
Abstract
Objectives
The aim of this study was to simulate the drug–drug interaction (DDI) between ritonavir-boosted atazanavir (ATV/r) and rifampicin (RIF) using physiologically based pharmacokinetic (PBPK) modelling, and to predict suitable dose adjustments for ATV/r for the treatment of people living with HIV (PLWH) co-infected with tuberculosis.
Methods
A whole-body DDI PBPK model was designed using Simbiology 9.6.0 (MATLAB R2019a) and verified against reported clinical data for all drugs administered alone and concomitantly. The model contained the induction mechanisms of RIF and ritonavir (RTV), the inhibition effect of RTV for the enzymes involved in the DDI, and the induction and inhibition mechanisms of RIF and RTV on the uptake and efflux hepatic transporters. The model was considered verified if the observed versus predicted pharmacokinetic values were within twofold. Alternative ATV/r dosing regimens were simulated to achieve the trough concentration (Ctrough) clinical cut-off of 150 ng/mL.
Results
The PBPK model was successfully verified according to the criteria. Simulation of different dose adjustments predicted that a change in regimen to twice-daily ATV/r (300/100 or 300/200 mg) may alleviate the induction effect of RIF on ATV Ctrough, with > 95% of individuals predicted to achieve Ctrough above the clinical cut-off.
Conclusions
The developed PBPK model characterized the induction-mediated DDI between RIF and ATV/r, accurately predicting the reduction of ATV plasma concentrations in line with observed clinical data. A change in the ATV/r dosing regimen from once-daily to twice-daily was predicted to mitigate the effect of the DDI on the Ctrough of ATV, maintaining plasma concentration levels above the therapeutic threshold for most patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The physiologically based pharmacokinetic (PBPK) framework proposed in this work was capable of simulating the interplay between the inhibition and induction of cytochrome P450 (CYP) 3A4 in the gut and liver as well as hepatic active transporters (uptake and efflux) for the prediction of the DDI between ritonavir-boosted atazanavir (ATV/r) and rifampicin. |
A twice-daily dose of ATV/r seems to provide the most clinically sustainable opportunity to overcome the DDI, rather than opting for a higher once-daily dose, considering the associated hepatotoxicity risk. |
1 Introduction
According to the World Health Organization (WHO), the risk of people living with HIV (PLWH) developing tuberculosis is 16–27 times greater than that of HIV-negative individuals [1]. In 2018, more than 37.9 million people were living with HIV, with one-third of AIDS-related deaths globally (251,000 of the overall 770,000 global HIV deaths in 2018) being linked to tuberculosis [1, 2]. The WHO recommends that all PLWH co-infected with tuberculosis should receive antiretroviral therapy (ART), regardless of their CD4 count, within 8 weeks of initiating antituberculosis treatment [3]. However, antiretroviral (ARV) drugs have a high potential for drug–drug interactions (DDIs), both as victims and perpetrators, and their coadministration with numerous drugs is contraindicated [4, 5]. Although global ART roll-out has been highly successful, with 23.3 million individuals estimated to be receiving ART [6], increasing numbers are failing first-line regimens and require second-line therapy [7]. WHO-recommended second-line regimens when failing first-line regimens with dolutegravir (DTG) are based on protease inhibitors (PI), with the ‘preferred’ choice being ritonavir-boosted atazanavir (ATV) because of its once-daily dosing regimen and improved clinical tolerability compared with older PIs such as lopinavir [7]. With increasing uptake of DTG, individuals failing non-nucleoside reverse transcriptase inhibitor (NNRTI)-based first-line therapy are recommended to switch to DTG-based second-line regimens [8]. However, as DTG is increasingly used as first-line throughout the world, future treatment failures that emerge will still require boosted PI-based second-line regimens.
PIs are known to be substrates of cytochrome P450 (CYP) 3A enzymes, efflux, and uptake transporters. Atazanavir is a P-glycoprotein (P-gp), organic anion transporting polypeptide (OATP), and CYP3A4 substrate extensively metabolized by the liver [9]. Although ATV can be used alone in the treatment of naive patients with HIV, the coadministration with a low dose of ritonavir (RTV) is recommended given the potent inhibition by RTV of CYP3A4 [10]. RTV ‘boosting’ of ATV (ATV/r) improves the pharmacokinetic and virologic activity of ATV [11]. The relationship between the ATV trough concentration (Ctrough) and effective ART response has been previously described, and a Ctrough of 150 ng/mL correlated with a plasma HIV RNA load < 50 copies/mL following 24 weeks of treatment with ATV 300 mg plus RTV 100 mg [10, 12].
Ritonavir is metabolized by CYP3A4 and CYP2D6 and is both a P-gp substrate and inhibitor [13,14,15,16]. It is also a weak inducer of CYP3A4 [17] and an inhibitor of OATPB1. The inhibition of this class of transporters can decrease the hepatic uptake and metabolism of many drugs [18]. Rifampicin (RIF), which is the mainstay of first-line tuberculosis treatment, is a strong inducer of many CYPs (CYP2A, CYP2B, CYP2C, and CYP3A) and transporters [19, 20] and an inhibitor of OATP1B1 [21]. The coadministration of ATV and RIF results in a substantial decrease in ATV plasma concentration [22, 23].
The investigation of clinically relevant DDIs can be limited by numerous ethical and logistical barriers. In vitro methodologies can provide a quantitative description of the induction and inhibition effect of drugs on metabolic enzymes and transporters; however, these approaches may have limitations depending on their experimental design. For example, the experiments may not reflect the concentrations used therapeutically, or they may not reproduce concurrent inhibition and inducing effects. Furthermore, the cell system used may not represent physiologically relevant enzyme or transporter expression and activity, hindering in vivo extrapolation. In silico techniques can be utilized to overcome some of the limitations mentioned above, specifically the prediction of multiple concurrent DDI mechanisms [24].
