
Abstract
Combined antiretroviral treatments have significantly improved the morbidity and mortality related to HIV infection, thus transforming HIV infection into a chronic disease; however, the efficacy of antiretroviral treatments is highly dependent on the ability of infected individuals to adhere to life-long drug combination therapies. A major milestone in HIV treatment is the marketing of the long-acting intramuscular antiretroviral drugs cabotegravir and rilpivirine, allowing for infrequent drug administration, with the potential to improve adherence to therapy and treatment satisfaction. Intramuscular administration of cabotegravir and rilpivirine leads to differences in pharmacokinetics and drug–drug interaction (DDI) profiles compared with oral administration. A notable difference is the long elimination half-life with intramuscular administration, which reaches 5.6–11.5 weeks for cabotegravir and 13–28 weeks for rilpivirine, compared with 41 and 45 h, respectively, with their oral administration. Cabotegravir and rilpivirine have a low potential to cause DDIs, however these drugs can be victims of DDIs. Cabotegravir is mainly metabolized by UGT1A1, and rilpivirine is mainly metabolized by CYP3A4, therefore these agents are susceptible to DDIs with inhibitors, and particularly inducers of drug-metabolizing enzymes. Intramuscular administration of cabotegravir and rilpivirine has the advantage of eliminating DDIs occurring at the gastrointestinal level, however interactions can still occur at the hepatic level. This review provides insight on the intramuscular administration of drugs and summarizes the pharmacology of long-acting cabotegravir and rilpivirine. Particular emphasis is placed on DDI profiles after oral and intramuscular administration of these antiretroviral drugs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Cabotegravir plus rilpivirine represents the first long-acting injectable monthly or bimonthly regimen for the treatment of HIV infection in adults who are virologically stable and suppressed. |
Intramuscular administration of cabotegravir and rilpivirine exhibits absorption limited kinetics (flip-flop kinetics), resulting in sustained plasma concentrations. |
Drug–drug interactions occurring at the gastrointestinal tract as a result of chelation, changes in gastric pH, or inhibition/induction of drug-metabolizing enzymes or transporters are avoided with the intramuscular administration of cabotegravir and rilpivirine. |
1 Introduction
The use of three-drug antiretroviral combinations has significantly reduced the morbidity and mortality associated with HIV infection, thus transforming HIV infection into a chronic disease. Treated people living with HIV (PLWH) nowadays have a life expectancy close to that of the general population [1,2,3]. In recent years, the efficacy of dual antiretroviral regimens has been investigated in an attempt to reduce life-long drug exposure and prevent toxicities. Some drug combinations (e.g. dolutegravir-rilpivirine, lamivudine-dolutegravir, lopinavir/ritonavir-lamivudine) have proved to be efficient in treatment-naïve HIV-infected individuals as well as in individuals with stable virologic suppression [4,5,6].
Treatment of HIV infection generally requires adherence to once-daily antiretroviral drug dosing. Incomplete treatment adherence can lead to the emergence of drug-resistant HIV strains resulting in loss of virologic control, and placing PLWH not only at risk of morbidity and mortality but also at risk of transmitting HIV. The concerns over both long-term adherence to treatment and dosing convenience have led to the development of injectable long-acting antiretroviral formulations allowing once-monthly or bimonthly administration. Antiretroviral drug attributes required for long-acting intramuscular administration include a high-level intrinsic antiviral activity, low systemic clearance, low aqueous solubility, and a high melting point (permitting micronization or nanomilling and the development of injectable nanoparticle suspensions) [7]. The integrase inhibitor cabotegravir and the non-nucleoside reverse transcriptase inhibitor (NNRTI) rilpivirine fulfil these requirements. The combination of these drugs successfully suppressed HIV with monthly or bimonthly intramuscular administration in a phase IIb trial [8], thus leading to their clinical development.
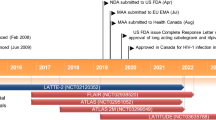
Long-acting intramuscular formulations of cabotegravir and rilpivirine were first approved in March 2020 in Canada for the treatment of HIV infection. These agents were subsequently approved by the European Medicines Agency (EMA) in December 2020 and by the US Food and Drug Administration (FDA) in January 2021. The intramuscular administration of antiretroviral drugs leads to differences in pharmacokinetics and drug–drug interaction (DDI) profiles compared with oral administration. This review provides insight on intramuscular administration of drugs and summarizes the pharmacology of long-acting cabotegravir and rilpivirine. Particular emphasis is placed on DDI profiles after oral and intramuscular administration of these antiretroviral drugs.
2 Intramuscular Administration of Drugs
An intramuscular injection of a suspension or lipophilic solution creates a drug ‘depot’ in the muscle from which the drug is released for an extended period, allowing for infrequent drug administration [9]. Unlike oral administration, the elimination rate of intramuscularly administered drugs far exceeds the absorption rate, making the latter the driver of the elimination half-life (t½; so-called flip-flop kinetics) [10].
The rich vascular supply of the muscle favours drug absorption [11]. A good intramuscular injection technique is therefore critical to ensure that the drug is not deposited in the subcutaneous adipose tissue. Adipose tissue is less vascular than muscle tissue, which may result in poor absorption and distribution of the drug, leading to suboptimal drug levels [12]. Thus, obesity may affect the exposure of drugs administered intramuscularly because of the difficulty in reaching the muscle through thicker layers of adipose tissue. Of note, the Canadian and US product information for Cabenuva (long-acting cabotegravir/rilpivirine) advises using a longer needle when administering the product to patients with a body mass index (BMI) > 30 kg/m2, to ensure intramuscular delivery as opposed to subcutaneous delivery [13, 14]. The ECLAIR2 and HPTN 077 trials used a needle of 2 inches for participants with a BMI > 30 kg/m2 and a needle of 1.5 inches for participants with a BMI < 30 kg/m2 [15, 16]; the ECLAIR2 trial noted changes in pharmacokinetic parameters associated with high BMI and/or longer needle length. Maximal concentration (Cmax) and the area under the curve (AUC) were higher in participants with a BMI below the median, while the concentration at the end of the dosing interval (Ctrough) was lower [16], although phase III trials have shown that lower Ctrough does not persist through week 48 [17]. There are also gender differences in adipose layer thickness, which may result in different pharmacokinetics between men and women, as reported for both intramuscular cabotegravir and rilpivirine [15, 18]. Other factors affecting the absorption and bioavailability of intramuscular injections include exercise and local blood flow (with greater drug absorption in the case of increased blood flow in the muscle) [11]. The absorption process in the muscle may also vary with aging, as indicated by differences in tobramycin pharmacokinetics in the young and elderly after intramuscular administration [19]. The pharmacokinetics of long-acting cabotegravir and rilpivirine have not yet been evaluated in elderly individuals.
Intramuscular administration allows a drug to bypass the gastrointestinal tract, thereby eliminating the risk of gut-based DDIs (Fig. 1). Orally administered drugs may be subject to DDIs at the intestinal level due to several mechanisms, including the following.
-
Changes in gastric pH Antacids markedly reduce the absorption of rilpivirine since a low pH is required for its solubility [20]. Oral rilpivirine is contraindicated with proton pump inhibitors, whereas antacids can be administered 2 h before or 4 h after rilpivirine due to their shorter acid-neutralizing effect.
-
Chelation All integrase inhibitors, including cabotegravir, possess a chelating motif allowing binding to a divalent cation in the active site of the HIV integrase. Thus, the divalent cations aluminium, calcium, magnesium, iron and zinc in antacids or mineral supplements can form a complex with integrase inhibitors at the intestinal level, thereby impairing their absorption [21]. The label recommends that products containing divalent cations are administered 2 h before or 4 h after oral cabotegravir [13].
-
Inhibition/induction of intestinal drug-metabolizing enzymes and/or intestinal transporters For instance, exposure of the CYP3A4 substrate rilpivirine is significantly increased by darunavir/ritonavir due to the inhibition of intestinal and hepatic CYP3A4 [22].

Mechanisms of drug–drug interaction after oral versus intramuscular administration of cabotegravir and rilpivirine
Intramuscular administration of drugs allows absorption into the systemic bloodstream, bypassing first-pass metabolism and in theory providing some level of protection against DDIs. In practice, no clinical studies have examined the difference in DDI magnitude between oral and intramuscular administration of antiretroviral drugs. DDI studies with intramuscular antiretrovirals are challenging in HIV-infected individuals. The risk of antiretroviral treatment (ART) toxicity/failure that cannot be readily reversed given the nature of the long-acting treatment must be avoided. Novel approaches are required when designing a DDI study with injectable antiretroviral drugs. Of interest, on coadministration of levonorgestrel subcutaneous implant with the inducer efavirenz, levonorgestrel exposure was 47% after 24 weeks and 57% after 48 weeks [23]. A comparable inducing effect was observed after oral administration since efavirenz reduced oral levonorgestrel exposure by 56% [24]. Furthermore, a modelling study predicted a comparable decrease in cabotegravir and rilpivirine exposures after both intramuscular and oral administration in the presence of rifampicin [25]. Together, these data suggest that bypassing the gastrointestinal tract does not mitigate the magnitude of DDIs with inducers. However, in case of inhibition, the magnitude of DDIs after intramuscular administration could possibly be less pronounced, particularly for rilpivirine, as bypassing gut-based inhibition of CYP3A4 may result in lower bioavailability.
3 Cabotegravir
3.1 Pharmacodynamics
A structural analogue of dolutegravir [26], cabotegravir inhibits the strand transfer step of viral complementary DNA (cDNA) integration [27] by binding to the active site of the HIV integrase [28]. Cabotegravir monotherapy was evaluated in treatment-naïve individuals at 5 mg and 30 mg once-daily for 10 days. The mean HIV RNA change from baseline was − 2.2 and − 2.3 log10 copies/mL for 5 mg and 30 mg once daily. Pharmacodynamic analyses found that the HIV-RNA change from baseline was associated with cabotegravir Ctrough, with a maximum effect (Emax) estimated to be 2.56 log10 and a 50% effective concentration (EC50) of 82 ng/mL [29]. Like other integrase inhibitors, the efficacy of cabotegravir relates to Ctrough [30].
