.
Allen KA, Brandon DH. Hypoxic ischemic encephalopathy: pathophysiology and experimental treatments. Newborn Infant Nurs Rev. 2011;11:125–33.
Epstein FH, McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312:159–63.
van Bel FV, Groenendaal F. Drugs for neuroprotection after birth asphyxia: pharmacologic adjuncts to hypothermia. Semin Perinatol. 2016;40:152–9.
Palmer C, Vannucci RC, Towfighi J. Reduction of perinatal hypoxic-ischemic brain damage with allopurinol. Pediatr Res. 1990;27:332–6.
Warner DS, Sheng H, Batinić-Haberle I. Oxidants, antioxidants and the ischemic brain. J Exp Biol. 2004;207:3221–31.
Moorhouse PC, Grootveld M, Halliwell B, Quinlan JG, Gutteridge JMC. Allopurinol and oxypurinol are hydroxyl radical scavengers. FEBS Lett. 1987;213:23–8.
Shadid M, Buonocore G, Groenendaal F, Moison R, Ferrali M, Berger HM, et al. Effect of deferoxamine and allopurinol on non-protein-bound iron concentrations in plasma and cortical brain tissue of newborn lambs following hypoxia-ischemia. Neurosci Lett. 1998;248:5–8.
Boda D. Results of and further prevention of hypoxic fetal brain damage by inhibition of xanthine oxidase enzyme with allopurinol. J Perinat Med. 2011;39:441–4.
Marro PJ, Mishra OP, Delivoria-Papadopoulos M. Effect of allopurinol on brain adenosine levels during hypoxia in newborn piglets. Brain Res. 2006;1073–1074:444–50.
Annink KV, Franz AR, Derks JB, Rüdiger M, van Bel F, Benders MJNL. Allopurinol: old drug, new indication in neonates? Curr Pharm Des. 2018;23:5935–42.
Maiwald CA, Annink KV, Rüdiger M, Benders MJNL, Van Bel F, Allegaert K, et al. Effect of allopurinol in addition to hypothermia treatment in neonates for hypoxic-ischemic brain injury on neurocognitive outcome (ALBINO): Study protocol of a blinded randomized placebo-controlled parallel group multicenter trial for superiority (phase III). BMC Pediatr. 2019;19:210.
Day RO, Graham GG, Hicks M, McLachlan AJ, Stocker SL, Williams KM. Clinical pharmacokinetics and pharmacodynamics of allopurinol and oxypurinol. Clin Pharmacokinet. 2007;46:623–44.
Murrell GAC, Rapeport WG. Clinical pharmacokinetics of allopurinol. Clin Pharmacokinet. 1986;11:343–53.
Mirzaei S, Taherpour AA, Mohamadi S. Mechanistic study of allopurinol oxidation using aldehyde oxidase, xanthine oxidase and cytochrome P450 enzymes. RSC Adv. 2016;6:109672–80.
Wright DFB, Stamp LK, Merriman TR, Barclay ML, Duffull SB, Holford NHG. The population pharmacokinetics of allopurinol and oxypurinol in patients with gout. Eur J Clin Pharmacol. 2013;69:1411–21.
Turnheim K, Krivanek P, Oberbauer R. Pharmacokinetics and pharmacodynamics of allopurinol in elderly and young subjects. Br J Clin Pharmacol. 1999;48:501–9.
Stocker SL, Mclachlan AJ, Savic RM, Kirkpatrick CM, Graham GG, Williams KM, et al. The pharmacokinetics of oxypurinol in people with gout. Br J Clin Pharmacol. 2012;74:477–89.
Spector T. Oxypurinol as an inhibitor of xanthine oxidase-catalyzed production of superoxide radical. Biochem Pharmacol. 1988;37:349–52.
Sierra H, Cordova M, Chen CSJ, Rajadhyaksha M. Confocal imaging-guided laser ablation of basal cell carcinomas: an ex vivo study. J Invest Dermatol. 2015;135:612–5.
McGaurn SP, Davis LE, Krawczeniuk MM, Murphy JD, Jacobs ML, Norwood WI, et al. The pharmacokinetics of injectable allopurinol in newborns with the hypoplastic left heart syndrome. Pediatrics. 1994;94:820–3.
van Kesteren C, Benders MJNL, Groenendaal F, Van Bel F, Ververs FFT, Rademaker CMA. Population pharmacokinetics of allopurinol in full-term neonates with perinatal asphyxia. Ther Drug Monit. 2006;28:339–44.