Physiologically based pharmacokinetic (PBPK) modelling is a ‘bottom-up’ approach in which the in vitro data of a drug (intrinsic clearance, blood-to-plasma ratio, protein binding, Caco-2 cell permeability, etc.), physicochemical properties (logP, pKa, polar surface area, hydrogen bond donors [HBDs]), anatomical, physiological, and demographic characteristics of a population are integrated into mathematical equations to describe absorption, distribution, metabolism, and excretion (ADME) [25]. PBPK modelling can be applied to simulate different clinical scenarios concerning DDIs and to identify potential dose adjustments [26].
The aim of this study was to develop and verify a PBPK framework to simulate the interplay between the inhibition and induction of CYP3A4 in the gut and liver as well as hepatic active transporters (uptake and efflux) for the prediction of the DDI between ATV/r and RIF. Alternative ATV/r dosing strategies were simulated to overcome the magnitude of RIF DDIs, suggesting potential ATV/r regimens achieving the clinical Ctrough cut-off of ATV required for the successful treatment of PLWH co-infected with tuberculosis.
2 Methods
2.1 Physiologically Based Pharmacokinetic (PBPK) Model
The model was designed using Simbiology® v.5.8.2, a product of MATLAB® v.R2019a (MathWorks, Natick, MA, USA; 2019). Some of the key assumptions considered in the simulations were first-order kinetics, blood flow-limited distribution, no drug reabsorption from the colon, and instant distribution of the drug in tissues and organs (well-stirred model), except in the liver where the distribution of ATV was refined by a mechanistic transport-based model that included active efflux (P-gp) and uptake mediated transport of the drug. For ATV and RTV, a full PBPK model was applied, while RIF was applied in a three-compartment PBPK model (absorption, distribution, and elimination).
2.2 Virtual Healthy Volunteers
A virtual cohort of 100 healthy individuals (50% female and 50% male) was created using a whole-body PBPK model. Virtual healthy volunteers aged between 18 and 60 years (39 ± 12 years), with a mean weight of 70 ± 17.6 kg and height of 1.75 ± 0.15 m were generated using equations that statistically described the physiological and anatomical parameters to represent the individuals from the observed clinical studies [22, 23] used to validate the model. Organ weights and volumes were calculated using anthropometric equations and organ density, previously described by Bosgra et al. [27] and Brown et al. [28], respectively. The regional blood flow and systemic circulation were based on the cardiac output [29]. Each simulation used random values based on the mean and standard deviation, ensuring variability among simulated individuals.
2.3 Oral Absorption
Oral absorption considered a seven-compartment transit model [30]. When an observed value was not available from the literature, the absorption constant rate (ka) was calculated using the effective permeability based on the Caco-2 cells in vitro permeability or using the polar surface area (PSA) and the number of HBDs [31]. PIs are known to be poorly water-soluble [32]; for this reason, the solubility of drugs in each intestinal compartment was considered to calculate the fraction absorbed in the model. Considering that RIF presents variability in both the rate and extent of absorption when administered orally, a lag time (tlag) was implemented in the model [33]. All parameters are presented in Table 1.
2.4 Intestinal Metabolism
The abundance of CYP3A4 in the intestine [34] and the in vitro intrinsic clearance (Clint,CYP3A4,gut) were used to calculate intestinal clearance (Clgut) [31]. The amount of the drug escaping intestinal metabolism (Fg) and arriving in the liver was determined by Eq. 1:
where Qgut is the flow of blood to the gut (L/h) and fu,gut is the fraction unbound of the drug in the gut, considered to have a value of 1 in the model [35].
The total Clgut (L/h) incorporated the DDI effect on the intestinal CYP3A4, as described in Eqs. 2–4:
where Clint,CYP3A4,gut, Emax, EC50, Kdeg, Kinact, Ki, and Igut represent the intrinsic clearance of each drug for the isoenzyme 3A4 (µL/min/pmol), maximum induction, the concentration of inducer producing 50% of Emax (µmol/L), the first-order degradation rate constant of CYP3A4, the maximal enzyme inactivation rate constant, the concentration of inhibitor producing 50% of maximum inhibition (µmol/L), and the unbound intestinal concentration of inducer or inhibitor (mg/L), respectively. The degradation rate constant of CYP3A4 was 0.02 h-1 (half-life time of 35 h) [36], and the total induction of CYP3A4 in the gut considered the sum of RTV and RIF induction. RTV was considered an irreversible inhibitor of CYP3A4.
2.5 Liver Metabolism
The hepatic clearance (Clliver,CYP) considered the intrinsic clearance of a given CYP in µL/min/pmol (Clint,CYP,liver), the abundance of the CYP in the liver per milligram of microsomal protein per gram of liver (MPPGL), the liver weight in grams (WLiver), and the DDI effect on the given CYP. The MPPGL was calculated as described previously by Barter et al. [37]. The induction and inhibition of liver enzymes were integrated into the Clliver,CYP (L/h) as described in Eq. 5:
The total hepatic clearance (Clhe) was considered as the sum of all enzymes involved in the metabolism. Additionally, Eqs. 6–8 were used when uptake and efflux transport was involved in ATV hepatic clearance:
where Clint,eff, Clint,up,act, Clint,up,pas, Hepatocytes, Indeff, Inhieff, Induptake and Inhiuptake represent the intrinsic efflux clearance of each drug for the given efflux transporter (µL/min/million hepatocytes), the intrinsic clearance of each drug for the given active uptake transporter (µL/min/million hepatocytes), the intrinsic clearance of each drug for the given passive uptake (µL/min/million hepatocytes), number of hepatocytes per gram of liver, induction and inhibition effect on the efflux transporter, and induction and inhibition effect on the active uptake transporter, respectively. The induction (\({\mathrm{Ind}}_{\mathrm{hep}})\) and inhibition (\({\text{Inhi}}_{\mathrm{hep}})\) of CYP3A4 hepatic enzymes were calculated the same way as described in Eqs. 3 and 4. As in the gut, the total induction of CYP3A4 in the liver was the sum of RTV and RIF induction. RTV was considered an irreversible inhibitor of CYP3A4.
