
Abstract
Background
BUP-XR (a.k.a. RBP-6000 or SUBLOCADE™) is an extended-release subcutaneous buprenorphine formulation for the treatment of opioid use disorder. BUP-XR was designed to provide sustained buprenorphine exposure throughout the monthly dosing interval, at concentrations sufficient to control all aspects of the disease (withdrawal, craving, and blockade of opioid subjective effects).
Objectives
To characterize the population pharmacokinetics of BUP-XR based on phase II and phase III data and to evaluate whether target therapeutic concentrations were reached with the dosing regimens evaluated in the phase III program.
Methods
The population pharmacokinetic analysis included 570 subjects with opioid use disorder who received up to 12 monthly BUP-XR injections following induction with sublingual buprenorphine.
Results
In phase III studies, target therapeutic concentrations of buprenorphine were achieved from the first injection and maintained over the entire treatment duration. Buprenorphine plasma concentration–time profiles were well described by a two-compartment model, with first-order absorption for sublingual buprenorphine and a dual absorption submodel for BUP-XR. A covariate analysis evaluated the effects of subjects’ demographic characteristics, laboratory data, and genetic status regarding buprenorphine-metabolizing enzymes. Only two covariates, body mass index and body weight, were retained in the final model. Overall, their effects were not of sufficient magnitude to justify a dose adjustment. Finally, pharmacokinetic simulations showed that buprenorphine plasma concentrations decreased slowly after discontinuation of treatment and that a 2-week occasional delay in dosing would not impact efficacy, which translated into labeling claims.
Discussion
In conclusion, the present analysis led to the development of a robust population pharmacokinetic model and confirms the ability of BUP-XR to deliver and maintain therapeutic plasma concentrations over the entire treatment duration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The current analysis confirms the ability of the BUP-XR formulation to deliver and maintain therapeutic plasma concentrations of buprenorphine throughout the monthly dosing interval. |
A robust population pharmacokinetic model was developed based on data from 570 patients covering up to 1 year of exposure. |
Model-based simulations translated into recommendations for treating physicians in the product prescribing information |
1 Introduction
Opioid use disorder (OUD) is a neurobehavioral syndrome characterized by the repeated compulsive seeking or use of an opioid despite adverse social, psychological, and physical consequences [1]. In the USA, this chronic relapsing disease has grown to epidemic proportions with high costs to individuals, families, and society [2,3,4]. Adding to the impact of OUD, deaths from opioid overdoses have resulted in a major public health crisis [5].
The mu-opioid receptor partial agonist buprenorphine has been shown to be efficacious and safe for the treatment of OUD [6, 7]. Until recently, buprenorphine was only available as daily transmucosal formulations (administered sublingually or buccally [8,9,10]), or as a 6-month subdermal implant [11]. Take-home transmucosal formulations present several limitations. First, they may not sustain systemic concentrations at therapeutic levels throughout the day [12]. Second, daily compliance may be an issue for some patients and serves as a constant reminder of the disease. Last, the use of transmucosal formulations may be associated with the risk of misuse, abuse, and diversion [13]. The 6-month subdermal implant delivers low buprenorphine plasma concentrations (0.5–1.0 ng/mL at steady-state) and is indicated for patients who have already achieved prolonged clinical stability on low-to-moderate doses of transmucosal buprenorphine (i.e., ≤ 8 mg per day); furthermore, treatment duration with the implant cannot exceed 1 year [11].
Buprenorphine formulated in the well-established ATRIGEL delivery system (BUP-XR a.k.a. RBP-6000 or SUBLOCADE™) is the first extended-release monthly formulation of buprenorphine approved by the US Food and Drug Administration for the treatment of moderate-to-severe OUD [14]. The ATRIGEL delivery system consists of a biodegradable poly(dl-lactide-co-glycolide) with a carboxylic acid end group polymer dissolved in a biocompatible solvent, N-methyl-2-pyrrolidone. Upon subcutaneous (SC) injection in the abdomen, the polymer precipitates to create a solid depot containing buprenorphine. After initial formation of the depot, buprenorphine is released via diffusion from, and biodegradation of, the depot, resulting in sustained buprenorphine plasma concentrations throughout the monthly dosing interval. BUP-XR can only be administered by a healthcare professional in a healthcare setting [14], which is expected to increase compliance and reduce abuse and diversion.