3.2 Dosing Recommendations
Cabotegravir is prescribed as an oral lead-in phase of 30 mg daily for at least 28 days to ensure tolerability, followed by an intramuscular loading dose of 600 mg, before administration can continue 4 weeks later at a maintenance dose of 400 mg once monthly. If dosing once every 2 months, a second 600 mg loading dose should be administered 4 weeks after the first dose before treatment can proceed at a maintenance dose of 600 mg every 8 weeks [31]. Delays and treatment interruptions have been evaluated in a population pharmacokinetic model, which has informed dosing recommendations [32]. Doses can be administered between 7 days before and 7 days after the monthly dose is due. If a monthly injection is missed by ≤ 2 months, then continuation of 400 mg every 4 weeks may continue; however, if the injection is missed by more than 2 months, another 600 mg loading dose must be administered before proceeding onto 400 mg every 4 weeks. Likewise, if an 8-weekly injection is missed by ≤ 2 months, treatment can be re-initialized as normal; however, if it is missed by > 2 months, a 600 mg dose must be administered as soon as possible, followed by a second dose after 4 weeks, before treatment may continue as normal. In cases where patients know they will miss a dose by 7 days, they can take oral therapy of 30 mg once daily to replace up to two once-monthly or one once every 2 months visits [31].
3.3 Pharmacokinetics
3.3.1 Absorption
A summary of the key pharmacokinetic parameters is presented in Table 1. As described in the previous section, the rate of absorption is a key difference between oral and intramuscular administration. Oral cabotegravir has a median time to maximal plasma concentration (Tmax) of 3 h, to be contrasted with that of 7 days for intramuscular administration [13]. Steady-state concentrations after intramuscular administration of cabotegravir are achieved after 44 weeks, much earlier than for rilpivirine (80% steady-state after 48 weeks) [17]. This difference may need to be accounted for when performing pharmacological studies (e.g. tissue pharmacology). The absolute bioavailability of oral cabotegravir has not been studied, however administering oral cabotegravir with a high-fat meal did not have a clinically significant effect on cabotegravir pharmacokinetics, thus cabotegravir can be administered regardless of food [33]. Cabotegravir is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), but owing to high passive permeability, inhibitors of these intestinal transporters are unlikely to affect cabotegravir intestinal absorption [34].
3.3.2 Distribution
Cabotegravir is highly protein-bound (> 99.8 %) and the blood-to-plasma ratio is 0.5. The volume of distribution following oral cabotegravir is 12.3 L [13, 31]. Of interest, the distribution of cabotegravir in the cerebrospinal fluid (CSF) was measured in HIV-infected individuals 7 days after the maintenance dose injection of cabotegravir 400 mg + rilpivirine 600 mg administered every 4 weeks (n = 3) or cabotegravir 600 mg + rilpivirine 900 mg administered every 8 weeks (n = 15) [35]. Median total cabotegravir CSF was 11 ng/mL (when administered every 4 weeks) and 13 ng/mL (when administered every 8 weeks), representing CSF/plasma ratios of 0.30% and 0.34%, respectively. CSF cabotegravir concentrations exceeded the in vitro EC50 for wild-type HIV (i.e. 0.10 ng/mL) and all patients maintained CSF viral RNA below 50 copies/mL This study demonstrates that therapeutic concentrations of cabotegravir can be achieved in the CSF following intramuscular administration.
3.3.3 Metabolism and Elimination
The main route of cabotegravir clearance is hepatic metabolism. A mass balance study of oral cabotegravir determined the main metabolite is a glucuronic acid conjugate (M1), which is excreted in the urine along with a minor metabolite, a glucose conjugate (M2). A small amount of unchanged cabotegravir is excreted via bile/faeces. There are no mass balance data for intramuscular cabotegravir, as the long t½ makes sample collection impractical. However, post hoc analysis of urine samples from a phase I trial has revealed that M1 accounted for > 90% of the drug-related material recovered, irrespective of route of administration (in this case, either subcutaneous or intramuscular). Thus, it is likely that hepatic clearance of cabotegravir is identical for both oral and intramuscular administration. In vitro analysis has revealed the primary hepatic enzyme responsible for cabotegravir metabolism is UGT1A1, with some additional contribution from UGT1A9 [36]. Cabotegravir does not inhibit or induce cytochromes (CYP) or glucuronidation enzymes. In vitro studies have also indicated that cabotegravir does not inhibit the transporters P-gp, BCRP, multidrug resistance protein (MRP) 2, MRP4, organic anion transporting polypeptide (OATP) 1B1, OATP1B3, organic cation transporter (OCT) 1, OCT2, bile salt export pump (BSEP), or the renal multidrug and toxin extrusion transporter (MATE) 1 and MATE2K at clinically relevant concentrations. On the other hand, cabotegravir inhibits the renal transporters OAT1 (half maximal inhibitory concentration [IC50] = 0.81 µM) and OAT3 (IC50 = 0.41 µM) [34]. Physiologically based pharmacokinetic (PBPK) studies showed a mean increase in OAT1 or OAT3 substrate (i.e. ciprofloxacin, tenofovir disoproxil fumarate, cidofovir, methotrexate, adefovir, oseltamivir and cefuroxime) exposure of < 25% with concomitant administration of oral cabotegravir [37]. PBPK modelling suggests that OAT1/OAT3 inhibition is not clinically relevant, however the European product label [31] recommends caution when coadministering with a narrow therapeutic index drug such as methotrexate.
Plasma concentration is a key consideration for an antiretroviral, be it for ART or pre-exposure prophylaxis (PrEP). Concentrations must be sufficient to not only prevent virologic failure (in ART) or de novo infection (PrEP) but also to guard against selecting for resistant HIV strains, as may occur when drug concentrations fall below the therapeutic range [38]. The long pharmacokinetic ‘tail’ of intramuscular cabotegravir, as driven by its greater t½ (see Table 1), may give rise to increased risk of HIV resistance after cessation of treatment. The ECLAIR2 reported an intramuscular cabotegravir t½ of 40 days (after multiple cabotegravir doses) in cisgender men. Seventeen percent of men had detectable cabotegravir after 52 weeks [16]. In a follow-up of the HPTN 077 trial, which studied intramuscular cabotegravir as PrEP in both men and women, showed that 78% of those assigned male at birth had undetectable cabotegravir 52 weeks after the final injection, as opposed to 37% of those assigned female at birth. Males had a t½ of 43.5 days compared with 60.4 days in females, an increase of 33%. These factors may need to be taken into consideration when addressing the potential for DDIs during a pharmacokinetic tail [15].
3.4 Pharmacokinetics in Special Populations
3.4.1 Renal Impairment
A phase I trial compared the pharmacokinetics of oral cabotegravir between healthy volunteers and patients with renal impairment (creatinine clearance < 30 mL/min). As is expected for a drug mainly metabolized by the liver, renal impairment did not affect cabotegravir Cmax, AUC, or t½. Dialysis is also unlikely to affect cabotegravir clearance as it is highly protein-bound [39]. The product monograph states that cabotegravir may be administered without dose adjustment in patients with mild–moderate renal impairment but advises monitoring for adverse events in patients with severe kidney impairment, end-stage kidney disease, or on dialysis [13].
3.4.2 Hepatic Impairment
The pharmacokinetics of oral cabotegravir was studied in another phase I trial comparing healthy volunteers with patients with moderate hepatic impairment (Child–Pugh 7-9). As cabotegravir is highly protein-bound (> 99.8%), particularly to albumin, hepatic impairment was expected to increase free plasma cabotegravir due to the associated decrease in the production of albumin. Indeed, unbound cabotegravir was 40% and 50% higher in hepatic insufficiency 2 and 24 h post-dose, respectively. This resulted in an increase in cabotegravir clearance in patients with moderate hepatic impairment, as signified by a decrease in AUC, Cmax and Ctrough of 27%, 31% and 27%, respectively. Neither the increase in free cabotegravir nor the increase in clearance seen in moderate hepatic impairment is clinically significant, given the wide therapeutic window of cabotegravir [40]. However, it should be noted that a 30% reduction in cabotegravir AUC could be relevant in patients receiving moderate inducers, or with high BMI or polymorphisms/resistance-associated mutations in the HIV integrase gene. The product monograph advises that cabotegravir may be administered without dose adjustment in mild or moderate hepatic impairment but warns that administration in severe hepatic impairment (Child–Pugh score C) has not been studied [13, 31].
3.4.3 Pregnancy
The Cabenuva product monograph states that cabotegravir has not been studied in pregnant women and therefore the associated risks, including neural tube defects, cannot be assessed [13]; however, 13 pregnancies were recorded during the HPTN077 trial, of which four were carried to term. Cabotegravir was discontinued after the discovery of pregnancy, but detectable levels of cabotegravir persisted throughout pregnancy. No birth defects occurred [41]. Currently, advice remains to not administer cabotegravir during pregnancy unless potential benefits to the patient outweigh the potential risks to the fetus [31].
3.5 Rilpivirine
3.5.1 Pharmacodynamics
Rilpivirine is a second-generation diarylpyrimidine NNRTI that binds to a hydrophobic pocket close to the active site of reverse transcriptase, causing conformational changes that inhibit the transcription of viral RNA to DNA [42]. When administered as monotherapy to treatment-naïve HIV-positive patients, 25–150 mg oral rilpivirine once daily decreased mean viral load by 1.2 log10 copies/mL over 7 days, without giving rise to any resistance mutations [43]. Pharmacodynamic analysis of both naïve and pretreated HIV patients estimated an EC50 of 65 ng/mL for 25 mg oral rilpivirine once daily (taken with 245 mg tenofovir disoproxil fumarate and 200 mg emtricitabine) [44].