Induction and inhibition of hepatic uptake and efflux transporters by RTV and RIF were calculated as described in Eqs. 9 and 10:
where Emax, EC50, Ki, and Iliver represent the maximum induction, the concentration of inducer producing 50% of Emax (µmol/L), the concentration of inhibitor producing 50% of maximum inhibition (µmol/L), and the unbound liver concentration of inducers or inhibitors (mg/L). RTV and RIF were considered reversible inhibitors of hepatic transporters.
Finally, the amount of the drug escaping the hepatic metabolism (Fh) and reaching the systemic circulation was determined by Eq. 11 for RTV.
where Qhe is the flow of blood to the liver (L/h), fup is the fraction unbound of the drug in the plasma, and R is the blood-to-plasma ratio.
2.6 Distribution
The volume of distribution (Vd) was calculated using the tissue-to-plasma ratio (TP) of each organ and the volume of each compartment [38], except for the liver, where a passive and active uptake and efflux transport was considered. A correction factor was applied to each organ TP in the ATV and RTV models to match observed Vd values of 109–187 L and 20–40 L, respectively [17, 39].
2.7 Drug Parameters
The PBPK model was developed using in vitro, in silico and in vivo data for ATV, RTV, and RIF. No renal clearance was considered in the simulation since all the drugs are metabolized almost entirely by the liver. The initial values of intrinsic hepatic passive uptake, active uptake, and P-gp efflux of ATV were based on a previous publication by Nicolaï et al. [40], however considering the limitations of in vitro/in vivo correlation, a correction factor of 0.4 and 0.02 was applied to Clint,up,act and Clint,eff, respectively, for the extrapolation of the in vitro transporter data via curve-fitting to reflect the observed ATV PK parameters area under the concentration time curve from time zero to 24 h (AUC24) and maximum concentration (Cmax) [12] (available in the electronic supplementary material [ESM]). RIF systemic clearance was used in the model as described in Table 1. All the physicochemical, in vitro parameters, and data describing the metabolism of the drugs by different enzyme isoforms are contained in Table 1.
2.8 Verification of the PBPK Model
To verify the model, simulated pharmacokinetic profiles of individual and coadministrated drugs were compared with clinical studies available in the literature [17, 22, 23]. The model verification was performed according to the European Medicines Agency (EMA) [41], and was considered validated when the mean of simulated PK parameters AUC24, Cmax, and Ctrough were less than twofold of the observed clinical mean and the absolute average fold error (AAFE) was below 2 [42]. The doses and regimens of the drugs were chosen to reflect the clinical studies used to validate the model [17, 22, 23]. The administration of the drugs was simulated considering a fed state, according to drug label recommendations [12, 17]. A sensitivity analysis of the model was performed considering the changes in simulated AUC24, Cmax, and Ctrough of ATV to a percentage range variation linearly spaced of 25%, 58%, and 75% in the gastrointestinal (GI) transit time, Emax and EC50 of RIF (CYP3A4), Kinact (CYP3A4) and Ki (efflux and active uptake transporters) of RTV, when simulating the administration of ATV/r 300/100 mg + RIF 600 mg once daily.
2.9 Predictions of Drug–Drug Interactions (DDIs) Between Rifampicin (RIF) and Ritonavir-Boosted Atazanavir (ATV/r)
Simulations were carried out to determine potential dose adjustments of ATV/r to overcome the DDI with RIF. Dosing regimens were selected based on the minimum concentration (Ctrough) necessary to observe the therapeutic efficacy of ATV, the protein binding-adjusted 90% inhibitory concentration (PBA-IC90) of 14 ng/mL [43, 44] and the established clinical ‘cut-off’ of 150 ng/mL [10]. Both were considered since the strains used for in vitro assays may present greater sensitivity than those observed in vivo. The pharmacokinetic parameters AUC24, Cmax, and Ctrough were calculated at steady state. The number of individuals below the PBA-IC90 and the clinical ‘cut-off’ were calculated as percentages.
3 Results
3.1 Verification of the PBPK Model
The simulated PKs at steady state of each drug and observed data are presented in Table 2. The PBPK model is assumed to be qualified for all drugs (alone or combined). Moreover, the simulated DDIs between RIF (600 mg once daily) and ATV and RTV (300 and 100 mg once daily, respectively) demonstrated results similar to those obtained clinically (an AUC24 reduction of 71.5% and 88.1% compared with 40–70% and 80%, and a Cmax decrease of 48.1% and 78.3% compared with 40–70% and 80% for ATV and RTV, respectively, were observed) [23]. A substantial decrease of > 90% (98.7%) and > 80% (85%) in the simulated Ctrough of ATV and RTV was predicted when coadministered with RIF, as reported in the clinical study [23]. Detailed pharmacokinetic plasma concentration-time plots are presented in the ESM.
The sensitivity analysis suggested that ATV plasma concentration is sensitive to changes in GI transit time, Emax and EC50 of RIF, and Kinact of RTV, but not for changes in Ki of RTV (Fig. 1). The AUC24 and Ctrough were more affected by changes of these parameters than the Cmax. In the fed state, AUC24 and Ctrough were higher compared with the fasted state (available in the ESM).