As reviewed by Volkow et al. [15], there are three well-described components to the disease: ‘binge and intoxication’ related to reward pathways (in the OUD context: related to the subjective effects produced by opioids); ‘withdrawal and negative effects’, and ‘preoccupation and anticipation’ (also commonly referred to as craving). Thus, treatment of OUD should adequately address or control these three aspects of the disease. Greenwald et al. [7, 12, 16] investigated the relationships between buprenorphine plasma concentrations, occupancy of mu-opioid receptors in the brain, and clinical outcomes. It was shown that different levels of mu-opioid receptor occupancy (and hence plasma concentrations) were needed to control different aspects of the disease. Withdrawal symptoms were controlled when at least 50% of mu-opioid receptors were occupied (corresponding to plasma levels ≥ 1 ng/mL), while subjective effects of an exogenous opioid agonist were controlled when more than 70–80% of mu-opioid receptors were occupied (corresponding to plasma levels ≥ 2–3 ng/mL; [7, 17]). These results were pivotal in defining target buprenorphine plasma concentrations of at least 2–3 ng/mL, which drove the clinical development of BUP-XR. Based on clinical data [18] and population pharmacokinetic (PK) modeling [19], two dosing regimens were selected for BUP-XR in the pivotal phase III efficacy study. Both dosing regimens started with two monthly doses of 300 mg to achieve target plasma concentrations rapidly. Then, subjects could receive either 100 mg monthly to maintain exposure levels reached with the two initial 300-mg doses (300/100-mg dosing regimen) or 300 mg monthly to provide higher concentrations hypothesized to be needed by some subjects depending on their drug-use history and clinical condition (300/300-mg dosing regimen) [20].
The objective of the present analysis was (1) to confirm, based on phase III data, the ability of BUP-XR to deliver and maintain target plasma exposure over the whole treatment duration, and (2) to refine our understanding of BUP-XR pharmacokinetics through the combined analysis of phase II and phase III studies. While the population PK models developed early in development proved extremely useful for dose selection and study design [17, 19], they were generated using a relatively small number of subjects and a limited number of monthly injections (generally four or fewer). In contrast, the present modeling provides an assessment of BUP-XR kinetics in a large representative set of 570 patients who received up to 12 monthly injections at the approved dosing regimens. With this analysis, we evaluated the variability in plasma exposure and the need for potential dose adjustments by assessing the impact of subjects’ demographic and genetic characteristics on BUP-XR pharmacokinetics. We also conducted simulations to inform buprenorphine long-term exposure after discontinuation of BUP-XR treatment and to evaluate the impact of occasional delays in dosing. The results of these simulations have translated into recommendations for physicians in the BUP-XR prescribing information [14].
2 Methods
2.1 Study Design and Patient Population
The population PK analysis included data from three prospective BUP-XR clinical studies: a phase IIa multiple-ascending-dose study (Study 1; NCT01738503), a phase III double-blind efficacy study (Study 2; NCT02357901), and a phase III, open-label long-term safety study (Study 3; NCT02510014). All studies were conducted in subjects with OUD who were seeking treatment. Table 1 summarizes the design of each study, with information on dosing, PK sampling, and the main inclusion criteria. Detailed inclusion/exclusion criteria can be found on https://clinicaltrials.gov using the NCT numbers provided above. In each study, BUP-XR administration was preceded by a run-in phase (4–14 days) during which subjects were inducted and dose stabilized with sublingual (SL) buprenorphine. The purpose of the run-in phase was to ensure that subjects could tolerate buprenorphine prior to receiving a monthly injection and to avoid precipitating opioid withdrawal. The run-in phase in Study 1 used SL buprenorphine tablets while Studies 2 and 3 (phase III) used SL buprenorphine/naloxone film formulation to better reflect clinical practice. Naloxone is a full antagonist at the µ-opioid receptor and is added to buprenorphine formulations to deter parenteral use. When administered sublingually, naloxone is poorly absorbed and has no impact on buprenorphine pharmacokinetics [21].
All studies were conducted in accordance with principles and requirements of the International Council for Harmonization Good Clinical Practice guidelines. Written informed consent was obtained from all participants before starting any study-related procedure. Clinical study protocols, informed consent forms, and all other appropriate study-related documents were reviewed and approved by institutional review boards.
2.2 Analysis of Plasma Samples
The same validated liquid chromatography and tandem mass-spectrometry method was used across the three studies to measure buprenorphine concentrations in plasma. The assay was fully validated for linearity, selectivity, recovery, matrix effect, accuracy, precision, and stability before application to the sample analysis. The calibration range for buprenorphine was 0.050–25.0 ng/mL.
2.3 Population Pharmacokinetic Modeling
Population PK analysis (non-linear mixed-effects modeling) was performed with NONMEM version 7.3.0, using the Stochastic Approximation Expectation-Maximization algorithm with interaction for model estimation. The importance sampling algorithm was used to calculate the − 2 log-likelihood value at the final parameter estimates and to obtain the asymptotic standard errors of estimates. Perl-speaks-NONMEM (PsN) version 4.4.0 [22] was used to operate NONMEM. R version 3.3.1 or older was used for data inspection/preparation, post-processing of NONMEM outputs, and to perform simulations.
Because of the extended-release nature of the formulation, the terminal phase of BUP-XR kinetics does not reflect drug elimination but reflects the slow release and absorption of buprenorphine from the SC depot (“flip-flop” kinetics). Therefore, to adequately characterize drug absorption, distribution, and elimination parameters, BUP-XR data were analyzed jointly with SL buprenorphine PK data measured during the run-in period of each study. Actual doses, dosing times, and sampling times were used for the analysis. In the phase III studies, SL buprenorphine intake at each scheduled visit was documented; however, the intake of SL buprenorphine between visits was not supervised and the records for dispensed units were not sufficient to accurately determine daily doses during that time period. Hence, it was decided to impute missing dosing records for SL buprenorphine in phase III studies by carrying backwards the dosing information collected at the scheduled visits.