3.6 Dosing Recommendations
As with cabotegravir, an oral lead-in phase of 25 mg rilpivirine daily for at least 28 days is administered to ensure tolerability. This is followed by an intramuscular loading dose of 900 mg, before administration can continue 4 weeks later at a maintenance dose of 600 mg once monthly. If dosing once every 2 months, a second 900 mg loading dose should be administered 4 weeks after the first dose, before treatment can proceed at a maintenance dose of 900 mg every 8 weeks. Population pharmacokinetic modelling has validated a similar oral bridging and re-initialization schedule as described for cabotegravir [45]. Rilpivirine injections are administered at the same time as cabotegravir injections (but into opposite-side gluteal muscles), and, likewise, doses can be administered between 7 days before and 7 days after the monthly dose is due. If a monthly injection is missed by ≤ 2 months, then continuation of 600 mg every 4 weeks may continue if the injection is administered as soon as possible. Again, if the injection is missed by more than 2 months, another 900 mg loading dose must be administered before proceeding onto 600 mg every 4 weeks. If an 8-weekly injection is missed by ≤ 2 months, treatment can be re-initialized as normal; however, if it is missed by more than 2 months, a 900 mg dose must be administered as soon as possible, again with a second 900 mg dose after 4 weeks, before treatment may continue as normal. In cases where patients know they will miss a dose by 7 days, they can take oral therapy of 25 mg once daily to replace up to two once-monthly or one once every 2 months visits [46].
3.7 Pharmacokinetics
3.7.1 Absorption
The key pharmacokinetic parameters are presented in Table 1. As with intramuscular administration of cabotegravir, the long-acting formation of rilpivirine displays flip-flop pharmacokinetics, with the absorption rate driving the t½ of intramuscular rilpivirine. Oral rilpivirine Tmax is 4 h, as opposed to 3–4 days for intramuscular rilpivirine, resulting in t½ of 45 h for oral rilpivirine and 13–28 weeks for intramuscular rilpivirine [13, 47]. Of note, rilpivirine half-life after oral administration has been shown to be lower in real-world cohorts compared with phase III trials (25 vs. 45 h) [48]. The absolute bioavailability of oral rilpivirine has not been studied. The effect of a high-fat meal increased the AUC of oral rilpivirine by approximately 45%, whereas administration with a high-protein drink decreased the AUC of oral rilpivirine by 50%, compared with administration with a solid meal. The product monograph therefore advises that rilpivirine must be taken with a meal [46]. Moreover, the bioavailability of oral rilpivirine is pH-dependent; coadministration with 20 mg omeprazole once daily decreased rilpivirine AUC by 40% (when rilpivirine was administered at a supratherapeutic dose of 150 mg) [49].
3.7.2 Distribution
Rilpivirine is also highly protein-bound (99.7%), primarily to albumin. The blood-to-plasma ratio is 0.7 [13]. The distribution of rilpivirine in the CSF was measured in HIV-infected individuals treated with a maintenance dose of intramuscular cabotegravir + rilpivirine administered every 4 or 8 weeks [35]. Median total rilpivirine CSF was 1.84 ng/mL (when administered every 4 weeks; n = 3) and 1.67 ng/mL (when administered every 8 weeks; n = 15), representing CSF/plasma ratios of 1.07% and 1.32%, respectively. CSF rilpivirine concentrations mostly exceeded the in vitro EC50 for wild-type HIV (i.e. 0.27 ng/mL) and all patients maintained CSF viral RNA below 50 copies/mL The CSF-to-plasma ratio of rilpivirine after intramuscular administration was comparable with the 1.4% ratio reported after oral administration of rilpivirine [50].
3.7.3 Metabolism and Elimination
Rilpivirine elimination occurs primarily via hepatic metabolism and CYP3A4 [46]. A mass balance study found that 85% of the administered oral rilpivirine dose was recovered in faeces, of which 26% was unchanged rilpivirine. No one major metabolite was identified, rather analysis revealed several oxidative metabolites. Only 6% of the dose was recovered in urine [51]. There are no reports of rilpivirine as a substrate of transporters, however in vitro analysis revealed rilpivirine can inhibit BCRP, P-gp, and OAT1B1/3, although this is not considered to be clinically significant [52]. Rilpivirine inhibits the renal transporter OCT2 involved in the tubular secretion of creatinine, thus leading to a slight increase in serum creatinine concentrations in individuals with normal renal function [53]. However, inhibition of this renal transporter did not alter the exposure of the antidiabetic metformin, which undergoes active renal elimination via OCT2 [46]. Thus, available data seem to indicate that rilpivirine has a low potential to cause renal DDIs.
Like long-acting cabotegravir, the long-acting intramuscular form of rilpivirine also displays a long pharmacokinetic tail. Rilpivirine was still detectable in samples collected 168 days after a single intramuscular injection of either 600 or 1200 mg [54]. In a pharmacokinetic study of a single dose of long-acting rilpivirine in healthy volunteers, rilpivirine was detectable in the last samples taken, 84 days after the dose. The elimination half-life was found to be 43, 39 and 38 days with 300, 600 and 1200 mg dosing, respectively [18]. The long elimination half-life raises the issue of drug resistance selection when stopping PrEP. The literature reports the case of one participant in the long-acting rilpivirine PrEP study (SSAT040 trial) who acquired HIV infection through unprotected intercourse 41 days following the administration of a single 300 mg intramuscular injection of rilpivirine. High levels of viral replication combined with low but detectable rilpivirine levels led to the selection of resistant virus [55]. This case highlights the need for a safe strategy to discontinue long-acting antiretroviral drugs.
3.8 Pharmacokinetics in Special Populations
3.8.1 Renal Impairment
Population pharmacokinetic analyses predict that mild renal impairment does not affect rilpivirine exposure [13]. Neither intramuscular nor oral rilpivirine have been studied in cases of moderate to severe renal impairment. The Cabenuva product monograph advises that no dosage adjustment is required in cases of renal impairment, but in patients with severe impairment, end-stage kidney disease, or on dialysis, increased monitoring is recommended [13].
3.8.2 Hepatic Impairment
The effect of mild and moderate hepatic impairment on oral rilpivirine exposure was studied in HIV negative volunteers. Mild impairment (Child–Pugh A) increased rilpivirine AUC by 47%, while moderate impairment (Child–Pugh B) increased rilpivirine AUC by 5%, compared with healthy controls [42]. No dosage adjustment of intramuscular rilpivirine is required in mild or moderate hepatic impairment, but extra caution is recommended when administering to patients with severe hepatic impairment [13].
3.8.3 Pregnancy
As stated in the previous section, long-acting cabotegravir/rilpivirine has not been studied in pregnant women and is therefore not recommended in this population [13]. Furthermore, several studies have shown decreased rilpivirine exposure during the third trimester of pregnancy, compared with postpartum, although this does not appear to lead to viral breakthrough [56,57,58].
4 Safety and Efficacy of Long-Acting Cabotegravir/Rilpivirine
The main efficacy endpoints determined by clinical trials are listed in Table 2. The phase IIb LATTE trial found that oral cabotegravir/rilpivirine was non-inferior to efavirenz plus two nucleos(t)ide reverse transcription inhibitors (NRTIs) in treatment-naïve HIV-positive patients [59]. LATTE-2 found that intramuscular cabotegravir/rilpivirine (both every 4 weeks and every 8 weeks) was non-inferior to oral cabotegravir with abacavir and lamivudine in treatment-naive HIV-positive patients [8]. In phase III trials, FLAIR showed that switching to intramuscular cabotegravir/rilpivirine was non-inferior to oral dolutegravir/abacavir/lamivudine in virally suppressed HIV-positive patients [60]. ATLAS demonstrated that switching to intramuscular cabotegravir/rilpivirine was non-inferior in virally suppressed HIV-positive patients compared with a variety of other treatments, including another integrase inhibitor, an NNRTI, a boosted protease inhibitor or unboosted atazanavir, together with two NRTIs [61]. Virologic suppression continued throughout the extension phase of ATLAS [62]. Furthermore, the phase IIIb ATLAS-2M trial demonstrated that long-acting cabotegravir/rilpivirine administered every 8 weeks was non-inferior to dosing every 4 weeks in virally suppressed HIV-positive patients [63]. Of interest, the extension phase of FLAIR found switching virally suppressed patients directly to intramuscular cabotegravir/rilpivirine from dolutegravir/abacavir/lamivudine (n = 111) was as efficacious as switching after oral lead-in (n = 121) and safety and tolerability were comparable between the two groups [64].
Resistance to cabotegravir has emerged in clinical trials, as is the case for other drugs in the integrase inhibitor class [30]. In phase III trials, three of four patients who developed virological failure in FLAIR had integrase inhibitor resistance, which reduced cabotegravir susceptibility by a factor of 5 [65]. During ATLAS, three patients experienced virological failure, one of whom developed the integrase inhibitor resistance mutation N155H, which reduced susceptibility to cabotegravir by a factor of 2.7 [61]. In vitro analysis demonstrated that cabotegravir has a higher genetic barrier to resistance than the first-line integrase inhibitor elvitegravir; however, it appears cabotegravir may have a lower genetic barrier than dolutegravir and bictegravir [66]. It is not known how this may manifest clinically, but care should be taken to ensure patient adherence to treatment and managing the pharmacokinetic tail after cessation of long-acting dosing [66].
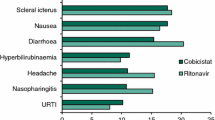
The most common adverse event seen with long-acting cabotegravir was injection site reactions, as well as headache and pyrexia. Twenty-five percent of injections resulted in an injection site reaction, the median duration of which was 3 days [60]. The following adverse reactions related to the trial regimen were seen in the ATLAS trial: injection site reaction (64%), pyrexia (4%), nausea (< 1%), diarrhoea (1%), headache (4%), and lipase increase (< 1%) [61]. Injection site reactions (most commonly pain, 91%) were also reported in the ECLAIR trial, which studied long-acting cabotegravir once every 12 weeks as PrEP for healthy men who have sex with men [16]. Despite these adverse effects, questionnaires about the participants’ experiences revealed they found them worthwhile, with many reporting they wished to continue with injections when the drug came to the market, and that they would recommend the treatment to others. Most participants experienced no difficulty attending the appointments every 12 weekly for dosing and preferred the intramuscular phase over the oral phase [67]. Patients in ATLAS-2M expressed a preference for treatment every 8 weeks over treatment every 4 weeks [63].