Sensitivity analysis of ATV plasma concentration to variations of ± 25, 58, and 75% of GI transit-time, Emax and EC50 of rifampicin for CYP3A4, Kinact of ritonavir for CYP3A4, Ki of ritonavir for efflux and active uptake transporter, respectively. The sensitivity analysis was performed at steady state, with simulation of administration of ATV/r 300/100 mg + 600 mg of RIF once daily. ATV atazanavir, GI gastrointestinal, Emax maximum induction, EC50 concentration of inducer producing 50% of maximum induction, CYP cytochrome P450, Kinact maximum inactivation rate (h−1), Ki concentration of inhibitor producing 50% of maximum inhibition, ATV/r ritonavir-boosted atazanavir, RIF rifampicin
3.2 Predictions of DDIs Between RIF and ATV/r
To overcome the DDI between RIF and ATV/r, different dosing regimens were simulated (ATV 300–800 mg, RTV 100–300 mg, once or twice daily). The model predictions at steady-state are presented in Table 3. Simulations of different doses within a twice-daily regimen predicted a relevant increase of the ATV Ctrough depending on the dose (Table 3). Additionally, as demonstrated in Fig. 2 and Table 3, the number of individuals who presented Ctrough above PBA-IC90 of 14 ng/mL [43, 44] and the clinical ‘cut-off’ of 150 ng/mL [10] are 100% and > 95%, respectively, when ATV/r is administered twice daily at all the tested doses with RIF 600 mg once daily. When simulating a higher dose of RIF (1200 mg once daily), a slight reduction in the ATV Ctrough and > 64% of simulated individuals above the clinical ‘cut-off’ was observed (Table 3).

Box plot for atazanavir Ctrough plasma concentration (ng/mL) when different doses of ATV/r twice daily and RIF 600 mg once daily. The dashed line represents the clinical Ctrough cut-off (150 ng/mL) of ATV. *Simulation of RIF 1200 mg once daily, ATV atazanavir, Ctrough trough concentration, ATV/r ritonavir-boosted atazanavir, RIF rifampicin
4 Discussion
The coadministration of RIF and the PIs ATV/r for the treatment of tuberculosis in PLWH is challenging, necessitating a switch to a less well-tolerated ART regimen with a higher pill burden and potential increased risk of therapeutic failure. There are currently no studies in the literature with RIF-mediated induction of both CYP3A4 and transporters incorporated in a PBPK model to predict the increased plasma clearance of boosted PIs. A PBPK model was developed herein to simulate the magnitude of the concurrent inhibition and induction of CYP3A4 and P-gp by the perpetrators RIF and RTV to determine the DDI effect on ATV. Virtual clinical studies were simulated, identifying dosing regimens of ATV/r to overcome the DDI magnitude. The selection of doses for further clinical investigation is based on a detailed evaluation of plasma concentrations concerning efficacy and toxicity. The simulation of a dose adjustment of ATV/r 300/100 mg to twice daily demonstrated a significant increase in the Ctrough of ATV (Fig. 3), with 100% of individuals above the PBA-IC90 of 14 ng/mL and 98% above the clinical cut-off of 150 ng/mL. A further dose increase to 600/200 mg twice daily was predicted to result in 100% of patients above the clinical cut-off of 150 ng/mL, but concomitantly expose the patients to a high Cmax and AUC24, increasing the potential risk of associated hepatotoxicity, as demonstrated previously with high doses of darunavir/ritonavir and RIF [45].

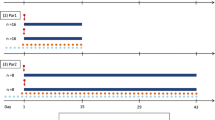
Simulated different regimens of ATV/r + RIF bid versus observed clinical data (ATV/r 300/100 + RIF mg od) [23]. Observed clinical data (solid dark circles), simulated mean: ATV/r 300/100 (dashed black line), ATV/r 300/200 (solid black line), ATV/r 400/100 (dashed dark grey line), ATV/r 400/100 (solid dark grey line), ATV/r 600/100 (dashed light grey line), ATV/r 600/200 (solid light grey line). ATV/r ritonavir-boosted atazanavir, RIF rifampicin, bid twice daily, od once daily
The selection of dosing strategy was driven by the objective of minimizing the risk of having a Ctrough below the clinical cut-off, as well as reducing the risk of hepatotoxicity. The twice-daily dose seems to provide the most clinically sustainable opportunity to overcome the DDI, rather than opting for a higher once-daily dose. Our findings indicate that higher doses of ATV/r once daily do not minimize the risk of failure and are likely to increase the risk of hepatotoxicity. ATV/r 800/300 mg + RIF 600 mg once daily was simulated. Despite a threefold dose increase, and despite resulting in higher Ctrough values compared with ATV/r 300/100 mg + RIF 600 mg once daily, the simulation resulted in 58% of simulated patients below the clinical cut-off. Additionally, higher Cmax could contribute to the higher risk of hepatotoxicity. Ritonavir super-boosting represents a dosing strategy to increase the exposure of ART concomitantly administered with RIF, as demonstrated by Rabie et al. [46]; however, it has poor acceptability owing to the hepatotoxicity risk, and, in this case, is not suitable considering the commercially available formulations of ATV and RTV.
Model predictions suggest concentrations at 12 h with twice-daily dosing are likely to be higher compared with concentrations observed at 24 h in the case of a once-daily, higher-dose regimen. Although no data with higher doses of ATV are available in the literature, studies with other PIs have highlighted different scenarios. According to Kendall et al. [47], a double dose of lopinavir/ritonavir + RIF was not associated with hepatotoxicity, while Nijland et al. [45] showed a high incidence of adverse events when a higher than standard dose of lopinavir/ritonavir tablets was combined with RIF. According to Cleijsen et al. [48], ATV plasma trough concentrations above 750 ng/mL were significantly related to the occurrence of increased total bilirubin concentrations. Future clinical studies should include a comprehensive evaluation of early signs of liver toxicity (transaminase elevations and bilirubin) and potential associations with PIs and RIF pharmacokinetics. The model predictions were applied for the design of a clinical trial protocol (DERIVE study; ATV/r 300/100 mg twice daily concurrently with a double dose of RIF [1200 mg] once daily) to evaluate the efficacy and toxicity of ATV when administered concomitantly with RIF in adults and special populations (NCT 04121195). Finally, patient adherence is a major factor necessary to achieve ART efficacy; consequently, three-times daily regimens are unlikely to represent suitable options.