The population PK model was developed in three steps (described below). Model selection was based on objective function values (OFV; the likelihood ratio test was applied to nested models), standard diagnostic plots, and visual predictive checks (VPCs).
2.3.1 Step 1: Selection of the Structural Pharmacokinetic Model
The rich PK data from Study 1 were used for selection of the structural model. Previous modeling work [17, 19] indicated that buprenorphine absorption following SC injection could be described by a dual absorption model including (i) a first-order process to characterize the early peak at 24 h post-dose and (ii) a transit compartment absorption model to mimic the slow release of buprenorphine from the SC depot. This model was refined in terms of the number of transit compartments and with the estimation of the fraction of the dose allocated to the fast and slow absorption pathways. Additionally, the bioavailability of buprenorphine for SL tablets relative to BUP-XR was estimated. The model was parameterized in terms of clearances and volumes. Based on prior knowledge and for mechanistic reasons, clearances and volumes of distributions were allometrically scaled by body weight, using the well-established power model and exponents of 0.75 for clearances and 1 for volumes [23]. A full variance–covariance matrix was estimated to quantify interindividual variability. Individual PK parameters were assumed to be log-normally distributed, except for the fraction of SC dose allocated to the fast absorption process for which a logit-normal distribution was used. A combined proportional and additive error model was used to describe residual variability.
2.3.2 Step 2: Development of the Population Pharmacokinetic Model with Covariates
The model developed in Step 1 was re-estimated based on the combined data from Study 1 and Study 2. Because Study 1 used buprenorphine SL tablet and Study 2 used buprenorphine/naloxone SL film, two additional PK parameters were added to estimate relative changes in bioavailability and the absorption rate constant between film and tablet formulations.
A covariate analysis was then conducted to assess the impact of subjects’ characteristics on the pharmacokinetics of BUP-XR. Covariates included subjects’ demographic characteristics (age, sex, body mass index [BMI], waist-to-hip ratio, race), genetic status for buprenorphine-metabolizing enzymes (cytochrome P450 [CYP] 3A4, CYP2C8, UGT1A1, UGT2B7), and laboratory data (aspartate aminotransferase, alanine aminotransferase, creatinine clearance). The single nucleotide polymorphisms investigated were CYP3A4*2 (rs55785340), CYP3A4*1B (rs2740574), CYP3A4*22 (rs35599367), CYP2C8*3 (rs10509681), UGT1A1*28 (rs8175347), and UGT2B7*3 (rs12233719).
The covariate analysis was performed in a stepwise manner. First, covariate effects on PK parameters were evaluated using the empirical Bayes estimates (EBEs) from the base model. The identification of potential relationships was based on visual inspection, statistical testing (Pearson correlation test for continuous covariates; chi-squared test for categorical covariates), and physiological relevance. Covariate relationships identified based on EBEs were further tested in NONMEM using the automated stepwise forward inclusion and backward elimination algorithm implemented in PsN (linearized stepwise covariate model building [24, 25]). Significance levels (alpha) of 0.05 and 0.01 were used for forward and backward selection, respectively. Covariate effects were added to the model in a multiplicative manner; linear and non-linear (power-model) relationships were tested for continuous covariates.
Genotype data and waist-to-hip ratio were only measured in Study 2 and hence were treated as missing in Study 1 for exploratory analyses on EBEs. In the formal covariate analysis in NONMEM, subjects with missing data were assumed to have the most common covariate value (for categorical covariates) or the median value (for continuous covariates). For covariates with repeated measures (weight, laboratory data), baseline values were used as no systematic trend was observed over time.
2.3.3 Step 3: Model Evaluation and Refinement
The final model developed in Step 2 was validated using Study 3 data. First, the model was used to predict buprenorphine plasma concentrations in Study 3, accounting for subjects’ dosing history and demographic characteristics. Model predictions were compared to observations using VPC plots. The model was then applied to Study 3 data with no estimation step, and goodness of fit was assessed with standard diagnostic plots. Finally, Study 3 data were added to the NONMEM dataset and the model was re-estimated based on the totality of the data.
2.4 Model Simulations
Simulations were conducted to derive steady-state secondary PK parameters (average concentration [Cavg], maximum concentration [Cmax], and minimum concentration [Cmin]) for each dosing regimen in the pivotal efficacy study (Study 2). Simulations also evaluated the impact of an occasional 2-week delay in dosing and predicted the long-term decrease in plasma concentrations following treatment discontinuation. For each simulation, a total of 5000 subjects were simulated by resampling from the distribution of subjects’ demographic characteristics across Studies 1, 2, and 3.
