
Abstract
Pyrimidine analogues can be considered as prodrugs, like their natural counterparts, they have to be activated within the cell. The intracellular activation involves several metabolic steps including sequential phosphorylation to its monophosphate, diphosphate and triphosphate. The intracellularly formed nucleotides are responsible for the pharmacological effects. This review provides a comprehensive overview of the clinical studies that measured the intracellular nucleotide concentrations of pyrimidine analogues in patients with cancer. The objective was to gain more insight into the parallels between the different pyrimidine analogues considering their intracellular pharmacokinetics. For cytarabine and gemcitabine, the intracellular pharmacokinetics have been extensively studied over the years. However, for 5-fluorouracil, capecitabine, azacitidine and decitabine, the intracellular pharmacokinetics was only very minimally investigated. This is probably owing to the fact that there were no suitable bioanalytical assays for a long time. Since the advent of suitable assays, the first exploratory studies indicate that the intracellular 5-fluorouracil, azacitidine and decitabine nucleotide concentrations are very low compared with the intracellular nucleotide concentrations obtained during treatment with cytarabine or gemcitabine. Based on their pharmacology, the intracellular accumulation of nucleotides appears critical to the cytotoxicity of pyrimidine analogues. However, not many clinical studies have actually investigated the relationship between the intracellular nucleotide concentrations in patients with cancer and the anti-tumour effect. Only for cytarabine, a relationship was demonstrated between the intracellular triphosphate concentrations in leukaemic cells and the response rate in patients with AML. Future clinical studies should show, for the other pyrimidine analogues, whether there is a relationship between the intracellular nucleotide concentrations and the clinical outcome of patients. Research that examined the intracellular pharmacokinetics of cytarabine and gemcitabine focused primarily on the saturation aspect of the intracellular triphosphate formation. Attempts to improve the dosing regimen of gemcitabine were aimed at maximising the intracellular gemcitabine triphosphate concentrations. However, this strategy does not make sense, as efficient administration also means that less gemcitabine can be administered before dose-limiting toxicities are achieved. For all pyrimidine analogues, a linear relationship was found between the dose and the plasma concentration. However, no correlation was found between the plasma concentration and the intracellular nucleotide concentration. The concentration–time curves for the intracellular nucleotides showed considerable inter-individual variation. Therefore, the question arises whether pyrimidine analogue therapy should be more individualised. Future research should show which intracellular nucleotide concentrations are worth pursuing and whether dose individualisation is useful to achieve these concentrations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Pyrimidine analogues can be considered as prodrugs. Understanding the plasma pharmacokinetics is therefore not enough. It is important to investigate the extent to which these agents enter the cell, are activated intracellularly and actually reach their target in the activated form. |
Intracellular pyrimidine nucleotide concentrations vary considerably between patients. This raises the question whether therapy with pyrimidine analogues should not be further individualised. |
Although the intracellular accumulation of nucleotides appears critical for the pharmacological effect of pyrimidine analogues, only a few clinical studies have actually investigated the relationship between intracellular nucleotide concentrations and the clinical outcome of patients. |
1 Introduction
Pyrimidine analogues form a classical group of cytostatic drugs that still have a very important place in the treatment of both solid and haematologic malignancies. Pyrimidine analogues are structural analogues of one of the physiological pyrimidine nucleobases: cytosine, uracil, or thymine or of their respective nucleosides: (deoxy)cytidine, uridine and thymidine (Fig. 1).

Chemical structures of the physiological pyrimidines (left) and the pyrimidine analogues used as an anticancer drug (right)
The first important pyrimidine analogue, 5-fluorouracil (5-FU), was synthesised in 1957 and still has an important place in the treatment of various solid tumours, including those originating into the gastrointestinal tract, breast, and head and neck region. After 5-FU, several other pyrimidine analogues followed. In 1969, cytarabine was introduced for the treatment of acute leukaemia. Today, cytarabine is still one of the cornerstones in the treatment of acute myeloid leukaemia (AML). Moreover, it has a place in the treatment of various other haematological malignancies. The success of cytarabine as an anti-leukaemic drug stimulated the research for other cytidine analogues that had anti-tumour activity and led to the first preclinical studies with azacitidine and decitabine in the 1960s [1]. Although azacitidine and decitabine initially seemed promising, the first clinical trials were not very impressive, limiting enthusiasm for these drugs as anti-tumour agents [2,3,4]. In 1995, gemcitabine was introduced. Although the structure of gemcitabine was similar to that of cytarabine, it had a different spectrum of anti-tumour activity [5]. Unlike cytarabine, gemcitabine exhibited activity in solid tumours. Today, gemcitabine has an important place in the treatment of non-small cell lung cancer, pancreatic cancer, bladder cancer and ovarian cancer. In the 1990s, capecitabine was developed as a more convenient oral alternative to intravenous 5-FU therapy. As a prodrug of 5-FU, capecitabine was rationally designed to mimic continuous infusion of 5-FU with a certain degree of tumour selectivity [6]. In this decade, there was also renewed interest in azacitidine and decitabine. This time because of their DNA demethylating properties in patients with myelodysplastic syndrome (MDS) [7,8,9,10]. Today, azacitidine and decitabine have gained their place in the treatment of MDS and AML [11].
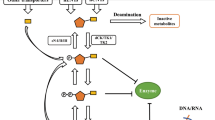
Pyrimidine analogues can be considered as prodrugs, like their natural counterparts, they have to be activated within the cell. The intracellular activation involves several metabolic steps including sequential phosphorylation of the pyrimidine analogue to its monophosphate, diphosphate and triphosphate (Fig. 2). Ultimately, these intracellularly phosphorylated anabolites, called nucleotides, are responsible for the pharmacological effects of the pyrimidine analogue. For instance, as the triphosphate form can compete with its natural counterpart for incorporation into the DNA or RNA, it thereby interferes with DNA or RNA synthesis and with their normal functions.

Intracellular metabolism of pyrimidine anticancer drugs. Pyrimidine analogues and their natural counterparts share the same metabolic pathways. 5-FUrd 5-fluorouridine, 5-FdUrd 5-fluoro-2ʹ-deoxyuridine, C cytidine, CMP cytidine monophosphate, CDP cytidine diphosphate, CTP cytidine triphosphate, dC 2ʹ-deoxycytidine, dCMP 2ʹ-deoxycytidine monophosphate, dCDP 2ʹ-deoxycytidine diphosphate, dCTP 2ʹ-deoxycytidine triphosphate, DNA deoxyribonucleic acid, dT 2ʹ-deoxythymidine, dTMP 2ʹ-deoxythymidine monophosphate, dTDP 2ʹ-deoxythymidine diphosphate, dTTP 2ʹ-deoxythymidine triphosphate, dU 2ʹ-deoxyuridine, dUMP 2ʹ-deoxyuridine monophosphate, dUDP 2ʹ-deoxyuridine diphosphate, dUTP 2ʹ-deoxyuridine triphosphate, hNTs human nucleoside transporters, RNA ribonucleic acid, U uridine, UMP uridine monophosphate, UDP uridine diphosphate, UTP uridine triphosphate
As the intracellularly formed nucleotides are ultimately responsible for the effect of the pyrimidine anticancer drugs, it would be useful to understand the intracellular pharmacokinetics (PK) of these nucleotides during treatment. In this review, we summarise the findings of clinical studies that measured the intracellular concentrations of pyrimidine nucleotides in patients with cancer. The objective of this review was to gain more insight into the parallels between the different pyrimidine analogues with regard to their intracellular PK. In this manner, we can learn the most from what is already known about the intracellular PK of the other drugs in this group. This information could ultimately be very useful to optimise treatment with these agents.
2 Literature Search
In this review, we focused on the six pyrimidine analogues that are currently considered the most important: 5-fluorouracil, capecitabine, cytarabine, gemcitabine, azacitidine and decitabine. All clinical studies were included in which intracellular nucleotide concentrations were measured in patients with cancer during or after treatment with one of these pyrimidine analogues.
Although much of our knowledge about the intracellular pharmacology of the pyrimidine analogues comes from in vitro and animal studies, these studies were not included in this review, as it remains difficult to extrapolate these results to the clinical situation. Knowledge derived from preclinical studies, which was considered relevant for the interpretation of the clinical studies, was briefly summarised at the beginning of each section.
More than 30 clinical studies were included in this review. The results of these studies are also summarised in Tables 1 and 2. The intracellular nucleotide concentrations measured in different studies are sometimes difficult to compare because different units were used. Concentrations were reported in µM, pmol/106 peripheral blood mononuclear cells (PBMCs) or ng/mg protein. In this review, data were presented as reported in the original publications. Units were not converted because not all required information, for instance the measured cell volume, was published in the original articles.
3 5-Fluorouracil and Capecitabine
5-Fluorouracil is a uracil analogue with a fluorine atom at the C-5 position instead of a hydrogen atom (Fig. 1). Approximately 80% of 5-FU is inactivated in the liver by the enzyme dihydropyrimidine dehydrogenase [12]. This means that only a small fraction of the administered 5-FU is available for intracellular anabolism to the active nucleotides. Capecitabine was rationally designed to mimic continuous infusion of 5-FU [13, 14]. After oral administration, capecitabine is rapidly and almost completely absorbed from the gastrointestinal tract. Subsequently, it is converted into 5-FU through a series of three metabolic steps [15, 16]. The third activation step preferentially takes place in tumour tissue rather than normal tissue, which confers a degree of tumour specificity to capecitabine [15,16,17,18].
3.1 Intracellular Metabolism and Mechanism of Action
5-Fluorouracil rapidly enters the cell using the same transport mechanism as uracil [12, 19]. Once in the cell, 5-FU has to be activated by ribosylation and sequential phosphorylation. The activation of 5-FU to its nucleotides can proceed via three pathways [20, 21]. First, 5-fluorouracil can be converted directly to 5-fluorouridine monophosphate by the enzyme orotate phosphoribosyl transferase. Second, 5-FU can be converted to 5-fluorouridine monophosphate in two steps, via 5-fluorouridine, through the sequential actions of uridine phosphorylase and uridine kinase. 5-Fluorouridine monophosphate is further phosphorylated to 5-fluorouridine diphosphate and the active metabolite 5-fluorouridine triphosphate (FUTP) by uridine monophosphate kinase and uridine diphosphate kinase, respectively. 5-Fluorouridine diphosphate might also be converted to 5-fluoro-2ʹ-deoxyuridine diphosphate by ribonucleotide reductase. The third activation pathway involves the conversion of 5-FU to 5-fluoro-2ʹ-deoxyuridine by thymidine phosphorylase. 5-Fluoro-2ʹ-deoxyuridine can then be phosphorylated to the active metabolite 5-fluoro-2ʹ-deoxyuridine monophosphate (FdUMP) by thymidine kinase. 5-Fluoro-2ʹ-deoxyuridine monophosphate can also be further phosphorylated to 5-fluoro-2ʹ-deoxyuridine diphosphate and the third active metabolite 5-fluoro-2′-deoxyuridine triphosphate (FdUTP). These reactions are catalysed by uridine monophosphate kinase and uridine diphosphate kinase, respectively. Conversely, the nucleotides may also be degraded to their inactive nucleosides by 5ʹ-nucleotidases and phosphatases [20, 21].