PBPK modelling has been previously applied to predict DDIs between ART, anticancer, antimicrobial, and other classes of drugs [49,50,51,52,53,54]. Moreover, there is no study in the literature with RIF-mediated induction of CYP3A4 and transporter incorporated at the same time in a PBPK model to predict the increased plasma clearance of boosted PIs.
Although the model successfully predicted the PK of ATV, RTV and RIF, the model is characterized by some limitations. When assuming a well-stirred perfusion-limited model, instantaneous equilibrium of the drug concentration between blood and tissue into organs is considered [38]; however, during passive diffusion, a time lag for drugs to permeate across the membrane would be expected before achieving equilibrium, and this duration should not change the total exposure of the drug in the body (AUC24) but could impact the plasma concentration at a determined time. Moreover, membrane transporters can have a relevant impact on tissue distribution. The mechanisms involved in the uptake and efflux of drugs are complex and difficult to be determined in vitro. ATV distribution is not driven by passive diffusion alone and this could explain the underestimation of ATV Ctrough; partitioning into the tissue is difficult to model as well as some drug or organ-specific interactions. Another important point to consider is the administration with or without food, as demonstrated in the sensitivity analysis (see the ESM). The oral administration of ATV/r with food increases bioavailability and decreases pharmacokinetic variability. Furthermore, Ctrough is expected to be increased due to delayed absorption and an increase in time to reach maximum concentration [46], confirming how complex it is to predict Ctrough or the concentration at a specific time.
Although, in some cases the predicted ATV Ctrough was underestimated for the coadministration of ATV/r and RIF, the model suggests that sufficient Ctrough could be achieved with the 300/100 mg twice-daily regimen. The presented PBPK model does not provide a mechanistic description of toxicity due to the limited understanding of the underlying concentration-dependent mechanisms of hepatotoxicity. The described model was developed considering a population of healthy individuals, and pharmacokinetic differences have been described between HIV-positive patients and healthy volunteers [55]. Studies investigating the DDI between lopinavir and RIF showed a higher rate of hepatoxicity in healthy volunteers compared with HIV-infected individuals [56, 57]. For this purpose, clinical studies should include HIV-positive individuals to fully capture pharmacokinetic variability and potential differences in ADME processes.
The described PBPK approach could be applied in investigating DDIs between RIF and ART in special populations, such as children, elderly, and kidney and liver impairment since physiological and metabolic specificities of each population have been characterized and implemented in PBPK models previously [58,59,60,61]. Additionally, potential novel formulations of ART (long-acting) [62], different routes of administration (intramuscular, subcutaneous, etc.), and other tuberculosis agents (moderate inducers—rifabutin) [63] could be used in future studies to predict possible DDIs.
5 Conclusions
The simulations confirm that coadministration of RIF with ATV/r 300/100 mg once daily will result in subtherapeutic drug concentrations in PLWH coinfected with TB. The predicted data suggest an increased Ctrough of ATV/r 300/100 mg when administered twice daily compared with the once-daily regimen. However, a rational clinical evaluation of efficacy and toxicity should be considered in selecting sustainable dose adjustments, since, depending on the cut-off considered (PBA-IC90 = 14 ng/mL, or clinical = 150 ng/mL), some patients may present subtherapeutic Ctrough values.
Change history
16 September 2022
A Correction to this paper has been published: https://doi.org/10.1007/s40262-022-01178-3
References
World Health Organization. Tuberculosis and HIV. https://www.who.int/hiv/topics/tb/about_tb/en/.
Global tuberculosis report 2019. https://www.who.int/tb/publications/global_report/en/.
Manosuthi W, Wiboonchutikul S, Sungkanuparph S. Integrated therapy for HIV and tuberculosis. AIDS Res Ther. 2016;13:22. https://doi.org/10.1186/s12981-016-0106-y.
Back D, Marzolini C. The challenge of HIV treatment in an era of polypharmacy. J Int AIDS Soc. 2020;23(2): e25449. https://doi.org/10.1002/jia2.25449.
Marzolini C, Battegay M, Back D. Mechanisms of drug interactions II: transport proteins. Drug interactions in infectious diseases. Totowa: Humana Press; 2011. https://doi.org/10.1007/978-3-319-72422-5_3.
World Health Organization. Global Health Observatory (GHO) data: Antiretroviral therapy (ART) coverage among all age groups. https://www.who.int/gho/hiv/epidemic_response/ART/en/.
World Health Organization. Updated recommendations on first-line and second-line antiretroviral regimens and post-exposure prophylaxis and recommendations on early infant diagnosis of HIV: interim guidelines: supplement to the 2016 consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection (No. WHO/CDS/HIV/18.51). World Health Organization; 2018. https://apps.who.int/iris/bitstream/handle/10665/277395/WHO-CDS-HIV-18.51-eng.pdf?ua=1.
World Health Organization. Policy brief: update of recommendations on first-and second-line antiretroviral regimens (No. WHO/CDS/HIV/19.15). World Health Organization; 2019. https://apps.who.int/iris/bitstream/handle/10665/325892/WHO-CDS-HIV-19.15-eng.pdf.
Tseng E, Walsky RL, Luzietti RA, et al. Relative contributions of cytochrome CYP3A4 versus CYP3A5 for CYP3A-cleared drugs assessed in vitro using a CYP3A4-selective inactivator (CYP3cide). Drug Metab Dispos. 2014;42(7):1163–73. https://doi.org/10.1124/dmd.114.057000.
Bertz RJ, Persson A, Chung E, et al. Pharmacokinetics and pharmacodynamics of atazanavir-containing antiretroviral regimens, with or without ritonavir, in patients who are HIV-positive and treatment-naïve. Pharmacotherapy. 2013;33(3):284–94. https://doi.org/10.1002/phar.1205.
Achenbach CJ, Darin KM, Murphy RL, Katlama C. Atazanavir/ritonavir-based combination antiretroviral therapy for treatment of HIV-1 infection in adults. Future Virol. 2011;6(2):157–77. https://doi.org/10.2217/fvl.10.89.