5 Drug–Drug Interactions (DDIs)
As mentioned previously, cabotegravir and rilpivirine have a low potential to cause DDIs as these antiretrovirals do not inhibit or induce CYPs, UGTs, or drug transporters to a clinically significant extent. Furthermore, there is no evidence that cabotegravir and rilpivirine cause clinically significant DDIs via protein displacement. However, oral cabotegravir and rilpivirine can be subject to DDIs at the intestinal and hepatic levels. Results from DDI studies between oral cabotegravir or rilpivirine and co-medications are summarized in Figs. 2 and 3. The interaction profiles of oral and intramuscular cabotegravir/rilpivirine, considering 725 co-medications listed in the Liverpool HIV website [68], are presented in Fig. 4. Rilpivirine has a higher potential for DDIs compared with cabotegravir, which is explained by not only their different metabolic pathways but also the fact that rilpivirine is associated with a risk of QT interval prolongation. Supratherapeutic doses of 75 mg and 300 mg oral daily rilpivirine resulted in QTc interval differences of 10.7 msec (95% upper confidence bound 15.3) and 23.3 msec (28.4), respectively [46]. The label recommends using rilpivirine with caution, particularly when combined with drugs with a known risk of Torsade de pointes [46]. Conversely, cabotegravir, at a supratherapeutic dose, did not affect cardiac repolarization [69].

a Impact of oral cabotegravir on co-medication pharmacokinetics from drug–drug interaction crossover studies in healthy volunteers. 1. Period 1: Oral midazolam 3 mg daily for 10 days. Period 2: Oral midazolam 3 mg + cabotegravir 30 mg daily for 14 days (n = 12). 2. Period 1: Levonorgestrel/ethinylestradiol 0.15/0.03 mg daily for 10 days. Period 2: Levonorgestrel/ethinylestradiol 0.15/0.03 mg + cabotegravir 30 mg daily for 10 days (n = 20). 3. Period 1: Cabotegravir 30 mg daily for 12 days. Period 2: Rilpivirine 25 mg daily for 12 days. Period 3: Cabotegravir 30 mg + rilpivirine 25 mg daily for 12 days (n = 11). b Impact of co-medications on oral cabotegravir pharmacokinetics from drug–drug interaction crossover studies in healthy volunteers. 1. Period 1: Cabotegravir 30 mg daily for 10 days. Period 2: Cabotegravir 30 mg + etravirine 200 mg daily for 14 days (n = 12) 2. Period 1: Cabotegravir 30 mg daily for 12 days. Period 2: Rilpivirine 30 mg daily for 12 days. Period 3: Cabotegravir 30 mg + rilpivirine 25 mg daily for 12 days (n = 11). 3. Cabotegravir 30 mg single-dose day 1. Rifampicin 600 mg daily days 8–28 with cabotegravir 30 mg single dose on day 21 (n = 15). 4. Period 1: Cabotegravir 30 mg daily for 14 days. Period 2: Cabotegravir 30 mg + rifabutin 300 mg for 14 days (n = 15). Data are expressed as area under the curve (squares) and trough plasma concentration (triangles) geometric mean ratios + 90% confidence intervals for cabotegravir with and without co-medication. The bioequivalence margin (0.8–1.25) is indicated by dashed vertical lines. EE ethinylestradiol, LNG levonorgestrel, sd single dose [34, 70, 71, 85, 86, 94]


a Impact of oral rilpivirine on co-medication pharmacokinetics from drug–drug interaction crossover studies in healthy volunteers. 1. Period 1: Atorvastatin 40 mg daily for 4 days. Period 2: Rilpivirine 150 mg daily for 15 days + atorvastatin 40 mg daily days 12–15 (n = 16). 2. Period 1: Rilpivirine 150 mg daily for 11 days. Period 2: Darunavir/ritonavir 800/100 mg daily for 22 days + rilpivirine 150 mg daily day 12–22 (n = 16). 3a. Period 1: Cabotegravir 30 mg daily for 12 days. Period 2: Rilpivirine 25 mg daily for 11 or 12 days. Period 3: Cabotegravir 30 mg daily + rilpivirine 25 mg daily, for 12 days (n = 11). 3b. Period 1: Dolutegravir 50 mg daily for 5 days. Period 2: Rilpivirine 25 mg daily for 11 or 12 days. Period 3: Dolutegravir 50 daily + rilpivirine 25 mg daily for 5 days (n = 16). 4. Period 1: Grazoprevir/elbasvir 200/50 mg daily for 8 days. Period 2: Rilpivirine 25 mg daily for 11 days. Period 3: Grazoprevir/elbasvir 200/50 mg daily + rilpivirine 25 mg daily for 9 days (n = 20). 5. Period 1: Rilpivirine 150 mg daily for 8 days. Period 2: Tenofovir disoproxil fumarate 300 mg daily for 16 days + rilpivirine 150 mg daily either days 1–8 or days 9–16 (n = 15). 6. Period 1: Norethindrone/ethinylestradiol 1/0.035 mg in three 21-day cycles + rilpivirine 25 mg daily on days 1–15 of the third cycle (n = 18). 7. Period 1: Rilpivirine 150 mg daily for 11 days. Period 2: Ketoconazole 400 mg daily for 22 days + rilpivirine 150 mg daily days 12–22. 8. Period 1: Rilpivirine 150 mg daily for 10 days. Period 2: Lopinavir/ritonavir 400/100 mg twice daily for 20 days + rilpivirine 150 mg days 11–20 (n = 14). 9. Period 1: Rilpivirine 150 mg daily for 11 days. Period 2: Omeprazole 20 mg daily for 22 days + rilpivirine 150 mg on days 12–22 (n = 16). 10. Period 1: Paracetamol 500 mg, single dose. Period 2: Rilpivirine 150 mg daily for 12 days + paracetamol 500 mg single dose on day 11 (n = 16). 11. Period 1: Rilpivirine 150 mg daily for 11 days. Period 2: Rifabutin 300 mg daily for 11 days. Period 3: Rilpivirine 150 mg + rifabutin 300 mg daily for 11 days (n = 18). 12. Period 1: Sildenafil 50 mg, single dose. Period 2: Rilpivirine 75 mg daily for 12 days + sildenafil 50 mg single dose on day 12 (n = 16). 13. Period 1: Sofosbuvir/velpatasvir 400/100 mg daily for 8 days. Period 2: Emtricitabine/rilpivirine/tenofovir disoproxil fumarate 200/25/300 mg + sofosbuvir/velpatasvir 400/100 mg daily for 8 days (n = 24). 14. Period 1: Rilpivirine 25 mg daily for 14 days. Period 2: Tenofovir alafenamide fumarate 25 mg daily for 14 days. Period 3: Rilpivirine 25 mg + tenofovir alafenamide fumarate 25 mg daily, for 14 days (n = 17). 15. Stable methadone dose (60–150 mg daily) for 25 days + rilpivirine 25 mg daily on days 15–25 (n = 13). (b) Impact of co-medications on oral rilpivirine pharmacokinetics from drug–drug interaction crossover studies in healthy volunteers. 1. Period 1: Atorvastatin 40 mg daily for 4 days. Period 2: Rilpivirine 150 mg daily for 15 days + atorvastatin 40 mg daily days 12–15 (n = 16). 2. Period 1: Rilpivirine 150 mg daily for 11 days. Period 2: Darunavir/ritonavir 800/100 mg daily for 22 days + rilpivirine 150 mg daily days 12–22 (n = 16). 3a. Period 1: Cabotegravir 30 mg daily for 12 days. Period 2: Rilpivirine 25 mg daily for 11 or 12 days. Period 3: Cabotegravir 30 mg daily + rilpivirine 25 mg daily for 12 days (n = 11). 3b. Period 1: Dolutegravir 50 mg daily for 5 days. Period 2: Rilpivirine 25 mg daily for 11 or 12 days. Period 3: Dolutegravir 50 daily + rilpivirine 25 mg daily for 5 days (n = 16). 4. Period 1: Grazoprevir/elbasvir 200/50 mg daily for 8 days. Period 2: Rilpivirine 25 mg daily for 11 days. Period 3: Grazoprevir/elbasvir 200/50 mg daily + rilpivirine 25 mg daily for 9 days (n = 20). 5. Period 1: Rilpivirine 150 mg daily for 8 days. Period 2: Tenofovir disoproxil fumarate 300 mg daily for 16 days + rilpivirine 150 mg daily either days 1–8 or days 9–16 (n = 15). 6. Period 1: Rilpivirine 150 mg single dose. Period 2: Rilpivirine 150 mg single dose 2 h after famotidine 40 mg single dose (n = 24). 7. Period 1: Rilpivirine 150 mg daily. Period 2: Ketoconazole 400 mg daily for 22 days + rilpivirine 150 mg daily days 12–22 (n = 14). 8. Period 1: Rilpivirine 150 mg daily for 10 days. Period 2: Lopinavir/ritonavir 400/100 mg twice daily for 20 days + rilpivirine 150 mg days 11–20 (n = 14). 9. Period 1: Rilpivirine 150 mg daily for 11 days. Period 2: Omeprazole 20 mg daily for 22 days + rilpivirine 150 mg on days 12–22 (n = 16). 10. Period 1: Rilpivirine 150 mg daily for 11 days. Period 2: Rifabutin 300 mg daily for 11 days. Period 3: Rilpivirine 150 mg + rifabutin 300 mg daily for 11 days (n = 18). 11. Period 1: Rilpivirine 150 mg daily. Period 2: Rilpivirine 150 mg daily + rifampicin 600 mg daily (n = 16). 12. Period 1: Sildenafil 50 mg, single dose. Period 2: rilpivirine 75 mg daily for 12 days + sildenafil 50 mg single dose on day 12 (n = 16). 13. Period 1: Sofosbuvir/velpatasvir 400/100 mg daily for 8 days. Period 2: Emtricitabine/rilpivirine/tenofovir disoproxil fumarate 200/25/300 mg + sofosbuvir/velpatasvir 400/100 mg daily for 8 days (n = 24). Data are presented as area under the curve (squares) and trough plasma concentration (triangles) geometric mean ratios + 90% confidence intervals for rilpivirine with and without co-medication. The bioequivalence margin (0.8–1.25) is indicated by dashed vertical lines. CAB cabotegravir, DRV/RTV darunavir/ritonavir, DTG dolutegravir, GS-331007 metabolite of sofosbuvir, GZR/EBR grazoprevir/elbasvir, LPV/RTV lopinavir/ritonavir, NOR/EE norgestrel/ethinylestradiol, RPV rilpivirine, sd single dose, SOF/VEL sofosbuvir/velpatasvir, TAF tenofovir alafenamide, TDF tenofovir disoproxil fumarate [49, 71,72,73,74,75,76,77,78,79,80,81,82,83, 88, 95, 96].