Ultimately, three intracellularly formed metabolites (nucleotides) are held responsible for the antineoplastic effects of 5-FU. These are FUTP, FdUTP and FdUMP. FUTP is incorporated into RNA and interferes with normal RNA processing and function. FdUTP is incorporated into DNA, leading to DNA damage and ultimately cell death. FdUMP inhibits thymidylate synthase, the enzyme that catalyses the methylation of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP). Inhibition of thymidylate synthase by FdUMP leads to accumulation of deoxyuridine triphosphate (dUTP) and depletion of deoxythymidine triphosphate (dTTP). This imbalance has deleterious consequences for DNA synthesis and repair, ultimately leading to cell death [14, 22, 23].
3.2 Clinical Studies Investigating the Intracellular Concentrations of the 5-Fluorouracil Nucleotides
Most of our knowledge about the intracellular metabolism of 5-FU and its mechanisms of action was derived from in vitro experiments and animal studies [24,25,26,27]. Little is known about the formation of 5-FU nucleotides in patients who are treated with 5-FU or capecitabine.
In the 1980s and 1990s, several studies investigated the intracellular FdUMP concentrations in patients who were treated with an intravenous 5-FU bolus [28,29,30]. Spears et al. [28, 29] and Peters et al. [30] determined the intracellular FdUMP concentrations in healthy liver and colon biopsies as well as tumour biopsies obtained from patients who had previously received an intravenous 5-FU bolus of 500 mg/m2 [28,29,30]. Surgical biopsies were obtained 20 min to 48 h after administration of 5-FU. For each patient, biopsies were obtained at a single time point after 5-FU administration. However, if the measured FdUMP concentrations for the total group of patients were expressed against the time interval between 5-FU administration and tissue biopsy, a graph was obtained that could be interpreted as a pharmacokinetic profile [28, 30].
The highest intracellular FdUMP concentrations were observed shortly after 5-FU administration, indicating that 5-FU is rapidly converted to FdUMP [28, 30]. At 1–5 h after 5-FU administration, the intracellular FdUMP concentrations measured in tumour tissue had a range of 10–1700 pmol/g [30]. The studies demonstrated that 5-FU plasma concentrations do not reflect intracellular FdUMP concentrations. Although 5-FU is rapidly eliminated from plasma, FdUMP is retained considerably longer in cells. At 19–27 h after 5-FU administration, FdUMP was still measurable in concentrations below 200 pmol/g [30]. After 48 h, FdUMP was measurable only in a few samples [30]. The concentrations of FdUMP measured in tissue samples obtained at the same time points after the 5-FU bolus showed large inter-individual variation [28,29,30]. Remarkably, the FdUMP concentrations in tumour tissue were higher than those observed in healthy tissue from the same patient [28, 30].
Spears et al. also measured, besides the FdUMP concentrations, the dUMP concentrations in the biopsy specimens and expressed these concentrations against the observed percentages of thymidylate synthase inhibition [29]. This suggested that a minimum FdUMP concentration of 75 pmol/g and a minimal FdUMP/dUMP ratio of 0.001 was needed to achieve maximal thymidylate synthase inhibition [29].
In the above studies, the intracellular FdUMP concentrations were determined using a competitive binding assay with bacterial thymidylate synthase as a binding protein. The quantification of FdUMP was based on the displacement of a known amount of radiolabelled [6-3H]-FdUMP, measured by scintillation counting [28,29,30,31]. This “isotope-dilution” assay for the quantification of FdUMP was available since 1979 [31]. However, a sufficiently sensitive assay for the quantification of the other two active nucleotides, FUTP and FdUTP, in cells of patients who were treated with 5-FU was lacking for a long time.
Therefore, in 2015, Derissen et al. developed an ultrasensitive liquid chromatography-tandem mass spectrometry assay for simultaneous quantification of the three pharmacologically active 5-FU nucleotides FUTP, FdUTP and FdUMP in PBMCs [32]. As a start, the intracellular 5-FU nucleotide concentrations were measured in PBMC samples of five patients who were treated with an intravenous 5-FU bolus of 400 mg/m2. Thirty minutes after the end of the infusion, FUTP concentrations between 4.7 and 11 µM were measured. 5-Fluoro-2ʹ-deoxyuridine monophosphate concentrations were much lower with a range of 0.02–0.6 µM. The FdUTP concentrations were below the detection limits of the assay [32].
In a subsequent study, the intracellular PK of the three active 5-FU nucleotides were explored during capecitabine treatment [33]. As usual in these studies, PBMCs were selected as a surrogate matrix to measure the intracellular nucleotide concentrations. After treatment with capecitabine, only FUTP could be measured in PBMCs (Table 2). Apparently, capecitabine is mainly converted into FUTP and to a lesser extent into FdUTP and FdUMP. No significant correlation was found between the plasma 5-FU exposure and the intracellular FUTP exposure. The FUTP concentration–time profiles demonstrated considerable inter-individual variation. The study also showed that FUTP remained much longer present in PBMCs than 5-FU in plasma. Twenty-four hours after a single capecitabine dose of 1000 mg, intracellular FUTP concentrations were on average still 41% of the maximum concentrations found. In accordance with this finding, after 14 days of treatment with capecitabine (850 mg/m2) twice daily, intracellular accumulation of FUTP was found in PBMCs. The area under the FUTP concentration–time curve was significantly increased on day 14 of the treatment compared with day 1. Remarkably, the intracellular FUTP concentrations measured on day 14 of capecitabine treatment were in the same range (0.6–14 µM) as the concentrations observed 30 min after an intravenous 5-FU bolus (400 mg/m2 in 30 min) [33]. It is important to realise that, by measuring in PBMCs, the tumour-specific activation of capecitabine is not taken into account and that the concentrations in tumour cells might be different.
So far, to the best of our knowledge, it has not been possible to measure FdUTP in cells of patients who were treated with 5-FU or capecitabine. Perhaps an even more sensitive assay is needed to measure these FdUTP concentrations.
4 Cytarabine
Cytarabine (1-β-d-arabinofuranosylcytosine, ara-C) is a deoxycytidine analogue as it combines a cytosine base with an arabinose sugar. The difference with respect to cytidine is that the 2ʹ hydroxyl group of cytarabine is oriented in the trans position, resulting in a metabolism similar to that of a deoxyribose nucleoside.
After administration, cytarabine is rapidly converted to 1-β-d-arabinofuranosyluracil (ara-U) by the enzyme cytidine deaminase. Cytidine deaminase is ubiquitous in the human body and catalyses the hydrolytic deamination of (deoxy)cytidine to (deoxy)uridine (Fig. 2). Cytidine deaminase is present in plasma, but also in the liver and kidneys, where high levels are found [34]. After cytarabine administration, plasma ara-U peak concentrations were generally 5–30 times higher than the peak concentrations of cytarabine [35,36,37].
4.1 Intracellular Metabolism and Mechanism of Action
Like the other pyrimidine analogues, cytarabine has to enter the tumour cell, where it has to be activated by phosphorylation (Fig. 2). Cytarabine is transported into the cell primarily by the human equilibrative nucleoside transporter (hENT) 1 [38]. Once in the cell, cytarabine is phosphorylated to its monophosphate (ara-CMP) by deoxycytidine kinase (dCK) and then to cytarabine diphosphate (ara-CDP) by pyrimidine nucleoside monophosphate kinase (UMP-CMP kinase). The final phosphorylation step to cytarabine triphosphate (ara-CTP) is catalysed by nucleoside diphosphate kinase. Ara-CTP competes with deoxycytidine triphosphate (dCTP) for incorporation into the DNA. Once incorporated into DNA, further extension of the DNA strand by DNA polymerase is inhibited. Correlations have been found between cytarabine incorporation into DNA and cytotoxicity [39,40,41].
Within the cell, cytarabine is also subject to deamination by cytidine deaminase. Additionally, ara-CMP can be deaminated by deoxycytidylate deaminase [42].
4.2 Clinical Studies Investigating the Intracellular Concentrations of Cytarabine Nucleotides
4.2.1 Relationship Between Cytarabine Dose, Plasma Concentrations and Intracellular Ara-CTP Concentrations
Several clinical studies investigated the relationship between the cytarabine dose, the plasma PK and the intracellular PK of ara-CTP in leukaemic cells [43,44,45,46]. A linear relationship was found between the cytarabine dose and the steady-state concentrations and area under the concentration–time curves (AUCs) of cytarabine and ara-U in plasma. However, the intracellular PK of ara-CTP was much more heterogeneous [44,45,46]. No correlation was found between cytarabine plasma concentrations and the intracellular concentrations or AUC of ara-CTP [43]. Patients who had all received an identical cytarabine dose demonstrated substantial differences in the intracellular concentrations and AUCs of ara-CTP [44, 45]. However, within the same patient, measurements of the ara-CTP concentrations were reproducible with subsequent treatments [47]. The ability of leukaemic cells to phosphorylate cytarabine is characteristic to each individual [43]. Inter-individual differences in the ability of patients to phosphorylate cytarabine and retain ara-CTP are related to the ability to transport cytarabine into the cell and the activities of deoxycytidine kinase, cytidine deaminase and dephosphorylating enzymes.
When examining the PK of intracellular ara-CTP, there are also commonalities. The accumulation of ara-CTP in leukaemic cells was dependent on the dose regime; however, for an individual patient, the cellular elimination rate of ara-CTP remained constant under all infusion conditions. The elimination of ara-CTP from leukaemic cells was monophasic. The elimination rate differed substantially between patients. In the study of Liliemark et al., a median elimination half-life of 3.4 h (range 1.25–18.9 h) was found [43].