3 Results
Figure 1 shows individual plasma concentrations of buprenorphine observed in the two phase III studies. The data indicate that administration of 2 monthly doses of 300 mg followed by 100 mg monthly (300/100-mg dosing regimen) achieved target concentrations of 2 ng/mL from the first injection. From the second injection onwards, plasma concentrations were sustained above 2 ng/mL over the entire treatment duration in the majority of subjects. Administration of 300 mg monthly (300/300-mg dosing regimen) provided higher buprenorphine plasma concentrations in the range of 5–10 ng/mL.

Buprenorphine plasma concentrations after multiple subcutaneous injections of BUP-XR in phase III studies. Top panel: compares concentrations in subjects receiving 300/100 mg of BUP-XR in Study 2 and 300/100-mg roll-over subjects after they received one or multiple subcutaneous injections of 100 mg of BUP-XR in Study 3. Bottom panel: compares concentrations in subjects receiving 300/300 mg of BUP-XR in Study 2 and 300/300-mg roll-over subjects who remained taking 300 mg of BUP-XR in Study 3. Rollovers: Study 2 participants who were enrolled in Study 3. Concentrations measured after sublingual dosing during the run-in are shown in green; concentrations measured after BUP-XR dosing are shown in black. The horizontal red dashed line indicates 2 ng/mL
The population PK analysis included a total of 19,686 buprenorphine plasma concentrations measured in 570 subjects. Table 2 summarizes demographics and other patient characteristics; genotype frequencies for SNPs assessed in Study 2 are provided in Table S1 of the Electronic Supplementary Material (ESM). Plasma concentrations below the lower limit of quantification represented a negligible fraction of the observed data (< 0.5%) and hence were excluded from the analysis. The results at each model development step are summarized below. Model parameter estimates at each step are displayed in Table S2 of the ESM.
3.1 Selection of Structural Pharmacokinetic Model
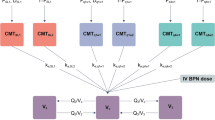
Buprenorphine plasma concentration data in Study 1 were adequately described by a two-compartment model, with first-order absorption for SL buprenorphine and a dual absorption model for BUP-XR (Fig. 2). This dual absorption model consisted of a first-order rate constant characterizing the rapid absorption associated with the early peak and a one-transit compartment model describing the slow release of buprenorphine from the SC depot. A relative change in buprenorphine bioavailability by the SL route was estimated for doses ≥ 16 mg compared with doses < 16 mg to account for the less than dose-proportional increase in plasma exposure following SL dosing.

Structural model for buprenorphine after sublingual (SL) and subcutaneous (SC) administration. A first-order absorption rate constant (k) was used to model SL absorption following repeated daily administration of buprenorphine SL tablets (k14). The bioavailability (F) of buprenorphine SL tablets relative to BUP-XR was estimated as F1. The relative changes in F and absorption rate for SL film vs SL tablet formulations were estimated (FRF1 and FRK14, respectively). Additionally, a relative change of F1 for doses ≥ 16 mg compared to doses < 16 mg was estimated (F1DOSE) to account for the less than dose-proportional increase in buprenorphine plasma exposure following SL dosing. abs. absorption, CL clearance, Q intercompartmental clearance, V volume
3.2 Development of Population Pharmacokinetic Model with Covariates
Re-estimation of the model based on combined data from Studies 1 and 2 led to similar parameter estimates, except for the variances of k14 and F1 (related to SL absorption) and variances for V5/F and Q/F (apparent peripheral volume and intercompartmental clearance, both informed by SL data), which were inflated. It was determined that this increase in variability resulted from the inclusion of SL film data from Study 2. As previously explained in “Methods”, SL film dosing in Study 2 was only assessed at scheduled visits during the run-in. Therefore, SL dosing between visits was imputed using the “next observation carried backward” methodology. To address this increase in variance estimates, two different approaches were considered. The first approach was to estimate k14 and F1 variances for SL tablet (Study 1) and SL film (Study 2) separately. The second approach was to fix both fixed effects and variances for k14, F1, Q/F, and V5/F to previous estimates obtained from the analysis of the rich PK data in Study 1 where SL dosing was accurately documented. Both approaches improved the VPCs for SL buprenorphine, but the second approach was retained as it was numerically more stable. Pharmacokinetic parameters specific to BUP-XR were not affected.
Exploratory analyses of EBEs vs covariates identified sex as a potential covariate on the fast absorption (k24) and slow absorption (k36, k64) of buprenorphine following SC injection. Body mass index was also identified as a potential covariate on k24, k36, k64, CL/F (apparent clearance), V4/F (apparent volume of central compartment), and F2 (fraction of SC dose allocated to fast absorption process). Finally, age was identified as a potential covariate on k36. Forward selection in NONMEM only retained BMI on CL/F (ΔOFV = 794.9) and k24 (ΔOFV = 648.5), and sex on k36 (ΔOFV = 514.5). Backward selection did not remove any of these covariates.