Bristol-Myers Squibb Company. Reyataz™ (atazanavir). US prescribing information. Bristol-Myers Squibb Company; 2017.
Koudriakova T, Iatsimirskaia E, Utkin I, et al. Metabolism of the human immunodeficiency virus protease inhibitors indinavir and ritonavir by human intestinal microsomes and expressed cytochrome P4503A4/3A5: mechanism-based inactivation of cytochrome P4503A by ritonavir. Drug Metab Dispos. 1998;26(6):552–61.
Elmeliegy M, Vourvahis M, Guo C, Wang DD. Effect of P-glycoprotein (P-gp) inducers on exposure of P-gp substrates: review of clinical drug-drug interaction studies. Clin Pharmacokinet. 2020;59(6):699–714. https://doi.org/10.1007/s40262-020-00867-1.
Greiner B, Eichelbaum M, Fritz P, et al. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest. 1999;104(2):147–53. https://doi.org/10.1172/JCI6663.
Gurley BJ, Swain A, Williams DK, Barone G, Battu SK. Gauging the clinical significance of P-glycoprotein-mediated herb-drug interactions: comparative effects of St. John’s wort, Echinacea, clarithromycin, and rifampin on digoxin pharmacokinetics. Mol Nutr Food Res. 2008;52(7):772–9. https://doi.org/10.1002/mnfr.200700081.
AbbVie Inc. Norvir™ (ritonavir). US prescribing information. Abbvie Inc.; 2019.
Williamson B, Dooley KE, Zhang Y, Back DJ, Owen A. Induction of influx and efflux transporters and cytochrome P450 3A4 in primary human hepatocytes by rifampin, rifabutin, and rifapentine. Antimicrob Agents Chemother. 2013;57(12):6366–9. https://doi.org/10.1128/AAC.01124-13.
Yamashita F, Sasa Y, Yoshida S, et al. Modeling of rifampicin-induced CYP3A4 activation dynamics for the prediction of clinical drug-drug interactions from in vitro data. PLoS ONE. 2013;8(9): e70330. https://doi.org/10.1371/journal.pone.0070330.
Mori D, Kimoto E, Rago B, et al. Dose-dependent inhibition of OATP1B by rifampicin in healthy volunteers: comprehensive evaluation of candidate biomarkers and OATP1B probe drugs. Clin Pharmacol Ther. 2020;107(4):1004–13. https://doi.org/10.1002/cpt.1695.
Guidelines for the Use of Antiretroviral Agents in adults and Adolescents with HIV. December 18, 2019. Department of Health and Human Services. https://clinicalinfo.hiv.gov/sites/default/files/inline-files/AdultandAdolescentGL.pdf.
Acosta EP, Kendall MA, Gerber JG, et al. Effect of concomitantly administered rifampin on the pharmacokinetics and safety of atazanavir administered twice daily. Antimicrob Agents Chemother. 2007;51(9):3104–10. https://doi.org/10.1128/AAC.00341-07.
Burger DM, Agarwala S, Child M, Been-Tiktak A, Wang Y, Bertz R. Effect of rifampin on steady-state pharmacokinetics of atazanavir with ritonavir in healthy volunteers. Antimicrob Agents Chemother. 2006;50(10):3336–42. https://doi.org/10.1128/AAC.00461-06.
Prueksaritanont T, Chu X, Gibson C, et al. Drug-drug interaction studies: regulatory guidance and an industry perspective. AAPS J. 2013;15(3):629–45. https://doi.org/10.1208/s12248-013-9470-x.
Moss DM, Marzolini C, Rajoli RK, Siccardi M. Applications of physiologically based pharmacokinetic modeling for the optimization of anti-infective therapies. Expert Opin Drug Metab Toxicol. 2015;11(8):1203–17. https://doi.org/10.1517/17425255.2015.1037278.
US FDA. Drug Interactions: Relevant Regulatory Guidance and Policy Documents. https://www.fda.gov/drugs/drug-interactions-labeling/drug-interactions-relevant-regulatory-guidance-and-policy-documents.
Bosgra S, van Eijkeren J, Bos P, Zeilmaker M, Slob W. An improved model to predict physiologically based model parameters and their inter-individual variability from anthropometry. Crit Rev Toxicol. 2012;42(9):751–67. https://doi.org/10.3109/10408444.2012.709225.
Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health. 1997;13(4):407–84. https://doi.org/10.1177/074823379701300401.
Birnbaum L, Brown R, Bischoff K, et al. Physiological parameter values for PBPK models. Washington, DC: International Life Sciences Institute, Risk Science Institute; 1994.
Yu LX, Amidon GL. A compartmental absorption and transit model for estimating oral drug absorption. Int J Pharm. 1999;186(2):119–25. https://doi.org/10.1016/S0378-5173(99)00147-7.
Gertz M, Harrison A, Houston JB, Galetin A. Prediction of human intestinal first-pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab Dispos. 2010;38(7):1147–58. https://doi.org/10.1124/dmd.110.032649.
Wuyts B, Brouwers J, Mols R, Tack J, Annaert P, Augustijns P. Solubility profiling of HIV protease inhibitors in human intestinal fluids. J Pharm Sci. 2013;102(10):3800–7. https://doi.org/10.1002/jps.23698.
Wilkins JJ, Savic RM, Karlsson MO, Langdon G, McIlleron H, Pillai G, et al. Population pharmacokinetics of rifampin in pulmonary tuberculosis patients, including a semimechanistic model to describe variable absorption. Antimicrob Agents Chemother. 2008;52(6):2138–48. https://doi.org/10.1128/AAC.00461-07.
Paine MF, Khalighi M, Fisher JM, et al. Characterization of interintestinal and intraintestinal variations in human CYP3A-dependent metabolism. J Pharmacol Exp Ther. 1997;283(3):1552–62.