4.2.2 Saturation of the Intracellular Accumulation of Ara-CTP
The accumulation of ara-CTP in leukaemic cells was also dependent on the dose regime. In vitro studies in human leukaemic cells demonstrated that the intracellular accumulation of ara-CTP was saturated at cytarabine concentrations above 10 µmol/L [42, 48, 49]. Plunkett et al. 1987 studied the saturation of intracellular ara-CTP formation in patients and investigated which cytarabine infusion rate produced steady-state plasma concentrations that saturate the intracellular ara-CTP accumulation [44]. To this end, the intracellular ara-CTP accumulation in leukaemic cells of 11 patients with refractory leukaemia was studied after different cytarabine doses infused over 2 h. Varying the cytarabine dose between 500 and 3000 mg/m2 infused over 2 h did not substantially change the accumulation rate and the AUC of ara-CTP in leukaemic cells. Only when the infusion rate was further reduced to 400 or 300 mg/m2 over 2 h, resulting in steady-state cytarabine plasma concentrations of less than 7 µM, the accumulation rate of ara-CTP was substantially reduced as was the AUC of intracellular ara-CTP [44]. This study demonstrated that infusion rates of 200–250 mg/m2/h produce cytarabine plasma concentrations that saturate the intracellular ara-CTP accumulation in leukaemic cells.
4.2.3 Optimisation of Dosing Regimes in Relation to the Intracellular Accumulation of Ara-CTP
Remarkably, for more than two decades, not much was done with the finding that cytarabine infusion rates above 200–250 mg/m2/h led to saturation of the intracellular ara-CTP formation. Until 2011, high-dose cytarabine regimes remained common practice for the treatment of patients with AML [50,51,52]. High cytarabine doses were given with a high infusion rate, typically 3000 mg/m2 over 1–3 h. These dosing regimes led to peak plasma concentrations that were about 5–15 times higher than the plasma concentrations that caused saturation of intracellular triphosphate formation in leukaemic cells [43, 53,54,55]. From a pharmacological perspective, this was not rational. Indeed, substantial toxicities were associated with these high-dose cytarabine regimes [56,57,58,59,60]. It is likely that plasma cytarabine in excess of the amount that can be phosphorylated by leukaemic cells will contribute to the observed toxicity, either directly or after deamination to ara-U [45, 61].
It was only in 2011 that clinical research by Löwenberg et al. showed that an intermediate-dose remission induction regime with an infusion rate just above the ‘saturation infusion rate’ was as effective as high-dose cytarabine, but was associated with less toxicity [62]. The idea is that these intermediate infusion rates still saturate the intracellular ara-CTP formation in leukaemic cells, but result in a substantial decrease of the peak plasma concentrations of cytarabine and ara-U leading to less toxicity [63].
In retrospect, you may wonder why it took so long before the findings from clinical pharmacological research were translated into clinical practice. Although many studies have been conducted over the last 30 years to find the most suitable cytarabine doses, little attention has been paid to the infusion rates. Yet, these infusion rates also determine how much cytarabine is converted intracellularly into ara-CTP, particularly in view of the short plasma half-life of cytarabine.
4.2.4 Relationship Between Intracellular Ara-CTP Concentrations and Efficacy
Several studies investigated the relationship between the accumulation of ara-CTP in leukaemic cells and the response to cytarabine therapy. Kantarjian et al. and Estey et al. investigated the relationship between the intracellular PK of ara-CTP in circulating blasts and the response of patients with refractory AML to remission induction therapy [47, 64]. Patients were treated with ‘high-dose cytarabine’ and received 3000 mg/m2 infused over 2 h, every 12 h for 4–12 doses. Intracellular ara-CTP concentrations were measured after the first dose and differed largely among patients. In the first study, a trend was observed and in the follow-up study a significant correlation was found between the AUC of intracellular ara-CTP and the complete remission rate. In addition to the AUC, a long half-life of ara-CTP and a high ara-CTP trough concentration correlated with a high complete remission rate. The number of patients achieving a complete remission was too small to investigate the association between the PK of ara-CTP and the remission duration [47, 64].
The subsequent study of Estey et al. did not confirm the correlation between the intracellular ara-CTP exposure and the remission rate [65]. However, this probably had to do with the fact that the PK of ara-CTP was investigated after a single gift of high-dose cytarabine for all patients, whereas these patients were subsequently treated according to three different treatment protocols probably resulting in different intracellular ara-CTP concentrations [65].
Rustum and Preisler [66] and Preisler et al. [67, 68] did not measure the intracellular ara-CTP concentrations during cytarabine therapy, but isolated leukaemic blasts from more than 80 patients with AML prior to therapy, incubated these blasts with [3H]-cytarabine and determined the percentage of ara-CTP that was retained after 4 h. Then, they studied the relationship between the in vitro retention of ara-CTP in the isolated blasts and patients’ response to subsequent therapy with cytarabine and an anthracycline. As the ara-CTP formation in leukaemic blasts was studied after incubation and not during treatment with cytarabine, this series of three studies did not meet the inclusion criteria of this review. Therefore, the results were not included in Tables 1 and 2. Nevertheless, we briefly mention this series here because it has contributed to a better understanding of the relationship between intracellular ara-CTP retention and the efficacy of cytarabine therapy. The design of the studies was not appropriate to examine the relationship between the initial ara-CTP formation and the probability of achieving a complete remission, as patients received continuous cytarabine infusions during induction therapy. However, all three studies did find a significant correlation between the in vitro retention of ara-CTP in blasts and the duration of remission of patients receiving cytarabine as part of their remission induction and maintenance therapy [66,67,68].
4.2.5 Role of Arabinosyluracil Triphosphate (ara-UTP) During Cytarabine Therapy
Gandhi et al. demonstrated that, during cytarabine (1-β-d-arabinosylcytosine; ara-C) therapy, arabinosyluracil 5ʹ-triphosphate (ara-UTP) was also formed in circulating leukaemic blasts [37]. The intracellular PK of ara-UTP was investigated in six patients with relapsed AML receiving cytarabine infusions of 500 mg/m2/h during 2–4 h. The intracellular peak concentrations of ara-UTP had a range of 6–50 µM and were between 2 and 10% of the peak concentrations that were measured for ara-CTP. Ara-UTP was maintained for several hours after the end of the cytarabine infusion. The intracellular ara-UTP peak concentrations were not related to the peak concentrations of cytarabine and ara-U in plasma. However, a linear relationship was found between the intracellular ara-CTP and ara-UTP peak concentrations. These data indicate that the formation of ara-UTP may be dependent on ara-CTP and not on the free nucleoside [37]. This is in line with previous in vitro studies. Incubation of leukaemic blasts with up to 100 µM [3H]-ara-U did not result in ara-UTP accumulation, indicating that ara-U is not phosphorylated directly in these cells [69]. In vitro studies, investigating the route of ara-UTP formation showed that ara-UTP was formed through the deamination of ara-CMP to ara-UMP by deoxycytidylate deaminase, followed by phosphorylation to ara-UTP [37].
The question remains as to whether ara-UTP also contributes to the cytotoxic effects of cytarabine. In vitro studies in leukaemic cells demonstrated that ara-UTP served as a good alternative substrate for deoxythymidine triphosphate for incorporation into DNA [70] and inhibited DNA polymerase [71, 72]. Hence, it could be postulated that ara-UTP may play a role in cytarabine-induced cytotoxicity [71, 73].
5 Gemcitabine
Gemcitabine (2ʹ,2ʹ-difluoro-2ʹ-deoxycytidine, dFdC) is a deoxycytidine analogue in which the two hydrogen atoms at the 2ʹ-position of the deoxyribose moiety are replaced by fluorine atoms. After administration, gemcitabine is rapidly cleared from plasma by deamination. More than 90% of the gemcitabine dose is converted into 2ʹ,2ʹ-difluoro-2ʹ-deoxyuridine (dFdU) by cytidine deaminase [74].
5.1 Intracellular Metabolism and Mechanism of Action
Like the other pyrimidine analogues, gemcitabine has to enter the tumour cell, where it has to be activated by phosphorylation (Fig. 2). Cellular uptake of gemcitabine is largely mediated by hENT 1 and, to a lesser extent, also by hENT2 and the human concentrative nucleoside transporters 1 and 3 [75, 76]. Once inside the cell, gemcitabine (dFdC) is phosphorylated in the cytoplasm by dCK to its monophosphate (dFdCMP) and then by pyrimidine nucleoside monophosphate kinase (UMP-CMP kinase) to gemcitabine diphosphate (dFdCDP). The enzyme responsible for the final phosphorylation step, the phosphorylation of dFdCDP to gemcitabine triphosphate (dFdCTP), is unclear, although nucleoside diphosphate kinase may play this role [77].
Intracellularly formed dFdCTP and dFdCDP are responsible for the cytotoxic effects of gemcitabine. dFdCTP competes with dCTP for incorporation into the DNA. Incorporation of dFdCTP into DNA leads to inhibition of the DNA synthesis and apoptosis of the cell [78, 79]. The mechanism of action of gemcitabine is self-potentiated by inhibition of two enzymes involved in the deoxynucleotide metabolism. dFdCDP inhibits the enzyme ribonucleotide reductase, which catalyses the reduction of ribonucleotides to deoxyribonucleotides [80]. By inhibiting this enzyme, the intracellular deoxyribonucleotide triphosphate pool is depleted. This increases the dFdCTP:dCTP ratio, which favours the incorporation of dFdCTP into the DNA. This effect is enhanced in several ways. dFdCTP directly inhibits the enzyme deoxycytidylate deaminase [81]. The same enzyme is also inhibited indirectly by the reduced intracellular deoxyribonucleotide triphosphate pool. As dCK activity is also regulated by dCTP, lowering the deoxyribonucleotide triphosphate pool also promotes gemcitabine phosphorylation [82, 83].
Like in plasma, also in the cell, a major form of elimination of gemcitabine is deamination to dFdU by cytidine deaminase. Likewise, dFdCMP is deaminated by deoxycytidylate deaminase [81, 84]. Gemcitabine can also become inactivated by dephosphorylation of the monophosphate form by 5ʹ-nucleotidases, converting nucleotides back to nucleosides [82].
5.2 Clinical Studies Investigating the Intracellular Concentrations of Gemcitabine Nucleotides
5.2.1 Saturation of the Intracellular Accumulation of dFdCTP
The intracellular metabolism of gemcitabine follows the same pathways as that of cytarabine (Fig. 2). Therefore, in the early 1990s, the intracellular accumulation of dFdCTP was expected to exhibit similar saturation kinetics as previously demonstrated for ara-CTP [85]. Grunewald et al. confirmed that the ability of PBMCs to convert gemcitabine into dFdCTP was saturable. In a phase I study, including 22 patients with solid tumours, gemcitabine doses between 53 and 1000 mg/m2 (infused over 30 min) were compared. The rate of dFdCTP accumulation in PBMCs initially increased with higher doses, but was found to be maximal at a dose of 350 mg/m2 infused over 30 min. At this dose rate, gemcitabine steady-state plasma concentrations of 15–20 µM were measured. Although the study demonstrated considerable inter-individual variation, above a gemcitabine steady-state concentration of 20 µM, no further increase of the dFdCTP AUC was found for the majority of patients [74, 85].