All fixed- and random-effects parameters were estimated with good precision with the exception of the effect of sex on k36 (140% RSE); however, this effect was marginal (+ 7.6% in female individuals) and not deemed clinically relevant. The effect of BMI on BUP-XR exposure was well described by the model and further supported by VPCs stratified by low, intermediate, and high BMI (Fig. S1 of the ESM). Although an increase in buprenorphine peak concentration was apparent at a lower BMI, this effect was not of sufficient magnitude to suggest that dose adjustments would be necessary.
3.3 Step 3: Model Evaluation and Refinement Using Study 3 Data
When the model was applied to describe Study 3 data with all parameters fixed, diagnostic plots indicated no deviations from expected patterns. Additionally, the model was able to correctly predict PK data for subjects who stayed at 300 mg across phase III studies (Fig. 3). Steady-state was reached after approximately six injections, and concentrations were consistent across BUP-XR repeated injections once steady-state was achieved.

Prediction of Study 3 data using the model developed from Studies 1 and 2. The plot shows predicted vs observed buprenorphine plasma concentrations for subjects who received 300 mg of BUP-XR throughout Studies 2 and 3 (e.g., subjects on 300/300 mg of BUP-XR in Study 2 who stayed on 300 mg of BUP-XR in Study 3). Dots represent observations. Predictions were generated using the model developed from Study 1 and Study 2 data and are summarized by the median (blue curve) and 90% prediction interval (gray shaded area) at each time point. Doses of sublingual buprenorphine were not simulated here as they have little impact on the pharmacokinetics of BUP-XR (owing to the more rapid decrease in plasma concentrations following sublingual dosing). The run-in period in Study 3 is not shown on the figure
Following re-estimation from the totality of the data (19,686 observations; 570 subjects), parameter estimates were similar to previous estimates (Table S2 of the ESM). As the effect of sex on k36 was small and estimated with great imprecision (282% RSE), it was removed from the model with negligible impact on other parameter estimates. Both VPCs (Fig. 4) and goodness-of-fit plots (Figure S2 of the ESM) confirm that the refined model adequately described the observed PK data across all studies, both in terms of central tendency and variability. Final model parameter estimates are displayed in Table 3.

Prediction-corrected (Pred-Corr) visual predictive checks for BUP-XR dosing regimens in Study 2 (phase III efficacy study). Data are shown for both sublingual (SL) buprenorphine and BUP-XR treatment phases. Time zero is the time of the first subcutaneous injection of BUP-XR. CI confidence interval, Conc concentration
3.4 Simulation Studies
Steady-state secondary PK parameters are summarized in Table 4 for each dosing regimen evaluated in the pivotal efficacy study (Study 2). Model predictions resulted in mean Cavg values of 3.0 ng/mL for 300/100 mg and 6.6 ng/mL for 300/300 mg; mean Cmin values were 2.6 ng/mL and 5.4 ng/mL, respectively (Table 4). Model predictions were very close to PK exposure parameters derived from observed data. Variability in exposure was low to moderate (~ 30%).
Simulations indicated that an occasional 2-week delay in dosing had a low impact on buprenorphine plasma concentrations (Fig. 5). Furthermore, simulations showed that buprenorphine plasma concentrations decreased slowly after discontinuation of BUP-XR treatment (Fig. 6). For the 300/100-mg dosing regimen, median plasma concentrations were predicted to stay above 2 ng/mL for approximately 2 months post the last BUP-XR injection vs 5 months for the 300/300-mg dosing regimen.

Simulated effect of a 2-week delay in BUP-XR dosing on buprenorphine plasma concentrations (300/100-mg dosing regimen). a Two-week delay in injection 2; b 2-week delay in injection 4. Predicted concentrations in 5000 subjects were summarized by the median (solid lines) and 90% prediction interval (shaded areas) at each time point for dosing on schedule (blue) and after an occasional 2-week delay (red)

Predicted decrease in buprenorphine plasma concentrations for BUP-XR dosing regimens following treatment interruption. a 300/100-mg dosing regimen 2; b 300/300-mg dosing regimen. Blue solid lines: median of the simulated data; gray shaded areas: 90% prediction intervals of simulated data. A total of nine subcutaneous injections were simulated in 5000 subjects. The horizontal red dashed line indicates the 2-ng/mL minimum concentration required for opioid blockade, as established from modeling and simulation and confirmed by clinical data (Nasser et al. [18])
4 Discussion
Overall, phase III data show that target therapeutic concentrations of ≥ 2–3 ng/mL were achieved and maintained over the entire treatment duration with the BUP-XR 300/100-mg dosing regimen. The 300/300-mg dosing regimen provided steady-state concentrations of ≥5 ng/mL, which were reached after approximately six injections. Because of the slow-release characteristics of BUP-XR, peak-to-trough fluctuations were low (75–78%, calculated as 100 × (Cmax − Cmin)/Cavg based on observed data in Table 4). By comparison, peak-to-trough fluctuations for SL buprenorphine were 223–271% based on observed data in Study 1. Furthermore, the variability in plasma exposure was approximately 30%, which is typically less than for SL buprenorphine products (variability of 31–84% [26, 27]). The maximal plasma concentration of buprenorphine at steady-state was 4.9 ng/mL for BUP-XR 300/100 mg and 10.1 ng/mL for BUP-XR 300/300 mg. Those concentrations were lower or comparable to the maximal concentrations achieved with the SL buprenorphine tablet at the highest dose of 24 mg/day (8.3 ng/mL [14]).