DDI profiles of oral cabotegravir and rilpivirine or intramuscular cabotegravir plus rilpivirine when considering non-HIV co-medications (n = 725) listed in the University of Liverpool HIV drug interaction website [68]. DDI drug–drug interaction
5.1 Impact of Cabotegravir and Rilpivirine on the Pharmacokinetics of Co-medications
Clinical DDI studies have demonstrated that oral cabotegravir does not have a clinically meaningful effect on the pharmacokinetics of the CYP3A4 probe substrate midazolam. The coadministration of midazolam 3 mg once daily with cabotegravir 30 mg once daily increased midazolam AUC and Cmax by 10% and 9%, respectively [34]. Cabotegravir minimally altered the pharmacokinetics of oral contraceptives [70] and rilpivirine [71], supporting in vitro findings that it is unlikely to impact the pharmacokinetics of other drugs via drug-metabolizing enzymes or transporter pathways (Fig. 2a).
Similarly, rilpivirine is devoid of inhibitory or inducing effects on drug-metabolizing enzymes or transporters and was therefore shown to minimally impact the pharmacokinetics of atorvastatin [72], paracetamol [73], sildenafil [74], oral contraceptives [75], omeprazole [49], rifabutin [76], cabotegravir [71], dolutegravir [71], lopinavir/ritonavir [77], darunavir/ritonavir [78], tenofovir disoproxil fumarate [79], tenofovir alafenamide [80], or the hepatitis C drugs grazoprevir and elbasvir [81] or sofosbuvir and velpatasvir [82] (Fig. 3a).
The coadministration of oral rilpivirine 25 mg once daily in patients receiving an individualized methadone dose (60–150 mg once daily) decreased the AUC of both R- and S-methadone by 16%. No a priori dose adjustment of methadone is needed, however clinical monitoring is recommended as methadone maintenance therapy may need to be adjusted in some patients [83]. Furthermore, caution is recommended as both drugs have the potential to prolong the QT interval [84].
Altogether, these DDI studies indicate that oral cabotegravir and rilpivirine have a low potential to cause pharmacokinetic interactions, which also holds true when these drugs are administered intramuscularly.
5.2 Impact of Co-medications on Cabotegravir and Rilpivirine Pharmacokinetics
The strong inducer rifampicin has been shown to reduce oral cabotegravir exposure by 59% [85], which may result in loss of virologic response, therefore coadministration with strong inducers (e.g. carbamazepine, oxcarbazepine, phenobarbital, phenytoin, rifampicin, rifapentine, or St John’s Wort) is contraindicated (Fig. 2b). Of interest, coadministration with the moderate inducer rifabutin was shown to have a more modest effect on oral cabotegravir exposure, with a decrease in Cmax, AUC and Ctrough of 17%, 23% and 26%, respectively [86]. This reduction is not clinically meaningful and therefore oral cabotegravir can be coadministered with moderate inducers. As for other inducers (see Sect. 2), rifabutin is expected to cause a similar DDI magnitude with both the intramuscular and oral forms of cabotegravir. Thus, coadministration with moderate inducers may be possible with intramuscular cabotegravir (used alone for HIV prevention), although further data are warranted, particularly in special populations. Since moderate inducers significantly alter rilpivirine pharmacokinetics, their use is contraindicated with intramuscular cabotegravir when combined with intramuscular rilpivirine for the treatment of HIV [68].
The effect of hormonal contraceptive use on intramuscular cabotegravir pharmacokinetics (maintenance dose) was evaluated in the framework of the HPTN077 study [87]. Geometric mean ratios (90% confidence interval) [intramuscular cabotegravir plus contraceptive vs. intramuscular cabotegravir alone] for cabotegravir Cmax, AUC and Ctrough were 0.75 (0.59–0.93), 0.81 (0.65–1.01) and 0.91 (0.72–1.15), respectively. Although the use of contraceptives caused a statistically significant reduction in cabotegravir Cmax concentration, no significant differences were observed for the other pharmacokinetic parameters, suggesting that this DDI is unlikely to be clinically meaningful. However, further studies are needed to fully characterize this interaction, particularly with gender-affirming hormonal therapies that use higher doses of estradiol.
As mentioned previously, rilpivirine is more susceptible to DDIs as it is primarily metabolized by CYP3A4 and can be impacted by CYP3A4 inhibitors or inducers (Fig. 3b). For instance, the coadministration of oral rilpivirine at a dose higher than the licensed dose (i.e. 150 mg once daily) and darunavir/ritonavir (800/100 mg once daily) increased rilpivirine Cmax, AUC and Ctrough by 79%, 130% and 178%, respectively [78]. However, given the good tolerability of rilpivirine, strong CYP3A4 inhibitors can be coadministered without the need for a dose adjustment, a recommendation that also applies to intramuscular rilpivirine [68].
Conversely, coadministration with the strong CYP3A4 inducer rifampicin decreased oral rilpivirine Cmax, AUC and Ctrough by 69%, 80% and 89%, respectively [88]. Due to the profound reduction in rilpivirine pharmacokinetics, rifampicin, along with other potent CYP3A4 inducers (e.g. anticonvulsants or St John’s wort), is contraindicated with both oral and intramuscular rilpivirine. The coadministration of oral rilpivirine (25 mg once daily) with the moderate CYP3A4 inducer rifabutin (300 mg once daily) reduced rilpivirine Cmax, AUC and Ctrough by 31%, 42% and 48%, respectively [76]. DDIs with moderate inducers can be managed by increasing the oral rilpivirine dose to 50 mg once daily (Fig. 3b). However, there are no recommendations on how to manage the DDI with moderate inducers with intramuscular administration of rilpivirine and therefore moderate inducers are contraindicated with the injectable form [68].
Besides moderate inducers, the main differences in the DDI profile of oral and intramuscular rilpivirine relate to proton pump inhibitors, H2 blockers or antacids, which have the potential to reduce rilpivirine absorption, whereas this DDI is prevented with the intramuscular form. Similarly, divalent cations contained in antacids or mineral supplements can reduce the absorption of oral cabotegravir due to chelation in the gastrointestinal tract, whereas this DDI is avoided with the intramuscular form.
5.3 Physiologically Based Pharmacokinetic Modelling to Simulate DDIs
Given the long half-life of intramuscular cabotegravir and rilpivirine, DDI studies cannot be easily conducted. Going forward, it is almost certain that data from oral DDI studies with cabotegravir and rilpivirine will be used to inform PBPK modelling. PBPK modelling is a bottom-up approach that predicts drug pharmacokinetics using anatomical, physiological and biological data for the population of interest and in vitro data describing the absorption, distribution, metabolism and elimination of the drug of interest [89]. PBPK modelling enables simulation of clinical scenarios and therefore constitutes a useful approach to predict DDIs with intramuscular cabotegravir and rilpivirine. Rifampicin was shown to reduce the AUC of oral cabotegravir by 59% [85] and oral rilpivirine by 80% [46]. PBPK models validated against the observed data for oral cabotegravir, rilpivirine and rifampicin predicted a reduction in AUC of 41% for intramuscular cabotegravir and 82% for intramuscular rilpivirine in the presence of rifampicin for the first maintenance dose of cabotegravir and rilpivirine [25]. Cabotegravir AUC was shown to be more profoundly reduced when simulating steady-state conditions. Thus, PBPK modelling supports the recommendation that rifampicin or any other strong inducers should also be avoided with intramuscular cabotegravir and rilpivirine due to the substantial reduction in their exposures, which may lead to treatment failure. Finally, PBPK models were able to show that the UGT1A1 inhibitor atazanavir and the UGT1A9 inhibitor mefenamic acid have a minimal effect, with 11% and 10% increases, respectively, in cabotegravir AUC [90]. This effect does not warrant a dose adjustment of cabotegravir in the presence of UGT inhibitors. This finding is also supported by the results of another study showing that genetic variations characterized by a reduced function of UGT1A1 resulted in a modest increase in cabotegravir AUC after oral (28–50%) or intramuscular (16–24%) administration [91].
6 Conclusions
The licensing of cabotegravir/rilpivirine as the first-ever long-acting regimen for the treatment of HIV in virologically supressed patients is an exciting advance for antiretroviral therapy. Reduced pill burden coupled with the long time between dose administrations (monthly or bimonthly) [92], affords PLWH an easy and well-tolerated treatment option. The possibility for long-acting cabotegravir/rilpivirine to also be used as PrEP promises new avenues, as adherence has been shown to be critical for optimal protection against HIV acquisition [93].
The pharmacokinetics of intramuscular administration of cabotegravir/rilpivirine are driven by the slow absorption of drug from the muscle depot, resulting in elimination half-lives measured in weeks rather than hours [13, 46]. While these pharmacokinetics are optimal for long-acting medication, it is important to consider the long pharmacokinetic tail of intramuscular cabotegravir/rilpivirine, which, if the patient is not switched to other ARV therapy, could lead to emergent treatment-resistant HIV strains.