Favié LMA, de Haan TR, Bijleveld YA, Rademaker CMA, Egberts TCG, Nuytemans DHGM, et al. Prediction of drug exposure in critically ill encephalopathic neonates treated with therapeutic hypothermia based on a pooled population pharmacokinetic analysis of seven drugs and five metabolites. Clin Pharmacol Ther. 2020;108:1098–106.
Van Bel F, Shadid M, Moison RMW, Dorrepaal CA, Fontijn J, Monteiro L, et al. Effect of allopurinol on postasphyxial free radical formation, cerebral hemodynamics, and electrical brain activity. Pediatrics. 1998;101:185–93.
Benders MJNL, Bos AF, Rademaker CMA, Rijken M, Torrance HL, Groenendaal F, et al. Early postnatal allopurinol does not improve short term outcome after severe birth asphyxia. Arch Dis Child Fetal Neonatal Ed. 2006;91:163–5.
Peeters C, Hoelen D, Groenendaal F, Van Bel F, Bär D. Deferoxamine, allopurinol and oxypurinol are not neuroprotective after oxygen/glucose deprivation in an organotypic hippocampal model, lacking functional endothelial cells. Brain Res. 2003;963:72–80.
Kim P, Yaksh TL, Romero SD, Sundt TM. Production of uric acid in cerebrospinal fluid after subarachnoid hemorrhage in dogs: Investigation of the possible role of xanthine oxidase in chronic vasospasm. Neurosurgery. 1987;21:39–44.
Keizer RJ, van Benten M, Beijnen JH, Schellens JHM, Huitema ADR. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101:72–9.
West GB, Brown JH, Enquist BJ. A general model for the origin of allometric scaling laws in biology. Science. 1997;276:122–6.
Rhodin MM, Anderson BJ, Peters AM, Coulthard MG, Wilkins B, Cole M, et al. Human renal function maturation: a quantitative description using weight and postmenstrual age. Pediatr Nephrol. 2009;24:67–76.
Rosenkrantz TS, Hussain Z, Fitch RH. Sex differences in brain injury and repair in newborn infants: clinical evidence and biological mechanisms. Front Pediatr. 2019;7:211.
Comets E, Brendel K, Mentré F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Programs Biomed. 2008;90:154–66.
Aoki Y, Nordgren R, Hooker AC. Preconditioning of nonlinear mixed effects models for stabilisation of variance-covariance matrix computations. AAPS J. 2016;18:505.
Ueckert S, Karlsson MO, Hooker AC. Accelerating Monte Carlo power studies through parametric power estimation. J Pharmacokinet Pharmacodyn J Pharmacokinet Pharmacodyn. 2016;43:223–34.
Dosne AG, Bergstrand M, Karlsson MO. An automated sampling importance resampling procedure for estimating parameter uncertainty. J Pharmacokinet Pharmacodyn. 2017;44:509–20.
Yang S, Roger J. Evaluations of Bayesian and maximum likelihood methods in PK models with below-quantification-limit data. Pharm Stat. 2010;9:313–30.
Hande K, Reed E, Chabner B. Allopurinol kinetics. Clin Pharmacol Ther. 1978;23:598–605.
Nelson DJ, Elion GB. Metabolic studies of high doses of allopurinol in humans. Adv Exp Med Biol. 1984;165 Pt A:167–70.
Beedham C. Aldehyde oxidase; new approaches to old problems. Xenobiotica. 2020;50:34–50.
Marro PJ, Baumgart S, Delivoria-Papadopoulos M, Zirin S, Corcoran L, McGaurn SP, et al. Purine metabolism and inhibition of xanthine oxidase in severely hypoxic neonates going onto extracorporeal membrane oxygenation. Pediatr Res. 1997;41:513–23.
Elion GB, Yü TF, Gutman AB, Hitchings GH. Renal clearance of oxipurinol, the chief metabolite of allopurinol. Am J Med. 1968;45:69–77.
Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, et al. Molecular identification of a renal urate-anion exchanger that regulates blood urate levels. Nature. 2002;417:447–52.
Iwanaga T, Kobayashi D, Hirayama M, Maeda T, Tamai I. Involvement of uric acid transporter in increased renal clearance of the xanthine oxidase inhibitor oxypurinol induced by a uricosuric agent, benzbromarone. Drug Metab Dispos. 2005;33:1791–5.