As demonstrated in the pivotal efficacy study (Study 2), target concentrations of ≥ 2–3 ng/mL translated into clinical efficacy, with substantial proportions of subjects achieving abstinence from opioids, relief of withdrawal symptoms, and control of opioid craving, without the use of supplemental buprenorphine [20]. There was no clinically relevant difference in outcomes between the two dosing regimens. However, subgroup analyses indicated that subjects injecting opioids by the intravenous route might benefit from the higher plasma concentrations delivered by the 300-mg maintenance dose [28]. Overall, BUP-XR administration was associated with improved health status, increased employment, decreased healthcare utilization, and increased medication satisfaction compared with placebo [29]. The safety profile was consistent with transmucosal buprenorphine products, except for injection-site reactions, which were mostly mild and not treatment limiting [20]. An integrated analysis of Study 2 and Study 3 data showed that the incidence of treatment-emergent adverse events decreased over time despite the maintenance of plasma concentrations [30].
The pharmacokinetics of BUP-XR was successfully characterized using a large set of 570 patients with OUD, covering multiple SC injections (up to 1 year of exposure) and a dose range of 50–300 mg. In contrast to previous modeling work [17, 19], BUP-XR and SL buprenorphine data were analyzed simultaneously to address identifiability issues associated with flip-flop kinetics. The relative bioavailability of the SL tablet vs BUP-XR was estimated to be 18.5%. The less than dose-proportional increase in SL buprenorphine exposure estimated by modeling, together with the higher bioavailability for SL film compared with the SL tablet (+ 47%) align with published data [26] and historical data on file.
The dual absorption model selected for BUP-XR is consistent with the release characteristics of the ATRIGEL delivery system. Indeed, after SC injection, there is a phase inversion process during which the biocompatible solvent dissipates, allowing the depot to solidify. During this period, a fraction of the buprenorphine diffuses with the dissipating solvent as there is no formed physical barrier at this stage. This early release or “initial burst” explains the early peak of buprenorphine observed at 24 h post-dose and is well characterized in the model using a first-order process. As the depot hardens, it entraps buprenorphine, which is then slowly released via diffusion from, and biodegradation of, the depot. This second stage is successfully described using a one-transit compartment absorption model. Modeling results indicate that the “initial burst” accounts for approximately 6.8% of the dose. High interindividual variability was estimated for SC absorption rate constants (coefficients of variation of 68–210%); however, this variability was not reflected in secondary PK parameters with coefficients of variation of ~ 30%.
The covariate analysis pointed towards BMI as a significant factor influencing buprenorphine initial release (k24) and CL/F following SC injection. This suggests that the presence of abdominal fat impacts buprenorphine absorption upon release from the SC depot. This result is consistent with previous findings for an extended-release SC formulation of risperidone that also uses the ATRIGEL delivery system [31]. Body weight was another covariate included in the model to account for allometric relationships on clearances and volumes [23]. Integrating the effects of body weight and BMI (which were highly correlated), there was overall little impact on buprenorphine plasma exposure (Cavg, Cmin). The main effect was seen on buprenorphine peak concentration, with a 24% decrease in median Cmax for subjects in the upper BMI quartile compared with subjects in the lower BMI quartile. These variations, however, were not of sufficient magnitude to justify a dosage adjustment.
Previous modeling from Study 2 alone [19] identified an effect of BMI on k36 (slow absorption phase) and not k24 (rapid absorption phase). No significant covariates were identified in the early population PK model developed from single-dose data [17]. These differences may reflect limitations of previous analyses, which did not address flip-flop kinetics and included a smaller number of subjects. Hence, the current results are considered more robust and relevant for the characterization of interindividual variability. Of note, no residual effects of covariates were identified on EBEs vs covariate plots based on the final model.
No effects were observed for genetic variants of CYP3A4, CYP2C8, UGT1A1, and UGT2B7, which are involved in buprenorphine metabolism [32,33,34]. It is possible that the low frequency of some genotypes limited the detection of an effect. Another explanation may be the lower metabolism of buprenorphine after SC administration compared with SL administration. Indeed, with SL buprenorphine, a fraction of the dose is typically swallowed and undergoes a first-pass effect in the intestines and the liver. No such first-pass effect occurs after SC administration. Given the sparse PK data for SL buprenorphine in Study 2 (where genotypes were measured), the effect of genetic covariates on SL bioavailability was not assessed.