The intramuscular administration of long-acting cabotegravir/rilpivirine effectively bypasses gut-based drug interactions, and the differing DDI profiles between the two routes of administration should be taken into account when administering these drugs. In particular, DDIs resulting from the chelation of cabotegravir by divalent cations in the stomach, or DDIs with rilpivirine due to pH changes, are avoided. Likewise, the effect on metabolizing enzymes is restricted to liver enzymes only, rather than in the gut. Both the oral and intramuscular formulations of cabotegravir and rilpivirine are included in the Liverpool drug interaction website (http://www.hiv-druginteractions.org), where interactions can be checked against more than 700 co-medications. Given that DDI studies have only been conducted with the oral form of cabotegravir and rilpivirine, this leads to inevitable questions on the relevance and potential management of DDIs with the intramuscular form during treatment, following cessation of treatment (i.e. the tail) and in the case of missed injection visits. Furthermore, differences in the pharmacokinetics after intramuscular administration have been observed in individuals with a high BMI or in females, during clinical trials. The clinical relevance of certain DDIs (notably moderate inducers administered with cabotegravir used as PrEP) have not been evaluated in these special populations. Other gaps relate to the effect of variation in the injection site or injection methods.
References
Gueler A, Moser A, Calmy A, Gunthard HF, Bernasconi E, Furrer H, et al. Life expectancy in HIV-positive Spersons in Switzerland: matched comparison with general population. AIDS. 2017;31(3):427–36.
Samji H, Cescon A, Hogg RS, Modur SP, Althoff KN, Buchacz K, et al. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS ONE. 2013;8(12):e81355.
Trickey A, May MT, Vehreschild J-J, Obel N, Gill MJ, Crane HM, et al. Survival of HIV-positive patients starting antiretroviral therapy between 1996 and 2013: a collaborative analysis of cohort studies. Lancet HIV. 2017;4(8):e349–56.
Cahn P, Andrade-Villanueva J, Arribas JR, Gatell JM, Lama JR, Norton M, et al. Dual therapy with lopinavir and ritonavir plus lamivudine versus triple therapy with lopinavir and ritonavir plus two nucleoside reverse transcriptase inhibitors in antiretroviral-therapy-naive adults with HIV-1 infection: 48 week results of the randomised, open label, non-inferiority GARDEL trial. Lancet Infect Dis. 2014;14(7):572–80.
Cahn P, Madero JS, Arribas JR, Antinori A, Ortiz R, Clarke AE, et al. Dolutegravir plus lamivudine versus dolutegravir plus tenofovir disoproxil fumarate and emtricitabine in antiretroviral-naive adults with HIV-1 infection (GEMINI-1 and GEMINI-2): week 48 results from two multicentre, double-blind, randomised, non-inferiority, phase 3 trials. Lancet. 2019;393(10167):143–55.
Orkin C, Llibre JM, Gallien S, Antinori A, Behrens G, Carr A. Nucleoside reverse transcriptase inhibitor-reducing strategies in HIV treatment: assessing the evidence. HIV Med. 2018;19(1):18–32.
Spreen WR, Margolis DA, Pottage JC Jr. Long-acting injectable antiretrovirals for HIV treatment and prevention. Curr Opin HIV AIDS. 2013;8(6):565–71.
Margolis DA, Gonzalez-Garcia J, Stellbrink HJ, Eron JJ, Yazdanpanah Y, Podzamczer D, et al. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet. 2017;390(10101):1499–510.
Alexander K. Dosage forms and their routes of administration. In: Hacker M, Messer W, Bachmann K, editors. Pharmacology: principles and practice. Academic Press; 2009. pp. 9–29.
Yanez JA, Remsberg CM, Sayre CL, Forrest ML, Davies NM. Flip-flop pharmacokineticsdelivering a reversal of disposition: challenges and opportunities during drug development. Ther Deliv. 2011;2(5):643–72.
Yin J, Collier AC, Barr AM, Honer WG, Procyshyn RM. Paliperidone palmitate long-acting injectable given intramuscularly in the deltoid versus the gluteal muscle: are they therapeutically equivalent? J Clin Psychopharmacol. 2015;35(4):447–9.
Chan VO, Colville J, Persaud T, Buckley O, Hamilton S, Torreggiani WC. Intramuscular injections into the buttocks: are they truly intramuscular? Eur J Radiol. 2006;58(3):480–4.
Vocabria/Cabenuva product monograph. Research Triangle Park, NC: ViiV Healthcare; 2020.
Cabenuva US product information. Research Triangle Park, NC: ViiV Healthcare; 2021.
Landovitz RJ, Li S, Eron JJ Jr, Grinsztejn B, Dawood H, Liu AY, et al. Tail-phase safety, tolerability, and pharmacokinetics of long-acting injectable cabotegravir in HIV-uninfected adults: a secondary analysis of the HPTN 077 trial. Lancet HIV. 2020;7(7):e472–81.
Markowitz M, Frank I, Grant RM, Mayer KH, Elion R, Goldstein D, et al. Safety and tolerability of long-acting cabotegravir injections in HIV-uninfected men (ECLAIR): a multicentre, double-blind, randomised, placebo-controlled, phase 2a trial. Lancet HIV. 2017;4(8):e331–40.
Patel P, Ford SL, Crauwels H, Han K, Rossenu S, Neyens M, et al. Pharmacokinetics of cabotegravir (CAB) and rilpivirine (RPV) long-acting (LA) injectables in HIV-infected individuals through 48 weeks in the FLAIR and ATLAS phase 3 studies [abstract no. 2495]. ID Week: 2–6 October 2019; Washington.
Jackson AG, Else LJ, Mesquita PM, Egan D, Back DJ, Karolia Z, et al. A compartmental pharmacokinetic evaluation of long-acting rilpivirine in HIV-negative volunteers for pre-exposure prophylaxis. Clin Pharmacol Ther. 2014;96(3):314–23.
Mayer PR, Brown CH, Carter RA, Welty TE, Millikan MD, Eberhard NK. Intramuscular tobramycin pharmacokinetics in geriatric patients. Drug Intell Clin Pharm. 1986;20(7–8):611–5.
Crauwels H, van Heeswijk RP, Stevens M, Buelens A, Vanveggel S, Boven K, et al. Clinical perspective on drug–drug interactions with the non-nucleoside reverse transcriptase inhibitor rilpivirine. AIDS Rev. 2013;15(2):87–101.
Kiser JJ, Bumpass JB, Meditz AL, Anderson PL, Bushman L, Ray M, et al. Effect of antacids on the pharmacokinetics of raltegravir in human immunodeficiency virus-seronegative volunteers. Antimicrob Agents Chemother. 2010;54(12):4999–5003.
Foca M, Yogev R, Wiznia A, Hazra R, Jean-Philippe P, Graham B, et al. Rilpivirine pharmacokinetics without and with darunavir/ritonavir once daily in adolescents and young adults. Pediatr Infect Dis J. 2016;35(9):e271–4.
Scarsi KK, Darin KM, Nakalema S, Back DJ, Byakika-Kibwika P, Else LJ, et al. Unintended pregnancies observed with combined use of the levonorgestrel contraceptive implant and efavirenz-based antiretroviral therapy: a three-arm pharmacokinetic evaluation over 48 weeks. Clin Infect Dis. 2016;62(6):675–82.
Carten ML, Kiser JJ, Kwara A, Mawhinney S, Cu-Uvin S. Pharmacokinetic interactions between the hormonal emergency contraception, levonorgestrel (Plan B), and Efavirenz. Infect Dis Obstet Gynecol. 2012;2012:137192.
Rajoli RKR, Curley P, Chiong J, Back D, Flexner C, Owen A, et al. Predicting drug-drug interactions between rifampicin and long-acting cabotegravir and rilpivirine using physiologically based pharmacokinetic modeling. J Infect Dis. 2019;219(11):1735–42.
Trezza C, Ford SL, Spreen W, Pan R, Piscitelli S. Formulation and pharmacology of long-acting cabotegravir. Curr Opin HIV AIDS. 2015;10(4):239–45.
Yoshinaga T, Kobayashi M, Seki T, Miki S, Wakasa-Morimoto C, Suyama-Kagitani A, et al. Antiviral characteristics of GSK1265744, an HIV integrase inhibitor dosed orally or by long-acting injection. Antimicrob Agents Chemother. 2015;59(1):397–406.
Chen AY, Adamek RN, Dick BL, Credille CV, Morrison CN, Cohen SM. Targeting metalloenzymes for therapeutic intervention. Chem Rev. 2019;119(2):1323–455.
Spreen W, Min S, Ford SL, Chen S, Lou Y, Bomar M, et al. Pharmacokinetics, Safety, and monotherapy antiviral activity of GSK1265744, an HIV integrase strand transfer inhibitor. HIV Clin Trials. 2013;14(5):192–203.
Scarsi KK, Havens JP, Podany AT, Avedissian SN, Fletcher CV. HIV-1 Integrase inhibitors: a comparative review of efficacy and safety. Drugs. 2020;80(16):1649–76.
Vocabria summary product characteristics. Research Triangle Park, NC: ViiV Healthcare; 2021.
Han K, Baker M, Patel P, Margolis D, Spreen W, Moore K, et al. Population pharmacokinetic (PPK) modeling and simulation of long-acting (LA) cabotegravir (CAB) to inform strategies following dosing interruptions in HIV-1 infected subjects [abstract no. 1532]. ID Week; 2–6 October 2019; Washington.
Patel P, Ford SL, Lou Y, Bakshi K, Tenorio AR, Zhang Z, et al. Effect of a high-fat meal on the pharmacokinetics of the HIV integrase inhibitor cabotegravir. Clin Pharmacol Drug Dev. 2019;8(4):443–8.