5.2.2 Increasing the Intracellular Accumulation of dFdCTP by Prolonging the Infusion Time
Subsequent studies attempted to maximise the intracellular formation of dFdCTP by giving more prolonged gemcitabine infusions at an infusion rate that would presumably saturate the intracellular dFdCTP formation. An infusion rate of 10 mg/m2/min was selected. This infusion rate was previously found to produce steady-state plasma concentrations above 15–20 µM [86].
Grunewald et al. conducted a phase I study with the administration of gemcitabine at this fixed dose rate and increased the dose intensity by prolonging the duration of the infusion (from 120 to 640 min in six steps) [87]. The mean steady-state plasma concentration of gemcitabine during infusion at 10 mg/m2/min was 26.5 µM, with a range of 13.0–53.5 µM. Only four of the 17 patients had steady-state plasma concentrations below 20 µM. For 12 of the 15 assessable patients, linear accumulation of intracellular dFdCTP was seen during the infusion of gemcitabine. In the remaining three patients, the intracellular dFdCTP accumulation reached a plateau during the infusion. The accumulation rate of dFdCTP in circulating leukaemic cells varied considerably among individuals, with a range of 9.7–789 µmol/L/h [87].
Since then, abundant evidence has been published that the intracellular accumulation of dFdCTP in PBMCs and leukaemic blasts indeed increased by prolonging the infusion time (Table 2) [88,89,90]. Patel et al. demonstrated that maximum intracellular dFdCTP concentrations were significantly higher if gemcitabine 1000 mg/m2 was given as a 150-min infusion compared with a standard 30-min infusion (median increase 1.4-fold, range 1.0- to 2.6-fold) [88]. Cattel et al. compared gemcitabine 300 mg/m2 infused over 1, 2 or 3 h with a conventional gemcitabine dose of 1000 mg/m2 infused in 30 min [90]. The intracellular dFdCTP accumulation in leucocytes was highest in patients who received gemcitabine 300 mg/m2 infused over 3 h. Indicating that, to achieve the highest possible dFdCTP concentrations, a prolonged infusion time was more important than a high dose administered as a short infusion [90].
Grimison et al. performed a cross-over study in which patients received a gemcitabine dose of 1000 mg/m2 over either 30 or 100 min [91]. At the second week, each patient received the alternate schedule. Intra-patient comparison showed that the intracellular dFdCTP exposure in leucocytes was significantly higher following the 100-min infusion. However, the effect was confounded by an increase in dFdCTP exposure in the second week compared to the first week, which was consistent with self-induction of the gemcitabine metabolism. Moreover, significant variations in intracellular dFdCTP exposure were observed both between and within individuals. We have to realise that self-induction of the gemcitabine metabolism (among others due to dCK activation) implicates that the optimal dose rate for saturation of the intracellular accumulation changes over time. Because of self-induction and inter-individual differences, a dose rate of 10 mg/m2/min will not be optimal for all patients all of the time [91].
5.2.3 Relationship between Intracellular dFdCTP Concentrations and Efficacy
Other than for cytarabine, for gemcitabine, there are no studies that investigated the relationship between the dFdCTP concentrations in cells of patients treated with a particular gemcitabine dose and treatment efficacy. Various clinical studies did compare the efficacy of prolonged administration of gemcitabine at a fixed-dose rate (FDR) (e.g. 10 mg/m2/min) with conventional 30-min infusion of gemcitabine (1000–1250 mg/m2) [92,93,94,95,96,97,98]. However, although these studies were all based on the principle that prolonged infusion would maintain gemcitabine plasma concentrations that result in saturation of the intracellular dFdCTP formation, only a few studies actually measured the intracellular concentrations of dFdCTP (Table 2) [91, 92, 95, 98].
One randomised phase II study showed a survival advantage for prolonged FDR infusion of gemcitabine in patients with advanced pancreatic cancer [92]. In this study, prolonged FDR infusion (1500 mg/m2 in 150 min) was compared with standard gemcitabine infusion (2200 mg/m2 in 30 min) in 92 patients [92]. The dose levels for both regimes were based on the maximum tolerated doses established in previous studies [99,100,101]. During the 150-min FDR infusion, the dFdCTP concentration in PBMCs increased linearly, whereas after the end of the 30-min infusion the accumulation rate of dFdCTP decreased markedly. Consistently, the maximum dFdCTP concentrations measured in PBMCs were significantly higher after the FDR infusion (median: 398 vs 188 µM). Median survival was longer for patients in the FDR arm than for those in the standard arm (8.0 vs 5.0 months; p = 0.013), and also the 1- and 2-year survival rates were significantly higher following the FDR infusion [92].
However, the confirmatory phase III trial in 832 patients with advanced pancreatic cancer failed to show a significant advantage with a prolonged infusion schedule [98]. In this study, three treatment arms were compared, namely standard gemcitabine 1000 mg/m2 infused in 30 min (GEM), FDR gemcitabine 1500 mg/m2 infused in 150 min (GEM-FDR) and gemcitabine 1000 mg/m2 infused in 100 min in combination with oxaliplatin (GEMOX). It should be noted that patients received a 1.5-fold higher gemcitabine dose in the GEM-FDR arm. The AUC of intracellular dFdCTP measured in PBMCs was significantly increased for both GEM-FDR and GEMOX compared with GEM (Table 2). However, neither GEM-FDR nor GEMOX resulted in a significantly improved overall survival, progression-free survival or response rate compared to GEM [98].
In the phase III study of Gridelli et al., 440 patients with advanced non-small cell lung cancer were treated with a combination of cisplatin and gemcitabine and the effect of FDR gemcitabine (1200 mg/m2) infusion in 120 min was compared with standard 30-min infusion. Also, in this patient population, FDR infusion of gemcitabine did not improve overall survival, progression-free survival or the response rate compared to standard infusion. Unfortunately, this study did not examine the PK of gemcitabine or dFdCTP [97].
5.2.4 Interpretation and Future Perspectives
Overall, there is thus no evidence that a prolonged infusion of gemcitabine at a fixed dose rate results in better treatment efficacy than conventional infusion in 30 min. This is not unexpected. Prolonged gemcitabine infusions at a fixed dose rate aim at a more efficient accumulation of the intracellular anabolites dFdCDP and dFdCTP. If both the antineoplastic effects and the toxic effects of gemcitabine are equally and exclusively mediated by dFdCDP and dFdCTP, more efficient administration of gemcitabine also means that less gemcitabine can be administered before dose-limiting toxicities are achieved. Looking at the phase I dose-escalation studies performed, the maximum tolerated dose for a prolonged FDR infusion of gemcitabine was indeed lower than the maximum tolerated dose for a conventional 30-min infusion due to myelotoxicity (1500–1800 vs 2200 mg/m2/week; for 3 weeks every 4 weeks) [99,100,101]. If a prolonged FDR infusion is adjusted to this lower maximum tolerated dose, and the total amount of gemcitabine administered is reduced, no increase in efficacy is to be expected.
If we really want to improve the dosing regimen of gemcitabine, we need to find out if there are places where the mechanisms underlying toxicity do not completely parallel the mechanisms that are responsible for the antineoplastic effects of gemcitabine. One could think of differences in the accumulation rate between target tissues and healthy tissues due to different membrane transporter capacities or enzyme capacities, differences in cell division rate, but also of other gemcitabine metabolites that might contribute to the dose-limiting toxicities. In this light, the role of the deaminated metabolite dFdU could be interesting.
5.2.5 Role of dFdU Nucleotides During Gemcitabine Therapy
Although dFdU was traditionally considered as an inactive metabolite, in vitro studies demonstrated that dFdU has cytotoxic activity in tumour cell lines [102,103,104]. The half-maximal inhibitory concentration (IC50) for dFdU in gemcitabine-sensitive tumour cell lines was at least 1000- to 2000-fold higher than for gemcitabine [102,103,104]. However, during gemcitabine treatment, the exposure to dFdU highly exceeds the exposure to gemcitabine [74, 105]. The question is therefore what the contribution of dFdU is to the cytotoxic effect of gemcitabine.
In vitro studies demonstrated that dFdU is taken up into cells, for instance by human concentrative nucleoside transporter 1 [103]. In the cell, dFdU can be phosphorylated to its monophosphate (dFdUMP), diphosphate (dFdUDP) and triphosphate (dFdUTP) [103]. Alternatively, dFdCMP can be deaminated to dFdUMP, by deoxycytidylate deaminase, and then further phosphorylated [81, 84]. In vitro studies also demonstrated that dFdUTP is incorporated into both DNA and RNA and that the extent of incorporation correlated with the cytotoxicity of dFdU [103].
Veltkamp et al. [106] performed a feasibility study with orally administered gemcitabine and demonstrated for the first time that, besides dFdCTP, dFdUTP was also formed in PBMCs of patients. After oral administration, the systemic gemcitabine exposure proved to be very low as a result of extensive first-pass metabolism to dFdU. Consistently, the intracellular dFdUTP exposure in PBMCs was significantly higher than the dFdCTP exposure. One patient died during the study experiencing grade 4 hepatic failure. It was noted that the exposure to dFdUTP was excessively high in this patient. This suggested that dFdUTP could have played a role in the toxicity experienced by this patient [106].
Derissen et al. [105] explored the intracellular PK of both the dFdC nucleotides and the dFdU nucleotides (i.e. dFdUMP, dFdUDP and dFdUTP) during intravenous gemcitabine treatment. Both in plasma and in PBMCs, dFdU was present in higher concentrations than gemcitabine. However, dFdU was only phosphorylated to a very limited extent in PBMCs. For all patients, intracellular dFdU nucleotide concentrations were much lower than the gemcitabine nucleotide concentrations. This suggests that the contribution of dFdU nucleotides to the cytotoxic effect of gemcitabine is limited [105].
6 Azacitidine and Decitabine
Azacitidine (aza-C) and decitabine (5-aza-2ʹ-deoxycytidine; aza-dC) are analogues of cytidine and deoxycytidine, respectively, in which the carbon atom at position 5 of the pyrimidine ring has been replaced by a nitrogen atom. Azacitidine and decitabine are rapidly cleared from plasma. Azacitidine undergoes spontaneous hydrolysis or is deaminated to 5-aza-uridine (aza-U) by cytidine deaminase [109, 110]. Decitabine is primarily eliminated through deamination to 5-aza-2ʹ-deoxyuridine (aza-dU) by cytidine deaminase [109, 111, 112].