Finally, model simulations were conducted to provide guidance regarding treatment discontinuation. They show a slow decrease in plasma concentrations after the last dose of BUP-XR, indicating that physicians may have to monitor patients for several weeks to several months for the re-emergence of withdrawal signs and symptoms. In phase III trials, withdrawal signs and symptoms were not observed in the month following treatment discontinuation [14]. Simulations also show that occasional delays in dosing up to 2 weeks would not have any clinically significant impact on treatment effect. Thus, while monthly dosing is needed for optimal exposure, the slow-release characteristics of the ATRIGEL delivery system allow coverage of the patient if he/she occasionally does not have an injection at the scheduled time. Both simulation results are described in the BUP-XR label [14].
5 Conclusions
The current analysis confirmed the ability of the BUP-XR formulation to deliver and maintain therapeutic plasma concentrations of buprenorphine throughout the monthly dosing interval and during the whole treatment period. A robust population PK model of BUP-XR was developed based on data from 570 patients covering up to 1 year of exposure. No dose adjustments appear to be required based on the investigated covariates, which included demographic characteristics, laboratory data, and genetic status for buprenorphine-metabolizing enzymes. Model-based simulations translated into recommendations for treating physicians in the product prescribing information.
References
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington: American Psychiatric Association; 2013.
United Nations Office on Drugs and Crime. World drug report 2018. https://www.unodc.org/wdr2018/prelaunch/WDR-2018-Methodology-web.pdf. Accessed Mar 2020.
Substance Abuse and Mental Health Services Administration (SAMHSA). Medications for opioid use disorder. Treatment improvement protocol (TIP) Series 63. Full Document, May 2020, Rockville, MD, USA. Available online from: https://store.samhsa.gov/sites/default/files/SAMHSA_Digital_Download/PEP20-02-01-006_508.pdf. Accessed 23 Oct 2020.
Reinhart M, Scarpati LM, Kirson NY, Patton C, Shak N, Erensen JG. The economic burden of abuse of prescription opioids: a systematic literature review from 2012 to 2017. Appl Health Econ Health Policy. 2018;16(5):609–32.
Gomes T, Tadrous M, Mamdani MM, Paterson JM, Juurlink DN. The burden of opioid-related mortality in the United States. JAMA Netw Open. 2018;1(2):e180217.
Walsh SL, Preston KL, Bigelow GE, Stitzer ML. Acute administration of buprenorphine in humans: partial agonist and blockade effects. J Pharmacol Exp Ther. 1995;274(1):361–72.
Greenwald MK, Comer SD, Fiellin DA. Buprenorphine maintenance and mu-opioid receptor availability in the treatment of opioid use disorder: implications for clinical use and policy. Drug Alcohol Depend. 2014;1(144):1–11.
Subutex [package insert]. North Chesterfield (VA): Indivior Inc., 2018.
Suboxone [package insert]. North Chesterfield (VA): Indivior Inc., 2018.
Bunavail [package insert]. Raleigh (NC): BioDelivery Sciences International, Inc., 2018.
Probuphine [package insert]. South San Francisco (CA): Titan Pharmaceuticals, Inc., 2018.
Greenwald M, Johanson CE, Bueller J, Chang Y, Moody DE, Kilbourn M, et al. Buprenorphine duration of action: mu-opioid receptor availability and pharmacokinetic and behavioral indices. Biol Psychiatry. 2007;61(1):101–10.
Reimer J, Wright N, Somaini L, Roncero C, Maremmani I, McKeganey N, et al. The impact of misuse and diversion of opioid substitution treatment medicines: evidence review and expert consensus. Eur Addict Res. 2016;22(2):99–106.
Sublocade [package insert]. North Chesterfield (VA): Indivior Inc., 2018.
Volkow ND, Koob GF, McLellan AT. Neurobiologic advances from the brain disease model of addiction. N Engl J Med. 2016;374(4):363–71.
Greenwald MK, Johanson CE, Moody DE, Woods JH, Kilbourn MR, Koeppe RA, et al. Effects of buprenorphine maintenance dose on μ-opioid receptor availability, plasma concentrations, and antagonist blockade in heroin-dependent volunteers. Neuropsychopharmacology. 2003;28(11):2000–9.
Nasser AF, Heidbreder C, Gomeni R, Fudala PJ, Zheng B, Greenwald MK. A population pharmacokinetic and pharmacodynamic modelling approach to support the clinical development of RBP-6000, a new, subcutaneously injectable, long-acting, sustained-release formulation of buprenorphine, for the treatment of opioid dependence. Clin Pharmacokinet. 2014;53(9):813–24.
Nasser AF, Greenwald MK, Vince B, Fudala PJ, Twumasi-Ankrah P, Liu Y, et al. Sustained-release buprenorphine (RBP-6000) blocks the effects of opioid challenge with hydromorphone in subjects with opioid use disorder. J Clin Psychopharmacol. 2016;36(1):18–26.