Yang J, Jamei M, Yeo KR, Tucker GT, Rostami-Hodjegan A. Prediction of intestinal first-pass drug metabolism. Curr Drug Metab. 2007;8(7):676–84. https://doi.org/10.2174/138920007782109733.
Ramsden D, Zhou J, Tweedie DJ. Determination of a degradation constant for CYP3A4 by direct suppression of mRNA in a novel human hepatocyte model, HepatoPac. Drug Metab Dispos. 2015;43(9):1307–15. https://doi.org/10.1124/dmd.115.065326.
Barter ZE, Chowdry JE, Harlow JR, Snawder JE, Lipscomb JC, Rostami-Hodjegan A. Covariation of human microsomal protein per gram of liver with age: absence of influence of operator and sample storage may justify interlaboratory data pooling. Drug Metab Dispos. 2008;36(12):2405–9. https://doi.org/10.1124/dmd.108.021311.
Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J Pharm Sci. 2002;91(1):129–56. https://doi.org/10.1002/jps.10005.
Goldsmith DR, Perry CM. Atazanavir. Drugs. 2003;63(16):1679–93.
Nicolaï J, De Bruyn T, Thevelin L, Augustijns P, Annaert P. Transport-metabolism interplay of atazanavir in rat hepatocytes. Drug Metab Dispos. 2016;44(3):389–97.
European Medicines Agency. Guideline on the qualification and reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation. https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-qualification-reporting-physiologically-based-pharmacokinetic-pbpk-modelling_en.pdf.
Abduljalil K, Cain T, Humphries H, Rostami-Hodjegan A. Deciding on success criteria for predictability of pharmacokinetic parameters from in vitro studies: an analysis based on in vivo observations. Drug Metab Dispos. 2014;42(9):1478–84. https://doi.org/10.1124/dmd.114.058099.
Bristol-Meyers Squibb. Reyataz (atazanavir). US prescribing information. Princeton: Bristol-Meyers Squibb; 2012.
Zhu L, Liao S, Child M, et al. Pharmacokinetics and inhibitory quotient of atazanavir/ritonavir versus lopinavir/ritonavir in HIV-infected, treatment-naive patients who participated in the CASTLE Study. J Antimicrob Chemother. 2012;67(2):465–8. https://doi.org/10.1093/jac/dkr490.
Nijland HM, L’homme RF, Rongen GA, et al. High incidence of adverse events in healthy volunteers receiving rifampicin and adjusted doses of lopinavir/ritonavir tablets. AIDS. 2008;22(8):931–5. https://doi.org/10.1097/QAD.0b013e3282faa71e.
Rabie H, Denti P, Lee J, et al. Lopinavir-ritonavir super-boosting in young HIV-infected children on rifampicin-based tuberculosis therapy compared with lopinavir-ritonavir without rifampicin: a pharmacokinetic modelling and clinical study. Lancet HIV. 2019. https://doi.org/10.1016/S2352-3018(18)30293-5.
Kendall MA, Lalloo U, Fletcher CV, Wu X, Podany AT, Cardoso SW, et al. Safety and pharmacokinetics of double-dose lopinavir/ritonavir + rifampin vs. lopinavir/ritonavir + daily rifabutin for treatment of HIV-TB coinfection. Clin Infect Dis. 2021. https://doi.org/10.1093/cid/ciab097.
Cleijsen RMM, Van de Ende ME, Kroon FP, et al. Therapeutic drug monitoring of the HIV protease inhibitor atazanavir in clinical practice. J Antimicrob Chemother. 2007;60(4):897–900. https://doi.org/10.1093/jac/dkm298.
Rajoli RKR, Curley P, Chiong J, et al. Predicting drug-drug interactions between rifampicin and long-acting cabotegravir and rilpivirine using physiologically based pharmacokinetic modeling. J Infect Dis. 2019;219(11):1735–42. https://doi.org/10.1093/infdis/jiy726.
Marzolini C, Rajoli R, Battegay M, Elzi L, Back D, Siccardi M. Physiologically based pharmacokinetic modeling to predict drug-drug interactions with efavirenz involving simultaneous inducing and inhibitory effects on cytochromes. Clin Pharmacokinet. 2017;56(4):409–20. https://doi.org/10.1007/s40262-016-0447-7.
Moltó J, Rajoli R, Back D, et al. Use of a physiologically based pharmacokinetic model to simulate drug-drug interactions between antineoplastic and antiretroviral drugs. J Antimicrob Chemother. 2017;72(3):805–11. https://doi.org/10.1093/jac/dkw485.
Hanke N, Frechen S, Moj D, et al. PBPK models for CYP3A4 and P-gp DDI prediction: a modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacomet Syst Pharmacol. 2018;7(10):647–59. https://doi.org/10.1002/psp4.12343.
Duan P, Zhao P, Zhang L. Physiologically based pharmacokinetic (PBPK) modeling of pitavastatin and atorvastatin to predict drug-drug interactions (DDIs). Eur J Drug Metab Pharmacokinet. 2017;42(4):689–705. https://doi.org/10.1007/s13318-016-0383-9.
Türk D, Hanke N, Wolf S, et al. Physiologically based pharmacokinetic models for prediction of complex CYP2C8 and OATP1B1 (SLCO1B1) drug-drug-gene interactions: a modeling network of gemfibrozil, repaglinide, pioglitazone, rifampicin, clarithromycin and itraconazole. Clin Pharmacokinet. 2019;58(12):1595–607. https://doi.org/10.1007/s40262-019-00777-x.
Stanley TL, Grinspoon SK. Body composition and metabolic changes in HIV-infected patients. J Infect Dis. 2012;205(Suppl 3):S383-390. https://doi.org/10.1093/infdis/jis205.
Decloedt EH, Maartens G, Smith P, Merry C, Bango F, McIlleron H. The safety, effectiveness and concentrations of adjusted lopinavir/ritonavir in HIV-infected adults on rifampicin-based antitubercular therapy. PLoS ONE. 2012;7(3): e32173. https://doi.org/10.1371/journal.pone.0032173.