6.1 Intracellular Metabolism and Mechanism of Action
Cellular uptake of azacitidine is mediated by all seven human nucleoside transporters, with human concentrative nucleoside transporter 3 showing the highest transport rates [107]. Decitabine is transported into cells by hENT1 and 2 [107]. As azacitidine is an analogue of cytidine and decitabine an analogue of deoxycytidine, different enzymes are involved in their intracellular metabolism (Fig. 2). Once in the cell, azacitidine is phosphorylated to 5-azacytidine monophosphate (aza-CMP) by uridine-cytidine kinase and then to 5-azacytidine diphosphate (aza-CDP) and 5-azacytidine triphosphate (aza-CTP) by UMP-CMP kinase and nucleoside diphosphate kinase, respectively. A small fraction, about 10–20%, of the aza-CDP is instead converted to 5-aza-2ʹ-deoxycytidine diphosphate (aza-dCDP) via the enzyme ribonucleotide reductase, followed by further phosphorylation to 5-aza-2ʹ-deoxycytidine triphosphate (aza-dCTP) [108,109,110]. Decitabine is, after uptake in the cell, converted to its monophosphate (aza-dCMP) by deoxycytidine kinase. dCMP kinase and nucleoside diphosphate kinase convert the monophosphate to its diphosphate form (aza-dCDP) and triphosphate form (aza-dCTP), respectively [111]. Like in plasma, azacitidine and decitabine are also in the cell primarily eliminated by deamination by cytidine deaminase. Likewise, aza-dCMP is deaminated to 5-aza-2ʹ-deoxyuridine monophosphate (aza-dUMP) by deoxycytidylate deaminase [111].
Intracellularly formed aza-CTP and aza-dCTP are responsible for the pharmacological effects of azacitidine and decitabine. Aza-CTP is incorporated into RNA. This ultimately disrupts messenger RNA metabolism and protein synthesis, leading to apoptosis [109, 110]. Furthermore, incorporation of aza-CTP into RNA inhibits ribonucleotide reductase, leading to a reduced deoxyribonucleotide pool, which has deleterious consequences for DNA synthesis and repair [109]. Aza-dCTP competes with endogenous dCTP for DNA incorporation. At high doses, aza-dCTP incorporation into DNA leads to inhibition of DNA synthesis and cell death [111, 112]. However, at lower doses, a smaller amount of aza-dCTP is incorporated into the DNA, which primarily causes inhibition of DNA methylation [111]. DNA methylation has an important role in the regulation of gene expression. Aberrant DNA methylation has a place in the pathogenesis of several malignancies including MDS and AML [113, 114]. The most studied change of DNA methylation is the silencing of tumour suppressor genes by hypermethylation of the CpG islands within their promoter regions [115]. The enzymes responsible for DNA methylation are DNA methyltransferases (DNMTs). When aza-dCTP is incorporated into the DNA, covalent bonds are formed between the aza-dC in the DNA and these DNMTs. At high doses, the DNA is not able to recover and cell death occurs. However, at lower doses, the formed DNA-DNMT adducts are degraded by the proteosome, after which the DNA is restored. DNA synthesis is then resumed in the absence of DNMTs. As a consequence, the aberrant DNA hypermethylation patterns, which play an important role in the pathogenesis of the mentioned hematologic abnormalities, can no longer be reproduced toward the daughter strands [116]. In this manner, a low dose of azacitidine or decitabine is able to induce re-expression of previously silenced genes. Reactivation of cell cycle-regulating genes (i.e. tumour suppressor genes), which were initially silenced due to hypermethylation, may restore cell differentiation and normal growth control in the daughter cells [110, 117, 118].
6.2 Clinical Studies Investigating the Intracellular Concentrations of Azacitidine and Decitabine Nucleotides
In the 1970s and 1980s, the anti-leukaemic effects of azacitidine and decitabine were studied in high doses based on the maximum tolerated dose. The mechanism of action at such high doses is based on cytotoxicity. From the 1990s, researchers also started to study low azacitidine and decitabine doses, based on the idea of their demethylating effect [119,120,121,122]. It was only at these low doses that the therapeutic benefits of azacitidine and decitabine were demonstrated [123, 124]. Both drugs were therefore registered in low doses.
Few clinical studies have examined the intracellular PK of the active nucleotides aza-CTP and aza-dCTP. Other than for the other pyrimidine analogues discussed in this review, for low-dose azacitidine and decitabine therapy, we are not interested in maximising intracellular triphosphate concentrations to maximise the effect. The optimal dose is more nuanced and comes down to the dose that maximises the demethylating effect without the cytotoxic effect gaining the upper hand.
Only at the beginning of this decade, three studies investigated the intracellular PK of the active metabolites aza-CTP and aza-dCTP in patients who were treated with azacitidine or decitabine [125,126,127]. The main objective of these studies was to gain insight into inter-individual differences in the intracellular PK, especially as some patients experience no effect from treatment with azacitidine or decitabine or become resistant to the treatment at some point [128].
Jansen et al. investigated the intracellular aza-dCMP, aza-dCDP and aza-dCTP concentrations in PBMCs obtained from three patients diagnosed with MDS who were treated with decitabine 15 mg/m2 administered as a 4-h infusion, repeated every 8 h for 3 days with a cycle duration of 6 weeks [125]. In contrast to plasma decitabine, intracellular aza-dCTP was found to accumulate during the treatment cycle of nine infusions. Two patients responded well to decitabine therapy. Remarkably, considerable aza-dCTP concentrations were measured in the PBMCs from these two patients, whereas only low concentrations were present in the third patient who did not respond. The authors also searched for the possible presence of deaminated decitabine nucleotides, i.e. 5-aza-2ʹ-deoxyuridine nucleotides, but these were not observed [125].
Wang et al. investigated the intracellular aza-dCTP concentrations in bone marrow cells and PBMCs from seven patients with AML who were treated with decitabine 20 mg/m2 infused in 1 h for 10 days [126]. Samples were collected on days 1 and 5 within 3 h after completion of the decitabine infusion. Other than in the study by Jansen et al., repeated dosing did not result in accumulation of intracellular aza-dCTP during the treatment cycle. Aza-dCTP concentrations were found to be relatively stable from day 1 to 5. In line with the study by Jansen et al., higher intracellular aza-dCTP concentrations also appeared to be associated with clinical response in this study. Patients who eventually achieved a complete response had higher aza-dCTP concentrations in both bone marrow and PBMCs than those who did not respond to the treatment. However, also in this explorative study, the number of patients was too small to draw definitive conclusions [126].
Following these two studies with decitabine triphosphate (aza-dCTP), Derissen et al. developed an assay to quantify azacitidine triphosphate (aza-CTP) in PBMCs [127]. This proved to be more complex than it had been for aza-dCTP. High-resolution mass spectrometry had to be used, because regular liquid chromatography-tandem mass spectrometry could not distinguish between aza-CTP and some of the endogenous nucleotides. The developed assay was applied to quantify aza-CTP in PBMCs of two patients diagnosed with MDS who were treated with azacitidine 75 mg/m2 subcutaneously for 7 days, every 4 weeks. The aza-CTP concentrations measured were about 10–25 times higher than the aza-dCTP concentrations found by Jansen et al. and Wang et al. (Table 2) [125,126,127].
We have not found any other studies that determined the intracellular nucleotide concentrations after azacitidine therapy. Nevertheless, the recent study by Unnikrishnan et al. is very interesting to mention in this light [128]. This study examined the successive steps in the pharmacology of azacitidine for eight patients diagnosed with MDS or chronic myelomonocytic leukaemia, and showed that responders to azacitidine therapy demonstrated more aza-dC incorporation into DNA and more DNA demethylation than non-responders. Unfortunately, the intracellular aza-CTP and aza-dCTP concentrations were not measured in this study. Instead, all intracellular nucleotides were enzymatically dephosphorylated during sample preparation, after which the total intracellular aza-C and aza-dC concentrations were measured. Interestingly, the non-responders, showing minimal DNA incorporation and minimal DNA demethylation, showed high intracellular aza-C and aza-dC concentrations (after dephosphorylation) as well as high aza-C incorporation into RNA. This means that the low aza-dC DNA incorporation could not be explained by limited cellular uptake and that the intracellular metabolism (at least the RNA pathway) was also intact. The authors suggest that aza-dC incorporation into DNA might be low in patients if an increased proportion of the bone marrow cells is quiescent and not undergoing DNA replication [128]. It would be interesting to measure the aza-CTP and aza-dCTP concentrations in these patients and examine if they correlate with DNA incorporation and response.
6.2.1 Interpretation and Future Perspectives
Only three small studies were found that examined the intracellular PK of azacitidine and decitabine nucleotides. Nevertheless, the two exploratory studies conducted so far for decitabine both suggest a relationship between intracellular aza-dCTP concentrations and clinical response to decitabine therapy. This relationship should be further investigated in larger numbers of patients. In addition, this relationship should also be investigated for azacitidine. If a relationship between intracellular triphosphate concentrations and response is confirmed, the intracellular concentrations of aza-CTP or aza-dCTP can serve as a biomarker for early identification of individuals who are unlikely to respond to treatment with azacitidine or decitabine. Further studies should then show whether these patients would benefit from a higher azacitidine or decitabine dose or whether they would be better off switching to another therapy.
7 Conclusions and Discussion
7.1 Differences Between Pyrimidine Analogues
For cytarabine and gemcitabine, the intracellular PK of the active nucleotides has been extensively studied over the years. However, for 5-FU, capecitabine, azacitidine and decitabine, the intracellular PK of the active nucleotides has only been studied to a very limited extent. This is remarkable. Of course, also for the latter agents, it is useful to gain insight into the intracellular PK of their active metabolites. For a long time, however, there were no suitable bioanalytical assays that made it possible to quantify the nucleotides of 5-FU, capecitabine, azacitidine and decitabine in cells.
In the past decade, using modern analytical techniques, we have managed to develop suitable bioanalytical assays to quantify also the nucleotides of these pyrimidine analogues [32, 125, 127]. Indeed, quantification of the active nucleotides of 5-FU, capecitabine, azacitidine and decitabine proved more difficult than quantifying cytarabine triphosphate and gemcitabine triphosphate [32, 125, 127]. Azacitidine and decitabine are very similar to endogenous nucleotides and also have the same molecular mass, which makes quantification by liquid chromatography-tandem mass spectrometry difficult [125, 127]. In addition, the intracellular nucleotide concentrations during treatments with 5-FU, capecitabine, azacitidine and decitabine were found to be very low compared with the intracellular nucleotide concentrations during treatment with cytarabine or gemcitabine (Table 2). Now that suitable bioanalytical assays are finally also available for quantifying the active nucleotides of 5-FU, capecitabine, azacitidine and decitabine, it is time that the intracellular PK of these pyrimidine analogues is also better studied.