Breithaupt H, Tittel M. Kinetics of allopurinol after single intravenous and oral doses—noninteraction with benzbromarone and hydrochlorothiazide. Eur J Clin Pharmacol. 1982;22:77–84.
Cheung KWK, van Groen BD, Spaans E, van Borselen MD, de Bruijn ACJM, Simons-Oosterhuis Y, et al. A comprehensive analysis of ontogeny of renal drug transporters: mRNA analyses, quantitative proteomics, and localization. Clin Pharmacol Ther. 2019;106:1083–92.
Stiburkova B, Bleyer AJ. Changes in serum urate and urate excretion with age. Adv Chronic Kidney Dis. 2012;19(6):372–6.
Acknowledgments
The ALBINO coordinating investigators were: Axel R. Franz (University Hospital Tübingen, Germany); Mario Rüdiger (University Hospital CG Carus, Medizinische Fakultät der TU Dresden, Germany). The beneficiaries and national coordinators were Axel R. Franz and Christian F. Poets (Tübingen, Germany), Mario Rüdiger (Dresden, Germany), Manon Benders and Frank van Bel (Utrecht, The Netherlands), Karel Allegaert and Gunnar Naulaers (Leuven, Belgium), Dirk Bassler (Zurich, Switzerland), Katrin Klebermass-Schrehof (Vienna, Austria), Maximo Vento (Valencia, Spain), Hercilia Guimaraes (Porto, Portugal), Tom Stiris (Oslo, Norway), Isabella Mauro (Udine, Italy), Marjo Metsäranta (Helsinki, Finland), Sampsa Vanhatalo (Helsinki, Finland), Jan Mazela (Poznan, Poland), Tuuli Metsvaht (Tartu, Estonia), and Yannique Jacobs (ACE Pharmaceuticals, Zeewolde, The Netherlands).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Funding
The ALBINO project is funded under the Horizon 2020 Framework EU Program call H2020-PHC-2015-two-stage, grant 667224. The research on the PK during whole-body hypothermia (KA) is further supported by the iPREDICT project (FWO senior research project, fundamental research, G0D0520N).
Conflicts of interest/Competing interests
Wan-Yu Chu, Kim Annink, Laura Nijstad, Christian Maiwald, Michael Schroth, Loubna el Bakkali, Frank van Bel, Manon Benders, Mirjam van Weissenbruch, Anja Hagen, Axel Franz, Thomas Dorlo, Karel Allegaert, and Alwin Huitema have no conflicts of interest to declare.
Ethics approval
The data used in this study were performed in accordance with the Declaration of Helsinki and the guidelines of Good Clinical Practice (GCP). The ALBINO study was granted by Institutional Review Boards and Independent Ethics Committees. The Medical Ethics Committee of the University Medical Centre Utrecht and the Central Committee on Human Research approved the study, including the specific PK substudy for The Netherlands (NL57237.041.16). The German Federal Authority (EudraCT 2016-000222-19), as well as the leading Ethics Committee Tübingen, approved the study for Germany.
Consent to participate
Short oral consent was obtained from at least one parent before administering the study medication. After the first dose, but before the (potential) second dose, full written parental consent was obtained. In case parents did not sign the informed consent, blood samples already collected shortly after birth were destroyed.
Availability of data and material
The raw data are available upon reasonable request by an e-mail to the corresponding author, while blinding for group allocation should be respected until the primary outcome analysis has been finalized.
Code availability
Not applicable.
Author contributions
Protocol development and study conduct: KVA, MS, CM, LEB, FVB, MB, MVW, AH, AF, KA. Ethics and study registration: KVA, FVB, MB, AF. Study recruitment and data collection: KVA, MS, CM, LEB, FVB, MB, MVW, AH, AF. Modelling: WYC, LN, TD, ADRH. Drafting of the manuscript: WYC, KVA, LN, TD, KA, ADRH. All authors have read and commented on the manuscript and have approved the submitted version.
Additional information
The members of the ALBINO Study Group are mentioned in the Acknowledgments section.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Chu, WY., Annink, K.V., Nijstad, A.L. et al. Pharmacokinetic/Pharmacodynamic Modelling of Allopurinol, its Active Metabolite Oxypurinol, and Biomarkers Hypoxanthine, Xanthine and Uric Acid in Hypoxic-Ischemic Encephalopathy Neonates. Clin Pharmacokinet 61, 321–333 (2022). https://doi.org/10.1007/s40262-021-01068-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01068-0