Shitara Y, Takeuchi K, Horie T. Long-lasting inhibitory effects of saquinavir and ritonavir on OATP1B1-mediated uptake. J Pharm Sci. 2013;102(9):3427–35. https://doi.org/10.1002/jps.23477.
Schlender JF, Meyer M, Thelen K, et al. Development of a whole-body physiologically based pharmacokinetic approach to assess the pharmacokinetics of drugs in elderly individuals. Clin Pharmacokinet. 2016;55(12):1573–89. https://doi.org/10.1007/s40262-016-0422-3.
Bunglawala F, Rajoli RKR, Mirochnick M, Owen A, Siccardi M. Prediction of dolutegravir pharmacokinetics and dose optimization in neonates via physiologically based pharmacokinetic (PBPK) modelling. J Antimicrob Chemother. 2020;75(3):640–7. https://doi.org/10.1093/jac/dkz506.
Johnson TN, Boussery K, Rowland-Yeo K, Tucker GT, Rostami-Hodjegan A. A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin Pharmacokinet. 2010;49(3):189–206. https://doi.org/10.2165/11318160-000000000-00000.
Sayama H, Takubo H, Komura H, Kogayu M, Iwaki M. Application of a physiologically based pharmacokinetic model informed by a top-down approach for the prediction of pharmacokinetics in chronic kidney disease patients. AAPS J. 2014;16(5):1018–28. https://doi.org/10.1208/s12248-014-9626-3.
Havlir D, Gandhi M. Implementation challenges for long-acting antivirals as treatment. Curr Opin HIV AIDS. 2015;10(4):282–9. https://doi.org/10.1097/COH.0000000000000158.
Horita Y, Doi N. Comparative study of the effects of antituberculosis drugs and antiretroviral drugs on cytochrome P450 3A4 and P-glycoprotein. Antimicrob Agents Chemother. 2014;58(6):3168–76. https://doi.org/10.1128/AAC.02278-13.
DrugBank. Atazanavir. https://www.drugbank.ca/drugs/DB01072.
DrugBank. Ritonavir. https://www.drugbank.ca/drugs/DB00503.
DrugBank. Rifampicin. https://www.drugbank.ca/drugs/DB01045.
National Center for Biotechnology Information. PubChem Compound Summary for CID 148192, Atazanavir. https://pubchem.ncbi.nlm.nih.gov/compound/Atazanavir.
Siccardi M, Marzolini C, Seden K, et al. Prediction of drug-drug interactions between various antidepressants and efavirenz or boosted protease inhibitors using a physiologically based pharmacokinetic modelling approach. Clin Pharmacokinet. 2013;52(7):583–92. https://doi.org/10.1007/s40262-013-0056-7.
Wilkins JJ, Savic RM, Karlsson MO, et al. Population pharmacokinetics of rifampin in pulmonary tuberculosis patients, including a semimechanistic model to describe variable absorption. Antimicrob Agents Chemother. 2008;52(6):2138–48. https://doi.org/10.1128/AAC.00461-07.
Mugundu GM, Hariparsad N, Desai PB. Impact of ritonavir, atazanavir and their combination on the CYP3A4 induction by efavirenz in primary human hepatocytes. Drug Metab Lett. 2010;4(1):45–50.
Kato M, Chiba K, Horikawa M, Sugiyama Y. The quantitative prediction of in vivo enzyme-induction caused by drug exposure from in vitro information on human hepatocytes. Drug Metab Pharmacokinet. 2005;20(4):236–43. https://doi.org/10.2133/dmpk.20.236.
Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 2008;9(4):310–22. https://doi.org/10.2174/138920008784220664.
Hirano M, Maeda K, Shitara Y, Sugiyama Y. Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab Dispos. 2006;34(7):1229–36.
Izumi S, Nozaki Y, Maeda K, Komori T, Takenaka O, Kusuhara H, Sugiyama Y. Investigation of the impact of substrate selection on in vitro organic anion transporting polypeptide 1B1 inhibition profiles for the prediction of drug-drug interactions. Drug Metab Dispos. 2015;43(2):235–47.
Telbisz Á, Ambrus C, Mózner O, Szabó E, Várady G, Bakos É, et al. Interactions of potential anti-COVID-19 compounds with multispecific ABC and OATP drug transporters. Pharmaceutics. 2021;13(1):81. https://doi.org/10.3390/pharmaceutics13010081.
Acknowledgements
This work was presented at the 20th International Workshop on Clinical Pharmacology of HIV, Hepatitis and Other Antiviral Drugs, Noordwijk, The Netherlands, 14–16 May 2019 (Poster 31).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This project was part of the EDCTP2 programme supported by the European Union (VirTUAL—RIA2016MC-1606).
Conflict of interest
Marco Siccardi has received research grant funding from Janssen and ViiV. Maiara Camotti Montanha, Francesc Fabrega, Alice Howarth, Nicolas Cottura, Hannah Kinvig, Fazila Bunglawala, Andrew Lloyd, Paolo Denti, and Catriona Waitt have no conflicts of interest to declare.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Code availability
The PBPK modelling code generated during the current study is available in the PBPKmodelling_DDI_ART_AdultHumans repository, persistent web link: https://github.com/UoLpharma/PBPKmodelling_DDI_ART_AdultHumans.
Author contributions
All authors contributed to the overall concept, design, verification, and application of the model. MCM and FF performed the modelling and analysis, and MCM and MS wrote the manuscript with support from all other authors. All authors reviewed and contributed to the final manuscript.
Additional information
The original online version of the article has been revised due to retrospective open access order.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Montanha, M.C., Fabrega, F., Howarth, A. et al. Predicting Drug–Drug Interactions between Rifampicin and Ritonavir-Boosted Atazanavir Using PBPK Modelling. Clin Pharmacokinet 61, 375–386 (2022). https://doi.org/10.1007/s40262-021-01067-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01067-1