Reese MJ, Bowers GD, Humphreys JE, Gould EP, Ford SL, Webster LO, et al. Drug interaction profile of the HIV integrase inhibitor cabotegravir: assessment from in vitro studies and a clinical investigation with midazolam. Xenobiotica. 2016;46(5):445–56.
Letendre SL, Mills A, Hagins D, Swindells S, Felizarta F, Devente J, et al. Pharmacokinetics and antiviral activity of cabotegravir and rilpivirine in cerebrospinal fluid following long-acting injectable administration in HIV-infected adults. J Antimicrob Chemother. 2020;75(3):648–55.
Bowers GD, Culp A, Reese MJ, Tabolt G, Moss L, Piscitelli S, et al. Disposition and metabolism of cabotegravir: a comparison of biotransformation and excretion between different species and routes of administration in humans. Xenobiotica. 2016;46(2):147–62.
Taskar KS, Patel A, Cozens SJ, Ford S, Spreen W, Baker M, et al. Prediction of renal OAT1 and OAT3 inhibition by cabotegravir using PBPK modelling [abstract no. 470]. Conference on Retrovirus and Opportunistic Infections; 4-7 March 2019: Seattle, WA.
McCluskey SM, Siedner MJ, Marconi VC. Management of virologic failure and HIV drug resistance. Infect Dis Clin N Am. 2019;33(3):707–42.
Parasrampuria R, Ford SL, Lou Y, Fu C, Bakshi KK, Tenorio AR, et al. A Phase I study to evaluate the pharmacokinetics and safety of cabotegravir in adults with severe renal impairment and healthy matched control participants. Clin Pharmacol Drug Dev. 2019;8(5):674–81.
Shaik JSB, Ford SL, Lou Y, Zhang Z, Bakshi KK, Tenorio AR, et al. A phase 1 study to evaluate the pharmacokinetics and safety of cabotegravir in patients with hepatic impairment and healthy matched controls. Clin Pharmacol Drug Dev. 2019;8(5):664–73.
Patel P, Thiagarajah S, Ford S, Margolis DA, Romach BH, Baker M, et al. Cabotegravir pharmacokinetic tail in pregnacy and neonatal outcomes [abstract no. 775]. Conference on Retroviruses and Opportunistic Infections; 8–11 March 2020: Boston, MA.
Sanford M. Rilpivirine. Drugs. 2012;72(4):525–41.
Goebel F, Yakovlev A, Pozniak AL, Vinogradova E, Boogaerts G, Hoetelmans R, et al. Short-term antiviral activity of TMC278—a novel NNRTI—in treatment-naive HIV-1-infected subjects. AIDS. 2006;20(13):1721–6.
Néant N, Solas C, Bouazza N, Lê MP, Yazdanpanah Y, Dhiver C, et al. Concentration-response model of rilpivirine in a cohort of HIV-1-infected naive and pre-treated patients. J Antimicrob Chemother. 2019;74(7):1992–2002.
Rossenu S, Neyens M, van Solingen-Ristea R, Baugh B, Crauwels H. Population pharmacokinetic (PopPK) modeling and simulation of monthly intramuscular (IM) long-acting rilpivirine (RPV LA) to inform strategies following dosing interruptions in HIV-infected subjects [abstract no. PEB0264]. 23rd AIDS International Conference; 2020: Virtual.
Rekambys summary of product characteristics. Research Triangle Park, NC: ViiV Healthcare; 2021.
Hoetelmans R, van Heeswijk R, Kestens D, Marien K, Stevens M, Peeters M, et al. Effect of food and multiple-dose pharmacokinetics of TMC278 as an oral tablet formulation: healthy volunteers [TuPe3.1B10]. International AIDS Conference; 24–27 July 2005. Rio de Janeiro.
Neant N, Gattacceca F, Le MP, Yazdanpanah Y, Dhiver C, Bregigeon S, et al. Population pharmacokinetics of rilpivirine in HIV-1-infected patients treated with the single-tablet regimen rilpivirine/tenofovir/emtricitabine. Eur J Clin Pharmacol. 2018;74(4):473–81.
Crauwels H, van Heeswijk R, Kestens D, Stevens M, Buelens A, Boven K, et al. The pharmacokinetic interaction between omeprazole and TMC278, a next generation non-nucleoside reverse transcriptase inhibitor [abstract no. P239]. 9th International Congress on Drug Therapy in HIV Infection; 9–13 November 2008: Glasgow.
Mora-Peris B, Watson V, Vera JH, Weston R, Waldman AD, Kaye S, et al. Rilpivirine exposure in plasma and sanctuary site compartments after switching from nevirapine-containing combined antiretroviral therapy. J Antimicrob Chemother. 2014;69(6):1642–7.
Lachau-Durand S, Mamidi NVS, Cuyckens S, Michlova V, Mannens G, Raoof A. Absorption, metabolism and excretion of TMC278, an NNRTI, after a single oral dose of 150 mg in healthy male volunteers [abstract no. PE7.1/3]. 12th European AIDS Conference; 11–14 November 2009: Cologne.
Weiss J, Haefeli WE. Potential of the novel antiretroviral drug rilpivirine to modulate the expression and function of drug transporters and drug-metabolising enzymes in vitro. Int J Antimicrob Agents. 2013;41(5):484–7.
Maggi P, Montinaro V, Mussini C, Di Biagio A, Bellagamba R, Bonfanti P, et al. Novel antiretroviral drugs and renal function monitoring of HIV patients. AIDS Rev. 2014;16(3):144–51.
McGowan I, Dezzutti CS, Siegel A, Engstrom J, Nikiforov A, Duffill K, et al. Long-acting rilpivirine as potential pre-exposure prophylaxis for HIV-1 prevention (the MWRI-01 study): an open-label, phase 1, compartmental, pharmacokinetic and pharmacodynamic assessment. Lancet HIV. 2016;3(12):e569–78.
Penrose KJ, Parikh UM, Hamanishi KA, Else L, Back D, Boffito M, et al. Selection of rilpivirine-resistant HIV-1 in a seroconverter from the SSAT 040 trial who received the 300 mg dose of long-acting rilpivirine (TMC278LA). J Infect Dis. 2016;13(6):1013–7.
Tran AH, Best BM, Stek A, Wang J, Capparelli EV, Burchett SK, et al. Pharmacokinetics of rilpivirine in HIV-infected pregnant women. J Acquir Immune Defic Syndr. 2016;72(3):289–96.
Osiyemi O, Yasin S, Zorrilla C, Bicer C, Hillewaert V, Brown K, et al. Pharmacokinetics, antiviral activity, and safety of rilpivirine in pregnant women with HIV-1 infection: results of a phase 3b, multicenter, open-label study. Infect Dis Ther. 2018;7(1):147–59.
Eke AC, Chakhtoura N, Kashuba A, Best BM, Sykes C, Wang J, et al. Rilpivirine plasma and cervicovaginal concentrations in women during pregnancy and postpartum. J Acquir Immune Defic Syndr. 2018;78(3):308–13.
Margolis DA, Brinson CC, Smith GHR, de Vente J, Hagins DP, Eron JJ, et al. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral-naive adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial. Lancet Infect Dis. 2015;15(10):1145–55.
Orkin C, Oka S, Philibert P, Brinson C, Bassa A, Gusev D, et al. Long-acting cabotegravir + rilpivirine for HIV treatment: FLAIR week 96 results [abstract no. 482]. Conference on Retroviruses and Opportunistic Infections; 8–11 March 2020: Boston, MA.
Swindells S, Andrade-Villanueva JF, Richmond GJ, Rizzardini G, Baumgarten A, Masiá M, et al. Long-acting cabotegravir and rilpivirine for maintenance of HIV-1 suppression. N Engl J Med. 2020;382(12):1112–23.
Swindells S, Lutz T, Van Zyl L, Porteiro N, Benn P, Huang JO, et al. Cabotegravir + rilpivirine long-acting as HIV-1 maintence therapy: ATLAS week 96 results [abstract no. P006]. HIV Drug Therapy Glasgow; 2020: Virtual.
Overton ET, Richmond G, Rizzardini G, Jaeger H, Orrell C, Nagimova F, et al. Long-acting cabotegravir and rilpivirine dosed every 2 months in adults with HIV-1 infection (ATLAS-2M), 48-week results: a randomised, multicentre, open-label, phase 3b, non-inferiority study. Lancet. 2021;396(10267):1994–2005.
D’Amico M, Orkin C, Bernal-Morell E, Tan D, Katner H, Singh Y, et al. Safety and efficacy of cabotegravir + rilpivirine long-acting with and without oral lead-in: FLAIR Week 124 results [abstract no. O414]. HIV Drug Therapy Glasgow; 2020: Virtual.
Orkin C, Arasteh K, Górgolas Hernández-Mora M, Pokrovsky V, Overton ET, Girard PM, et al. Long-acting cabotegravir and rilpivirine after oral induction for HIV-1 infection. N Engl J Med. 2020;382(12):1124–35.
Oliveira M, Ibanescu RI, Anstett K, Mésplède T, Routy JP, Robbins MA, et al. Selective resistance profiles emerging in patient-derived clinical isolates with cabotegravir, bictegravir, dolutegravir, and elvitegravir. Retrovirology. 2018;15(1):56.
Murray MI, Markowitz M, Frank I, Grant RM, Mayer KH, Hudson KJ, et al. Satisfaction and acceptability of cabotegravir long-acting injectable suspension for prevention of HIV: Patient perspectives from the ECLAIR trial. HIV Clin Trials. 2018;19(4):129–38.
HIV Drug Interactions. https://www.hiv-druginteractions.org. Accessed 15 Nov 2020.
Lou Y, Buchanan AM, Chen S, Ford SL, Gould E, Margolis D, et al. Effect of cabotegravir on cardiac repolarization in healthy subjects. Clin Pharmacol Drug Dev. 2016;5(6):509–16.