7.2 Relationship Between the Intracellular Nucleotide Concentrations and the Antineoplastic Effect
Based on pharmacology, a relationship is expected between the intracellular concentrations of the active nucleotides and the cytotoxic effects of pyrimidine analogues. For cytarabine and gemcitabine, this relationship has also been demonstrated in in vitro studies and animal studies [41, 129]. However, not many clinical studies have investigated the relationship between intracellular nucleotide concentrations and the clinical outcome in patients. Only for cytarabine, a relationship was demonstrated between the intracellular triphosphate concentrations in leukaemic cells and the response rate in patients with AML [64]. Future clinical studies should show, for the other pyrimidine analogues, whether there is a relationship between the intracellular nucleotide concentrations and the therapeutic effect in patients.
7.3 Optimisation of Dosing Regimes
Research that examined the intracellular PK of cytarabine and gemcitabine focused primarily on the saturation aspect of the intracellular triphosphate formation [44, 45, 74, 85]. Attempts to improve the dosing regimen of gemcitabine were aimed at maximising the intracellular gemcitabine triphosphate concentrations [88,89,90,91,92, 95, 98]. However, if the dose is based on the maximum tolerated dose and the mechanisms underlying the therapeutic effect and toxicity are exactly the same, this strategy does not make sense. Efficient administration of gemcitabine, aimed at maximum triphosphate formation, also means that less gemcitabine can be administered before dose-limiting toxicities are achieved [99,100,101]. In all cases, the dosage is limited by the ‘maximum tolerated intracellular triphosphate exposure’.
If we really want to improve the dosing regimens of pyrimidine analogues, we will have to examine if there are areas where the mechanism of action and the mechanism of toxicity are not completely parallel. One could think of differences in cellular uptake or phosphorylation capacity between healthy cells and tumour cells. It is also useful to investigate the effects of high plasma peaks, which far exceed plasma concentrations that saturate the intracellular triphosphate formation. What happens to the excess of pyrimidine analogue that is not phosphorylated intracellularly? What is, for instance, the effect of deamination products such as ara-U and dFdU?
A recent study investigating the intracellular PK of gemcitabine showed that dFdU was only phosphorylated to a very limited extent in PBMCs [105]. Future studies should show whether dFdU is also hardly phosphorylated in tumour cells. If this is the case, dFdU is unlikely to contribute substantially to the cytotoxic effects of gemcitabine. In this light, it would be rational to pay more attention to inter-individual differences in the deamination rate of gemcitabine to dFdU. Some clinical studies have shown that gemcitabine therapy was more effective and more toxic in patients with low cytidine deaminase activity, i.e. a decreased deamination of gemcitabine to dFdU [50,51,52]. Future studies should show whether individualisation of the gemcitabine dose, based on the measured cytidine deaminase activity, leads to better treatment results.
7.4 Inter-Individual Differences
For all pyrimidine analogues, a linear relationship was found between the dose and the plasma concentration [18, 43,44,45,46, 74, 130]. However, no correlation was found between the plasma concentration and the intracellular nucleotide concentration [33, 43, 85]. The concentration–time curves for the intracellular nucleotides showed considerable inter-individual variation. This is not unexpected as many different proteins are involved in cellular uptake and the extensive intracellular metabolism of the pyrimidine analogues. The activity of all these membrane transporters and enzymes might vary from patient to patient.
Given the considerable inter-individual differences observed for the intracellular nucleotide concentrations, the question arises whether pyrimidine analogue therapy should be more individualised. Future research should show which intracellular nucleotide concentrations are worth pursuing and whether dose individualisation is useful to achieve these concentrations. In addition, the possibility of using the intracellular nucleotide concentration as a predictive marker for the therapeutic effect might be investigated. In this way, potential non-responders, owing to inadequate intracellular metabolism, could be recognised early.
References
de Vos D, van Overveld W. Decitabine: a historical review of the development of an epigenetic drug. Ann Hematol. 2005;84(Suppl. 1):3–8.
McCredie KB, Bodey GP, Burgess MA, et al. Treatment of acute leukemia with 5-azacytidine (NSC-102816). Cancer Chemother Rep. 1973;57:319–23.
Vogler WR, Miller DS, Keller JW. 5-Azacytidine (NSC 102816): a new drug for the treatment of myeloblastic leukemia. Blood. 1976;48:331–7.
Abele R, Clavel M, Dodion P, et al. The EORTC Early Clinical Trials Cooperative Group experience with 5-aza-2′-deoxycytidine (NSC 127716) in patients with colo-rectal, head and neck, renal carcinomas and malignant melanomas. Eur J Cancer Clin Oncol. 1987;23:1921–4.
Merriman RL, Hertel LW, Schultz RM, et al. Comparison of the antitumor activity of gemcitabine and ara-C in a panel of human breast, colon, lung and pancreatic xenograft models. Investig New Drugs. 1996;14:243–7.
Hoff PM, Cassidy J, Schmoll HJ. The evolution of fluoropyrimidine therapy: from intravenous to oral. Oncologist. 2001;6(Suppl. 4):3–11.
Wijermans PW, Krulder JW, Huijgens PC, et al. Continuous infusion of low-dose 5-Aza-2′-deoxycytidine in elderly patients with high-risk myelodysplastic syndrome. Leukemia. 1997;11(Suppl. 1):S19–23.
Wijermans P, Lübbert M, Verhoef G, et al. Low-dose 5-aza-2′-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol. 2000;18:956–62.
Silverman LR, Holland JF, Weinberg RS, et al. Effects of treatment with 5-azacytidine on the in vivo and in vitro hematopoiesis in patients with myelodysplastic syndromes. Leukemia. 1993;7(Suppl. 1):21–9.
Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–40.
Derissen EJB, Beijnen JH, Schellens JHM. Concise drug review: azacitidine and decitabine. Oncologist. 2013;18:619–24.
Diasio RB, Harris BE. Clinical pharmacology of 5-fluorouracil. Clin Pharmacokinet. 1989;16:215–37.
De Bono JS, Twelves CJ. The oral fluorinated pyrimidines. Investig New Drugs. 2001;19:41–59.
Álvarez P, Marchal JA, Boulaiz H, et al. 5-Fluorouracil derivatives: a patent review. Expert Opin Ther Pat. 2012;22:107–23.
Miwa M, Ura M, Nishida M, et al. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer. 1998;34:1274–81.
Ishikawa T, Utoh M, Sawada N, et al. Tumor selective delivery of 5-fluorouracil by capecitabine, a new oral fluoropyrimidine carbamate, in human cancer xenografts. Biochem Pharmacol. 1998;55:1091–7.
Schüller J, Cassidy J, Dumont E, et al. Preferential activation of capecitabine in tumor following oral administration to colorectal cancer patients. Cancer Chemother Pharmacol. 2000;45:291–7.
Reigner B, Blesch K, Weidekamm E. Clinical pharmacokinetics of capecitabine. Clin Pharmacokinet. 2001;40:85–104.
Wohlhueter RM, McIvor RS, Plagemann PG. Facilitated transport of uracil and 5-fluorouracil, and permeation of orotic acid into cultured mammalian cells. J Cell Physiol. 1980;104:309–19.
Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–8.
Nies AT, Magdy T, Schwab M, et al. Role of ABC transporters in fluoropyrimidine-based chemotherapy response. Adv Cancer Res. 2015;125:217–43.
Grem J. Mechanisms of action and modulation of fluorouracil. Semin Radiat Oncol. 1997;7:249–59.
Wilson PM, Danenberg PV, Johnston PG, et al. Standing the test of time: targeting thymidylate biosynthesis in cancer therapy. Nat Rev Clin Oncol. 2014;11:282–98.
Peters GJ, Laurensse E, Leyva A, et al. Sensitivity of human, murine, and rat cells to 5-fluorouracil and 5′-deoxy-5-fluorouridine in relation to drug-metabolizing enzymes. Cancer Res. 1986;46:20–8.
Ciccolini J, Peillard L, Evrard A, et al. Enhanced antitumor activity of 5-fluorouracil in combination with 2ʹ-deoxyinosine in human colorectal cell lines and human colon tumor xenografts. Clin Cancer Res. 2000;6:1529–35.
Ciccolini J, Peillard L, Aubert C, et al. Monitoring of the intracellular activation of 5-fluorouracil to deoxyribonucleotides in HT29 human colon cell line: application to modulation of metabolism and cytotoxicity study. Fundam Clin Pharmacol. 2000;14:147–54.
Kamm YJL, Peters GJ, Hull WE, et al. Correlation between 5-fluorouracil metabolism and treatment response in two variants of C26 murine colon carcinoma. Br J Cancer. 2003;89:754–62.
Spears CP, Gustavsson BG, Mitchell MS, et al. Thymidylate synthetase inhibition in malignant tumors and normal liver of patients given intravenous 5-fluorouracil. Cancer Res. 1984;44:4144–50.
Spears CP, Gustavsson BG, Berne M, et al. Mechanisms of innate resistance to thymidylate synthase inhibition after 5-fluorouracil. Cancer Res. 1988;48:5894–900.
Peters GJ, Lankelma J, Kok RM, et al. Prolonged retention of high concentrations of 5-fluorouracil in human and murine tumors as compared with plasma. Cancer Chemother Pharmacol. 1993;31:269–76.
Moran RG, Spears CP, Heidelberger C. Biochemical determinants of tumor sensitivity to 5-fluorouracil: ultrasensitive methods for the determination of 5-fluoro-2ʹ-deoxyuridylate, 2′-deoxyuridylate, and thymidylate synthetase. Proc Natl Acad Sci USA. 1979;76:1456–60.
Derissen EJB, Hillebrand MJX, Rosing H, et al. Development of an LC–MS/MS assay for the quantitative determination of the intracellular 5-fluorouracil nucleotides responsible for the anticancer effect of 5-fluorouracil. J Pharm Biomed Anal. 2015;110:58–66.
Derissen EJB, Jacobs BAW, Huitema ADR, et al. Exploring the intracellular pharmacokinetics of the 5-fluorouracil nucleotides during capecitabine treatment. Br J Clin Pharmacol. 2016;81:949–57.
Maring JG, Wachters FM, Slijfer M, et al. Pharmacokinetics of gemcitabine in non-small-cell lung cancer patients: impact of the 79A > C cytidine deaminase polymorphism. Eur J Clin Pharmacol. 2010;66:611–7.
DeAngelis LM, Kreis W, Chan K, et al. Pharmacokinetics of ara-C and ara-U in plasma and CSF after high-dose administration of cytosine arabinoside. Cancer Chemother Pharmacol. 1992;29:173–7.
Burk M, Heyll A, Arning M, et al. Pharmacokinetics of high-dose cytarabine and its deamination product: a reappraisal. Leuk Lymphoma. 1997;27:321–7.