Laffont CM, Gomeni R, Heidbreder C, Jones JP III, Nasser AF. Population pharmacokinetic modeling after repeated administrations of RBP-6000, a new, subcutaneously injectable, long-acting, sustained-release formulation of buprenorphine, for the treatment of opioid use disorder. J Clin Pharmacol. 2016;56(7):806–15.
Haight BR, Learned SM, Laffont CM, Fudala PJ, Zhao Y, Garofalo AS, et al. Efficacy and safety of a monthly buprenorphine depot injection for opioid use disorder: a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2019;393(10173):778–90.
Harris DS, Jones RT, Welm S, Upton RA, Lin E, Mendelson J. Buprenorphine and naloxone co-administration in opiate-dependent patients stabilized on sublingual buprenorphine. Drug Alcohol Depend. 2000;61(1):85–94.
Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit: a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–57.
Anderson BJ, Holford NH. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet. 2009;24(1):25–36.
Khandelwal A, Harling K, Jonsson EN, Hooker AC, Karlsson MO. A fast method for testing covariates in population PK/PD models. AAPS J. 2011;13(3):464–72.
Svensson EM, Karlsson MO. Use of a linearization approximation facilitating stochastic model building. J Pharmacokinet Pharmacodynam. 2014;41(2):153–8.
Chiang CN, Hawks RL. Pharmacokinetics of the combination tablet of buprenorphine and naloxone. Drug Alcohol Depend. 2003;70(2):S39-47.
Elkader A, Sproule B. Buprenorphine. Clin Pharmacokinet. 2005;44(7):661–80.
Fox NL, Laffont CM, Zhao S, Gray F, Andorn A, Learned SM, et al. Risk-adjusted abstinence rates are higher with greater buprenorphine plasma exposure among patients who inject opioids. In: 81st annual scientific meeting of the college on problems of drug dependence (CPDD); 15–19 June 2019; San Antonio (TX).
Ling W, Nadipelli VR, Solem CT, Ronquest NA, Yeh YC, Learned SM, et al. Patient-centered outcomes in participants of a buprenorphine monthly depot (BUP-XR) double-blind, placebo-controlled, multicenter, phase 3 study. J Addict Med. 2019;13(6):442–9.
Andorn AC, Haight BR, Shinde S, Fudala PJ, Zhao Y, Heidbreder C, et al. Treating opioid use disorder with a monthly subcutaneous buprenorphine depot injection: 12-month safety, tolerability, and efficacy analysis. J Clin Psychopharmacol. 2020;40(3):231–9.
Laffont CM, Gomeni R, Zheng B, Heidbreder C, Fudala PJ, Nasser AF. Population pharmacokinetic modeling and simulation to guide dose selection for RBP-7000, a new sustained-release formulation of risperidone. J Clin Pharmacol. 2015;55(1):93–103.
Kobayashi K, Yamamoto T, Chiba K, Tani M, Shimada N, Ishizaki T, Kuroiwa Y. Human buprenorphine N-dealkylation is catalyzed by cytochrome P450 3A4. Drug Metab Dispos. 1998;26(8):818–21.
Picard N, Cresteil T, Djebli N, Marquet P. In vitro metabolism study of buprenorphine: evidence for new metabolic pathways. Drug Metab Dispos. 2005;33(5):689–95.
Chang Y, Moody DE. Glucuronidation of buprenorphine and norbuprenorphine by human liver microsomes and UDP-glucuronosyltransferases. Drug Metab Lett. 2009;3(2):101–7.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was funded by Indivior Inc.
Conflict of interest
Celine M. Laffont and Malcolm A. Young are employees of Indivior, Inc. Aksana K. Jones was an employee of Indivior, Inc. at the time the work was performed. Eliford Ngaimisi and Mathangi Gopalakrishnan have received funding for performing part of the modeling work.
Ethics approval
All studies were performed in line with the principles of the Declaration of Helsinki. Clinical study protocols, informed consent forms, and all other appropriate study-related documents were reviewed and approved by institutional review boards (IRBs) [Midlands IRB and University of Michigan IRB for Study 1; Quorum Review IRB for Studies 2 and 3].
Consent to participate
Written informed consent was obtained from all individual participants before starting any study-related procedure.
Consent for publication
The consent for publication was included in the written informed consent.
Data availability
The modeling code is available upon request.
Code availability
The modeling code is available upon request.
Author contributions
AJ, CL, MG, and MY made substantial contributions to the conception or design of the work; AJ, CL, and MY participated in the acquisition/collection of data; AJ, CL, EN, MG, and MY participated in the analysis or interpretation of data. All authors drafted the manuscript or revised it critically for important intellectual content and approved the version to be submitted. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Jones, A.K., Ngaimisi, E., Gopalakrishnan, M. et al. Population Pharmacokinetics of a Monthly Buprenorphine Depot Injection for the Treatment of Opioid Use Disorder: A Combined Analysis of Phase II and Phase III Trials. Clin Pharmacokinet 60, 527–540 (2021). https://doi.org/10.1007/s40262-020-00957-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00957-0