Trezza C, Ford SL, Gould E, Lou Y, Huang C, Ritter JM, et al. Lack of effect of oral cabotegravir on the pharmacokinetics of a levonorgestrel/ethinyl oestradiol-containing oral contraceptive in healthy adult women. Br J Clin Pharmacol. 2017;83(7):1499–505.
Ford SL, Gould E, Chen S, Margolis D, Spreen W, Crauwels H, et al. Lack of pharmacokinetic interaction between rilpivirine and integrase inhibitors dolutegravir and GSK1265744. Antimicrob Agents Chemother. 2013;57(11):5472–7.
Van Heeswijk R. The pharmacokinetic interaction between atorvastatin and TMC278, a next generation non-nucleoside reverse transcriptase inhibitor, in HIV-negative volunteers [abstract no. P4.3/04]. 11th European AIDS Conference; 24–27 October 2007: Madrid.
van Heeswijk R, Hoetelmans R, Kestens D, Stevens M, Peeters M, Williams P, et al. The effects of TMC278, a next generation non-nucleoside reverse transcriptase inhibitor, on the pharmacokinetics of acetaminophen and CYP2E1 activity in HIV-negative volunteers [abstract no. 67]. 8th International Workshop on Clinical Pharmacology of HIV Therapy; 16–18 April 2007: Budapest.
Crauwels H, van Heeswijk R, Stevens M, Buelens A, Boven K, Hoetelmans R. TMC278, a next-generation non-nucleoside reverse transcriptase inhibitor, does not alter the pharmacokinetics of sildenafil [abstract no. P22]. 10th International Workshop on Clinical Pharmacology of HIV Therapy; 15–17 April 2009: Amsterdam.
Crauwels H, van Heeswijk R, Cornelis L, McNeeley D, Buelens A, Clark A, et al. Pharmacokinetic interaction study between TMC278, a next generation non-nucleoside reverse transcriptase inhibitor, and the contraceptives norethindrone plus ethinylestradiol [abstract no. P4.3/]. 12th European AIDS Conference; 11–14 November 2009: Cologne.
Crauwels H, van Heeswijk R, Kestens D, Stevens M, Buelens A, Boven K, et al. The pharmacokinetic interaction between rifabutin and TMC278, a next generation non-nucleoside reverse transcriptase inhibitor [abstract no. TUPE0080]. 17th International AIDS Conference; 3–8 August 2008: Mexico.
Hoetelmans R, van Heeswijk R, Kestens D, El Malt M, Stevens M, Peeters M, et al. Pharmacokinetic interaction between TMC278, an investigational non-nucleoside reverse transcriptase inhibitor (NNRTI), and Lopinavir/Ritonavir (LPV/r) in healthy volunteers [abstract no. P4.3/1]. 10th European AIDS Conference; 17–20 November 2005: Dublin.
van Heeswijk R, Hoetelmans R, Kestens D. The pharmacokinetic interaction between TMC278, a next generation non-nucleoside reverse transcriptase inhibitor, and once daily darunavir/ritonavir in HIV-negative volunteers [abstract no. 18]. 47th Interscience Conference on Antimicrobial Agents and Chemotherapy; 17–20 September 2007: Chicago; IL.
Hoetelmans R, Kestens D, Stevens MC, Peeters M, Williams PE, Bastiaanse L, et al. Pharmacokinetic interaction between TMC278, an investigational non-nucleoside reverse transcriptase inhibitor, and tenofovir in healthy volunteers [abstract no. WePe3.C15]. Conference on HIV Pathogenesis, Treatment and Prevention; 24–27 July 2005: Rio de Janeiro.
Begley R, Das M, Zhong L, Ling J, Kearney BP, Custodio JM. Pharmacokinetics of tenofovir alafenamide when coadministered with other HIV antiretrovirals. J Acquir Immune Defic Syndr. 2018;78(4):465–72.
Yeh WW, Feng HP, Auger P, Guo Z, Stypinski D, Bredja J, et al. No pharmacokinetic interaction between HCV inhibitors grazoprevir/elbasvir with rilpivirine [abstract no. 63]. 16th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy; 26–28 May 2015: Washington.
Mogalian E, Stamm LM, Osinusi A, Brainard DM, Shen G, Ling KHJ, et al. Drug-drug interaction studies between hepatitis C virus antivirals sofosbuvir/velpatasvir and boosted and unboosted human immunodeficiency virus antiretroviral regimens in healthy volunteers. Clin Infect Dis. 2018;67(6):934–40.
Crauwels H, van Heeswijk R, Vandevoorde A, McNeeley D, Buelens A, Boven K, et al. Pharmacokinetic interaction study between TMC278, a next-generation non-nucleoside reverse transcriptase inhibitor (NNRTI), and methadone [abstract no. 33]. 11th International Workshop on Clinical Pharmacology of HIV Therapy; 7–9 April 2010: Sorrento.
Woosley RL HC, Gallo T, Tate J, Woosley D, Romero KA. QTdrugs List. https://www.crediblemeds.org. Accessed 15 Nov 2020.
Ford SL, Sutton K, Lou Y, Zhang Z, Tenorio A, Trezza C, et al. Effect of rifampin on the single-dose pharmacokinetics of oral cabotegravir in healthy subjects. Antimicrob Agents Chemother. 2017;61(10):e00487-e517.
Ford SL, Lou Y, Lewis N, Kostapanos M, D’Amico R, Spreen W, et al. Effect of rifabutin on the pharmacokinetics of oral cabotegravir in healthy subjects. Antivir Ther. 2019;24(4):301–8.
Blair CS, Li S, Chau G, Cottle L, Richardson P, Marzinke MA, et al. Hormonal contraception use and cabotegravir pharmacokinetics in HIV-uninfected women enrolled in HPTN 077. J Acquir Immune Defic Syndr. 2020;85(1):93–7.
van Heeswijk R, Hoetelmans R, Kestens D, Stevens M, Peeters M, Boven K, et al. The effects of CYP3A4 modulation on the pharmacokinetics of TMC278, an investigational non-nucleoside reverse transcriptase inhibitor [abstract no. 74]. 7th International Workshop on Clinical Pharmacology of HIV Therapy; 20–22 April 2006: Lisbon.
Stader F, Penny MA, Siccardi M, Marzolini C. A comprehensive framework for physiologically based pharmacokinetic modelling in Matlab®. CPT Pharmacomet Syst Pharmacol. 2019;8(7):444–59.
Taskar K, Patel P, Cozens S, Tracey H, Bloomer J, Han K, et al. Utilization of physiologically based pharmacokinetic modelling (PBPK) to predict the effect of UGT enzyme inhibition and induction on the systemic exposure of cabotegravir [abstract no. 18]. International Workshop on Clinical Pharmacology of HIV, Hepatitis, and Other Antiviral Drugs; 14–16 May 2019: Noordwijk.
Patel P, Xue Z, King KS, Parham L, Ford S, Lou Y, et al. Evaluation of the effect of UGT1A1 polymorphisms on the pharmacokinetics of oral and long-acting injectable cabotegravir. J Antimicrob Chemother. 2020;75(8):2240–8.
Overton ET, Richmond GJ, Rizzardini G, Jaeger H, Orrell C, Nagimova F, et al. Cabotegravir + rilpivirine every 2 months is noninferior to monthly: ATLAS-2M study [abstract no. 34]. Conference on Retroviruses and Opportunistic Infections; 8–11 March 2020: Boston, MA.
Landovitz RJ, Donnell D, Clement M, Hanscom B, Coelho L, Cabello R, et al. HPTN083 interim results: Pre-exposure prophylaxis (PrEP) containing long-acting injectable cabotegravir (CAB-LA) is safe and highly effective for cisgender men and transgender women who have sex with men (MSM,TGW) (OAXLB0101) [abstract no. OAXLB0101]. 23rd International AIDS Conference; 2020: Virtual.
Ford SL, Gould E, Chen S, Lou Y, Dumont E, Spreen W, et al. Effects of etravirine on the pharmacokinetics of the integrase inhibitor S/GSK1265744. Antimicrob Agents Chemother. 2013;57(1):277–80.
van Heeswijk R, hoetelmans R, Kestens D, Stevens M, Peeters M, Williams P, et al. The pharmacokinetic interaction between famotidine and TMC278, a next generation non-nucleoside reverse transcriptase inhibitor, in HIV-negative volunteers [abstract TUPDB01]. 4th IAS Conference on HIV Pathogenesis, Treatment and Prevention; 22–25 July 2007: Sydney.
van Heeswijk R, Hoetelmans R, Kestens D, Stevens M, Peeters M, Williams P, et al. The pharmacokinetic interaction between ketoconazole and TMC278, an investigational non-nucleoside reverse transcriptase inhibitor (NNRTI), in healthy HIV-negative subjects [abstract no. TUPE0087]. 16th International AIDS Conference; 13–18 August 2006: Toronto, ON.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding provided by Universität Basel (Universitätsbibliothek Basel).
Conflict of interest
Daryl Hodge and Sara Gibbons have no conflicts of interest to declare. David Back and Saye Khoo have received educational grants for the Liverpool drug interaction website (http://www.hiv-druginteractions.org) from Frontier, Gilead Sciences, MSD, Janssen, and ViiV Healthcare. Saye Khoo has also received speakers’ honoraria from ViiV Healthcare, Gilead Sciences, and AbbVie; consultancy fees from ViiV Healthcare and Merck; and research funding from Gilead Sciences and ViiV Healthcare. David Back has also received honoraria for speakers’ bureau or advisory boards received from Gilead Sciences, MSD, and ViiV Healthcare. Catia Marzolini has received research funding from Gilead and honoraria for lectures from MSD.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
DH carried out the literature search. DH prepared the figures and tables. DH and CM wrote the manuscript. DJB, SG and SHK critically reviewed the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Hodge, D., Back, D.J., Gibbons, S. et al. Pharmacokinetics and Drug–Drug Interactions of Long-Acting Intramuscular Cabotegravir and Rilpivirine. Clin Pharmacokinet 60, 835–853 (2021). https://doi.org/10.1007/s40262-021-01005-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01005-1