Gandhi V, Xu YZ, Estey E. Accumulation of arabinosyluracil 5ʹ-triphosphate during arabinosylcytosine therapy in circulating blasts of patients with acute myelogenous leukemia. Clin Cancer Res. 1998;4:1719–26.
Clarke ML, Mackey JR, Baldwin SA, et al. The role of membrane transporters in cellular resistance to anticancer nucleoside drugs. Cancer Treat Res. 2002;112:27–47.
Kufe DW, Major PP, Egan EM, et al. Correlation of cytotoxicity with incorporation of ara-C into DNA. J Biol Chem. 1980;255:8997–9000.
Major PP, Egan EM, Beardsley GP, et al. Lethality of human myeloblasts correlates with the incorporation of arabinofuranosylcytosine into DNA. Proc Natl Acad Sci USA. 1981;78:3235–9.
Kufe D, Spriggs D, Egan EM, et al. Relationships among Ara-CTP pools, formation of (Ara-C)DNA, and cytotoxicity of human leukemic cells. Blood. 1984;64:54–8.
Chou TC, Arlin Z, Clarkson BD, et al. Metabolism of 1-beta-d-arabinofuranosylcytosine in human leukemic cells. Cancer Res. 1977;37:3561–70.
Liliemark JO, Plunkett W, Dixon DO. Relationship of 1-beta-d-arabinofuranosylcytosine in plasma to 1-beta-d-arabinofuranosylcytosine 5ʹ-triphosphate levels in leukemic cells during treatment with high-dose 1-beta-d-arabinofuranosylcytosine. Cancer Res. 1985;45:5952–7.
Plunkett W, Liliemark JO, Adams TM, et al. Saturation of 1-beta-d-arabinofuranosylcytosine 5ʹ-triphosphate accumulation in leukemia cells during high-dose 1-beta-d-arabinofuranosylcytosine therapy. Cancer Res. 1987;47:3005–11.
Plunkett W, Liliemark JO, Estey E, et al. Saturation of ara-CTP accumulation during high-dose ara-C therapy: pharmacologic rationale for intermediate-dose ara-C. Semin Oncol. 1987;14:159–66.
Hiddemann W, Schleyer E, Unterhalt M, et al. Differences in the intracellular pharmacokinetics of cytosine arabinoside (AraC) between circulating leukemic blasts and normal mononuclear blood cells. Leukemia. 1992;6:1273–80.
Kantarjian HM, Estey EH, Plunkett W, et al. Phase I–II clinical and pharmacologic studies of high-dose cytosine arabinoside in refractory leukemia. Am J Med. 1986;81:387–94.
Harris AL, Grahame-Smith DG. Variation in sensitivity of DNA synthesis to ara-C in acute myeloid leukaemia. Br J Haematol. 1980;45:371–9.
Köhl U, Schwabe D, Montag E, et al. Formation of cytosine arabinoside-5ʹ-triphosphate in different cultured lymphoblastic leukaemic cells with reference to their drug sensitivity. Eur J Cancer. 1995;31A:209–14.
Farag SS, Ruppert AS, Mrózek K, et al. Outcome of induction and postremission therapy in younger adults with acute myeloid leukemia with normal karyotype: a cancer and leukemia group B study. J Clin Oncol. 2005;23:482–93.
Büchner T, Berdel WE, Schoch C, et al. Double induction containing either two courses or one course of high-dose cytarabine plus mitoxantrone and postremission therapy by either autologous stem-cell transplantation or by prolonged maintenance for acute myeloid leukemia. J Clin Oncol. 2006;24:2480–9.
Döhner H, Estey EHE, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–74.
Breithaupt H, Pralle H, Eckhardt T, et al. Clinical results and pharmacokinetics of high-dose cytosine arabinoside (HD ARA-C). Cancer. 1982;50:1248–57.
Early AP, Preisler HD, Slocum H, et al. A pilot study of high-dose 1-beta-d-arabinofuranosylcytosine for acute leukemia and refractory lymphoma: clinical response and pharmacology. Cancer Res. 1982;42:1587–94.
Capizzi RL, Yong JL, Cheng E. Alteration of the pharmacokinetics of high-dose Ara-C by its metabolite, high Ara-U in patients with acute leukemia. J Clin Oncol. 1983;1:763–71.
Lazarus HM, Herzig RH, Herzig GP, et al. Central nervous system toxicity of high-dose systemic cytosine arabinoside. Cancer. 1981;48:2577–82.
Salinsky MC, Levine RL, Aubuchon JP, et al. Acute cerebellar dysfunction with high-dose ARA-C therapy. Cancer. 1983;51:426–9.
Andersson BS, Cogan BM, Keating MJ, et al. Subacute pulmonary failure complicating therapy with high-dose Ara-C in acute leukemia. Cancer. 1985;56:2181–4.
Hwang TL, Yung WK, Estey EH, et al. Central nervous system toxicity with high-dose Ara-C. Neurology. 1985;35:1475–9.
Nand S, Messmore HL, Patel R, et al. Neurotoxicity associated with systemic high-dose cytosine arabinoside. J Clin Oncol. 1986;4:571–5.
Lopez JA, Agarwal RP. Acute cerebellar toxicity after high-dose cytarabine associated with CNS accumulation of its metabolite, uracil arabinoside. Cancer Treat Rep. 1984;68:1309–10.
Löwenberg B, Pabst T, Vellenga E, et al. Cytarabine dose for acute myeloid leukemia. N Engl J Med. 2011;364:1027–36.
Mantovani L, Hasenclever D, Krahl R, et al. Intermediate-dose cytarabine treatment delivered at moderate infusion rates for de novo acute myeloid leukemia: results of a phase I–II study. Leuk Lymphoma. 2002;43:265–74.
Estey E, Plunkett W, Dixon D, et al. Variables predicting response to high dose cytosine arabinoside therapy in patients with refractory acute leukemia. Leukemia. 1987;1:580–3.
Estey EH, Keating MJ, McCredie KB, et al. Cellular ara-CTP pharmacokinetics, response, and karyotype in newly diagnosed acute myelogenous leukemia. Leukemia. 1990;4:95–9.
Rustum YM, Preisler HD. Correlation between leukemic cell retention of 1-beta-d-arabinofuranosylcytosine 5ʹ-triphosphate and response to therapy. Cancer Res. 1979;39:42–9.
Preisler HD, Rustum Y, Priore RL. Relationship between leukemic cell retention of cytosine arabinoside triphosphate and the duration of remission in patients with acute non-lymphocytic leukemia. Eur J Cancer Clin Oncol. 1985;21:23–30.
Preisler HD, Rustum YM, Azarnia N, et al. Abrogation of the prognostic significance of low leukemic cell retention of cytosine arabinoside triphosphate by intensification of therapy and by alteration in the dose and schedule of administration of cytosine arabinoside. Cancer Chemother Pharmacol. 1987;19:69–74.
Gandhi V, Huang P, Plunkett W. Metabolism and action of arabinosyluracil 5ʹ-triphosphate in acute myelogenous leukemia blasts. Proc Am Assoc Cancer Res. 1992;33:545.
Gandhi V, Chapman A, Huang P, et al. Sequence specific inhibition of DNA polymerase α by 5ʹ-triphosphates of 1-β-d-arabinofuranosylcytosine and arabinosyluracil. Proc Am Assoc Cancer Res. 1993;34:349.
Müller WE, Zahn RK. Metabolism of 1-beta-d-arabinofuranosyluracil in mouse L5178Y cells. Cancer Res. 1979;39:1102–7.
Ono K, Ohashi A, Ogasawara M, et al. Inhibition of deoxyribonucleic acid polymerases from murine cells and oncornavirus by 5-alkylated derivatives of 1-beta-d-arabinofuranosyluracil 5ʹ-triphosphate: substituent effects on inhibitory action. Biochemistry. 1981;20:5088–93.
Yang JL, Cheng EH, Capizzi RL, et al. Effect of uracil arabinoside on metabolism and cytotoxicity of cytosine arabinoside in L5178Y murine leukemia. J Clin Investig. 1985;75:141–6.
Abbruzzese JL, Grunewald R, Weeks EA, et al. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol. 1991;9:491–8.
Mackey JR, Mani RS, Selner M, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58:4349–57.
Ritzel MW, Ng AM, Yao SY, et al. Recent molecular advances in studies of the concentrative Na+-dependent nucleoside transporter (CNT) family: identification and characterization of novel human and mouse proteins (hCNT3 and mCNT3) broadly selective for purine and pyrimidine nucleosides. Mol Membr Biol. 2001;18:65–72.
Wong A, Soo RA, Yong W-P, et al. Clinical pharmacology and pharmacogenetics of gemcitabine. Drug Metab Rev. 2009;41:77–88.
Gandhi V, Legha J, Chen F, et al. Excision of 2ʹ,2ʹ-difluorodeoxycytidine (gemcitabine) monophosphate residues from DNA. Cancer Res. 1996;56:4453–9.
Huang P, Chubb S, Hertel L, et al. Action of 2′, 2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51:6110–7.
Heinemann V, Xu YZ, Chubb S, et al. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2ʹ,2ʹ-difluorodeoxycytidine. Mol Pharmacol. 1990;38:567–72.
Heinemann V, Xu YZ, Chubb S, et al. Cellular elimination of 2ʹ,2ʹ-difluorodeoxycytidine 5ʹ-triphosphate: a mechanism of self-potentiation. Cancer Res. 1992;52:533–9.
de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol. 2014;741:8–16.
Hunsucker SA, Mitchell BS, Spychala J. The 5′-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol Ther. 2005;107:1–30.
Xu Y-Z, Plunkett W. Modulation of deoxycytidylate deaminase in intact human leukemia cells: action of 2ʹ,2ʹ-difluorodeoxycytidine. Biochem Pharmacol. 1992;44:1819–27.
Grunewald R, Abbruzzese JL, Tarassoff P, et al. Saturation of 2ʹ,2ʹ-difluorodeoxycytidine 5ʹ-triphosphate accumulation by mononuclear cells during a phase I trial of gemcitabine. Cancer Chemother Pharmacol. 1991;27:258–62.
Grunewald R, Kantarjian H, Keating MJ, et al. Pharmacologically directed design of the dose rate and schedule of 2ʹ,2ʹ-difluorodeoxycytidine (gemcitabine) administration in leukemia. Cancer Res. 1990;50:6823–6.
Grunewald R, Kantarjian H, Du M, et al. Gemcitabine in leukemia: a phase I clinical, plasma, and cellular pharmacology study. J Clin Oncol. 1992;10:406–13.
Patel SR, Gandhi V, Jenkins J, et al. Phase II clinical investigation of gemcitabine in advanced soft tissue sarcomas and window evaluation of dose rate on gemcitabine triphosphate accumulation. J Clin Oncol. 2001;19:3483–9.
Gandhi V, Plunkett W, Du M, et al. Prolonged infusion of gemcitabine: clinical and pharmacodynamic studies during a phase I trial in relapsed acute myelogenous leukemia. J Clin Oncol. 2002;20:665–73.
Cattel L, Airoldi M, Delprino L, et al. Pharmacokinetic evaluation of gemcitabine and 2ʹ,2ʹ-difluorodeoxycytidine-5ʹ-triphosphate after prolonged infusion in patients affected by different solid tumors. Ann Oncol. 2006;17(Suppl. 5):v142–7.
Grimison P, Galettis P, Manners S, et al. Randomized crossover study evaluating the effect of gemcitabine infusion dose rate: evidence of auto-induction of gemcitabine accumulation. J Clin Oncol. 2007;25:5704–9.
Tempero M, Plunkett W, Ruiz van Haperen VW, et al. Randomized phase II comparison of dose-intense gemcitabine: thirty-minute infusion and fixed dose rate infusion in patients with pancreatic adenocarcinoma. J Clin Oncol. 2003;21:3402–8.
Guan Z, Wang Y, Maoleekoonpairoj S, et al. Prospective randomised phase II study of gemcitabine at standard or fixed dose rate schedule in unresectable hepatocellular carcinoma. Br J Cancer. 2003;89:1865–9.
Ceribelli A, Gridelli C, De Marinis F, et al. Prolonged gemcitabine infusion in advanced non-small cell lung carcinoma: a randomized phase II study of two different schedules in combination with cisplatin. Cancer. 2003;98:337–43.
Soo RA, Wang LZ, Tham LS, et al. A multicentre randomised phase II study of carboplatin in combination with gemcitabine at standard rate or fixed dose rate infusion in patients with advanced stage non-small-cell lung cancer. Ann Oncol. 2006;17:1128–33.
Cappuzzo F, Novello S, De Marinis F, et al. A randomized phase II trial evaluating standard (50 mg/min) versus low (10 mg/min) infusion duration of gemcitabine as first-line treatment in advanced non-small-cell lung cancer patients who are not eligible for platinum-based chemotherapy. Lung Cancer. 2006;52:319–25.
Gridelli C, Gallo C, Ceribelli A, et al. Factorial phase III randomised trial of rofecoxib and prolonged constant infusion of gemcitabine in advanced non-small-cell lung cancer: the GEmcitabine-COxib in NSCLC (GECO) study. Lancet Oncol. 2007;8:500–12.
Poplin E, Feng Y, Berlin J, et al. Phase III, randomized study of gemcitabine and oxaliplatin versus gemcitabine (fixed-dose rate infusion) compared with gemcitabine (30-minute infusion) in patients with pancreatic carcinoma E6201: a trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2009;27:3778–85.
Fossella FV, Lippman SM, Shin DM, et al. Maximum-tolerated dose defined for single-agent gemcitabine: a phase I dose-escalation study in chemotherapy-naive patients with advanced non-small-cell lung cancer. J Clin Oncol. 1997;15:310–6.
Brand R, Capadano M, Tempero M. A phase I trial of weekly gemcitabine administered as a prolonged infusion in patients with pancreatic cancer and other solid tumors. Investig New Drugs. 1997;15:331–41.
Touroutoglou N, Gravel D, Raber MN, et al. Clinical results of a pharmacodynamically-based strategy for higher dosing of gemcitabine in patients with solid tumors. Ann Oncol. 1998;9:1003–8.
Ruiz van Haperen VW, Veerman G, Eriksson S, et al. Development and molecular characterization of a 2ʹ,2ʹ-difluorodeoxycytidine-resistant variant of the human ovarian carcinoma cell line A2780. Cancer Res. 1994;54:4138–43.
Veltkamp SA, Pluim D, van Eijndhoven MAJ, et al. New insights into the pharmacology and cytotoxicity of gemcitabine and 2ʹ,2ʹ-difluorodeoxyuridine. Mol Cancer Ther. 2008;7:2415–25.
Honeywell RJ, Ruiz Van Haperen VWT, Veerman G, et al. Inhibition of thymidylate synthase by 2ʹ,2ʹ-difluoro-2ʹ-deoxycytidine (gemcitabine) and its metabolite 2ʹ,2ʹ-difluoro-2ʹ-deoxyuridine. Int J Biochem Cell Biol. 2015;60:73–81.
Derissen EJB, Huitema ADR, Rosing H, et al. Intracellular pharmacokinetics of gemcitabine, its deaminated metabolite 2ʹ,2ʹ-difluorodeoxyuridine and their nucleotides. Br J Clin Pharmacol. 2018;84:1279–89.
Veltkamp SA, Jansen RS, Callies S, et al. Oral administration of gemcitabine in patients with refractory tumors: a clinical and pharmacologic study. Clin Cancer Res. 2008;14:3477–86.
Damaraju VL, Mowles D, Yao S, et al. Role of human nucleoside transporters in the uptake and cytotoxicity of azacitidine and decitabine. Nucleosides, Nucleotides Nucleic Acids. 2012;31:236–55.
Li LH, Olin EJ, Buskirk HH, et al. Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer Res. 1970;30:2760–9.
Aimiuwu J, Wang H, Chen P, et al. RNA-dependent inhibition of ribonucleotide reductase is a major pathway for 5-azacytidine activity in acute myeloid leukemia. Blood. 2012;119:5229–38.
Kaminskas E, Farrell AT, Wang Y-C, et al. FDA drug approval summary: azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist. 2005;10:176–82.
Momparler RL. Pharmacology of 5-Aza-2′-deoxycytidine (decitabine). Semin Hematol. 2005;42:S9–16.
Momparler RL. Molecular, cellular and animal pharmacology of 5-aza-2′-deoxycytidine. Pharmacol Ther. 1985;30:287–99.
Uchida T, Kinoshita T, Nagai H, et al. Hypermethylation of the p15INK4B gene in myelodysplastic syndromes. Blood. 1997;90:1403–9.
Christiansen DH, Anderson MK, Pedersen-Bjergaard J. Methylation of p15 INK4B is common, is associated with deletion of genes on chromosome arm 7q and predicts a poor prognosis in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2003;17:1813–9.
Issa J-P. Epigenetic changes in the myelodysplastic syndrome. Hematol Oncol Clin N Am. 2010;24:317–30.
McCabe MT, Brandes JC, Vertino PM. Cancer DNA methylation: molecular mechanisms and clinical implications. Clin Cancer Res. 2009;15:3927–37.
Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–95.
EPAR Dacogen. European Medicines Agency. EMA/620205/2012. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002221/WC500133571.pdf. Accessed 17 Aug 2020.
Kantarjian HM, Issa J-PJ. Decitabine dosing schedules. Semin Hematol. 2005;42:S17–22.
Oki Y, Aoki E, Issa JPJ. Decitabine: bedside to bench. Crit Rev Oncol Hematol. 2007;61:140–52.
Jabbour E, Issa JP, Garcia-Manero G, et al. Evolution of decitabine development: accomplishments, ongoing investigations, and future strategies. Cancer. 2008;112:2341–51.
O’Dwyer K, Maslak P. Azacitidine and the beginnings of therapeutic epigenetic modulation. Expert Opin Pharmacother. 2008;9:1981–6.
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–32.
Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. 2012;30:2670–7.
Jansen RS, Rosing H, Wijermans PW, et al. Decitabine triphosphate levels in peripheral blood mononuclear cells from patients receiving prolonged low-dose decitabine administration: a pilot study. Cancer Chemother Pharmacol. 2012;69:1457–66.
Wang H, Chen P, Wang J, et al. In vivo quantification of active decitabine-triphosphate metabolite: a novel pharmacoanalytical endpoint for optimization of hypomethylating therapy in acute myeloid leukemia. AAPS J. 2013;15:242–9.
Derissen EJB, Hillebrand MJX, Rosing H, et al. Quantitative determination of azacitidine triphosphate in peripheral blood mononuclear cells using liquid chromatography coupled with high-resolution mass spectrometry. J Pharm Biomed Anal. 2014;90:7–14.
Unnikrishnan A, Vo ANQ, Pickford R, et al. AZA-MS: a novel multiparameter mass spectrometry method to determine the intracellular dynamics of azacitidine therapy in vivo. Leukemia. 2018;32:900–10.
Van Haperen VWTR, Veerman G, Boven E, et al. Schedule dependence of sensitivity to 2ʹ,2ʹ-difluorodeoxycytidine (gemcitabine) in relation to accumulation and retention of its triphosphate in solid tumour cell lines and solid tumours. Biochem Pharmacol. 1994;48:1327–39.
Laille E, Goel S, Mita AC, et al. A phase I study in patients with solid or hematologic malignancies of the dose proportionality of subcutaneous azacitidine and its pharmacokinetics in patients with severe renal impairment. Pharmacotherapy. 2014;34:440–51.
Kroep JR, Giaccone G, Voorn DA, et al. Gemcitabine and paclitaxel: pharmacokinetic and pharmacodynamic interactions in patients with non-small-cell lung cancer. J Clin Oncol 1999;17:2190–7.
van Moorsel CJ, Kroep JR, Pinedo HM, et al. Pharmacokinetic schedule finding study of the combination of gemcitabine and cisplatin in patients with solid tumors. Ann Oncol 1999;10:441–448.
Nieto Y, Aldaz A, Rifón J, et al. Phase I and Pharmacokinetic Study of Gemcitabine Administered at Fixed-Dose Rate, Combined with Docetaxel/Melphalan/Carboplatin, with Autologous Hematopoietic Progenitor-Cell Support, in Patients with Advanced Refractory Tumors. Biol Blood Marrow Transplant 2007;13:1324–1337.
Caffo O, Fallani S, Marangon E, et al. Pharmacokinetic study of gemcitabine, given as prolonged infusion at fixed dose rate, in combination with cisplatin in patients with advanced non-small-cell lung cancer. Cancer Chemother Pharmacol 2010;65:1197–202.
Author information
Authors and Affiliations
Contributions
ED performed the literature review, acquired the data and wrote the manuscript; JB reviewed the contents of the manuscript. Both authors have read and agree to the published version of the manuscript.
Corresponding author
Ethics declarations
Funding
No sources of funding were used to assist with the preparation of this review.
Conflicts of interest
Ellen J. B. Derissen and Jos H. Beijnen have no conflicts of interest that are directly relevant to the content of this review.
Declarations
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Derissen, E.J.B., Beijnen, J.H. Intracellular Pharmacokinetics of Pyrimidine Analogues used in Oncology and the Correlation with Drug Action. Clin Pharmacokinet 59, 1521–1550 (2020). https://doi.org/10.1007/s40262-020-00934-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00934-7