
Abstract
Medication use during pregnancy in the absence of pharmacokinetic and safety data is common, particularly for antiretrovirals, as pregnant women are not usually included in clinical trials leading to drug licensure. To date, data are typically generated through opportunistic pregnancy studies performed in the postmarketing setting, leading to a substantial time-lag between initial regulatory approval of a drug and availability of essential pregnancy-specific pharmacokinetic and safety data. During this period, health care providers lack key information on human placental transfer, fetal exposure, optimal maternal dosing in pregnancy, and maternal and fetal drug toxicity, including teratogenicity risk. We discuss new approaches that could facilitate the acquisition of these critical data earlier in the drug development process, aiding clinicians and patients in making informed decisions on drug selection and dosing during pregnancy. An integrated approach utilizing multiple novel methodologies (in vitro, ex vivo, in silico and in vivo) is needed to accelerate the availability of pharmacology data in pregnancy and lactation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Novel approaches should be applied in pharmacology studies in pregnant and lactating women. |
Artificial intelligence is an important cornerstone of these novel approaches. |
An integrated approach of in vitro, ex vivo, in silico and in vivo studies is key to enable acceleration of availability of pharmacology data in pregnancy and lactation. |
1 Introduction
The majority of pregnant women use one or more medications through the course of pregnancy, and, on average, 2.6–4.6 different prescriptions are used per pregnancy [1, 2]. Indications for medication use during pregnancy include chronic pre-existing medical conditions (e.g. pre-gestational diabetes), pregnancy-specific diseases (e.g. hyperemesis gravidarum, intrahepatic cholestasis of pregnancy, HELLP syndrome), and medication therapy to treat fetal conditions during pregnancy (e.g. maternal digoxin therapy to treat fetal tachyarrhythmias) [3]. Fetal and infant safety concerns have led to the exclusion of pregnant and lactating women from clinical trials during drug development programs for licensure, unless the drug is intended for a pregnancy-specific condition [4, 5]. Despite this exclusion, many drugs are subsequently prescribed off-label during pregnancy and lactation, as only a small percentage of drugs contain a pregnancy or breastfeeding contraindication in their label. Moreover, even when a product label includes information about the use of medications during pregnancy and lactation, it is sometimes insufficient to guide clinicians on efficacy and safety [6].
For some chronic diseases, it is not possible to avoid drug exposure to the fetus during pregnancy, as exemplified in women living with HIV who must take antiretrovirals (ARVs) during pregnancy and lactation for their own health and to prevent transmission of HIV to their infants. Even for ARVs commonly used during pregnancy and lactation, data on toxicity, pharmacokinetics (PK), and neonatal exposure during breastfeeding are collected sporadically, mainly through postmarketing surveillance and opportunistic studies of women who become pregnant while receiving ARVs. As a result, availability of pregnancy safety and PK data become available years after drug licensure, if at all. For example, it has been demonstrated that drug exposure is reduced during pregnancy (primarily during the third trimester) with the licensed adult dose of specific ARVs during pregnancy [7,8,9,10]. This PK information, in combination with clinical data, leads to dose adjustments or a recommendation that specific ARVs or ARV combinations should not to be used in pregnancy, but only became available years after marketing [11,12,13]. A specific example is cobicistat-boosted ARVs (cobicistat–darunavir, cobicistat–elvitegravir, cobicistat–atazanavir), whose concentrations were demonstrated to be reduced by 50–90% during the second and third trimesters of pregnancy compared with postpartum, potentially reducing efficacy [14,15,16]. Information describing ARV PK in pregnancy and lactation has depended largely on data generated within opportunistic studies of women receiving these drugs as part of clinical care, and ARV PK and safety data have become available after a median of 6 years (pregnancy) and > 8 years (lactation) after marketing [17]. To balance the concerns of fetal and infant exposure to new and potentially toxic medications, with the pressing need for PK and safety data earlier in the drug development process, new approaches to study the PK and pharmacodynamics (PD) of drugs during pregnancy and lactation to enable acceleration of the drug development process for pregnant women are needed.
In this article, we have summarized novel approaches (Table 1) that could be implemented during the development phase of new drugs in order to close the time lag between initial regulatory approval and availability of pregnancy and lactation information. Data for this manuscript were obtained from a comprehensive review of HIV, pregnancy, lactation, and pharmacology literature; expert opinions; and from the workshop on ‘Approaches to Optimize and Accelerate Pharmacology Studies in Pregnant and Lactating Women’, convened by the International Maternal Pediatric Adolescent AIDS Clinical Trials (IMPAACT) Network and the World Health Organization (WHO) in June 2019.
2 Innovative approaches
2.1 Innovative Preclinical Approaches to Assess Placental Transfer and Infant Exposure Through Breastmilk
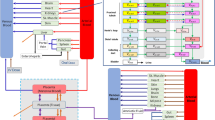
Accurate knowledge of mechanisms involved in the placental transfer of drugs is pivotal to studying drug safety and effectiveness in pregnant and lactating women as part of the drug development process [18]. Innovative approaches to study the placental and feto-maternal interface, such as in vitro, ex vivo human cotyledon perfusion, placental drug transport-on-a-chip, and in silico models, are increasingly being utilized to characterize maternal/fetal drug transfer prior to human administration during pregnancy (Fig. 1). Human trophoblastic placental cell line studies (in vitro placental studies), such as the use of BeWo [19], Jeg-3 [20], JAr [20], and ACH-3P [20] cell lines, are methods used to facilitate the study of placental influx and efflux transport systems, active, passive and facilitated diffusion, and drug metabolism in the placenta [21, 22]. Among these placental cell lines, the JAr and Jeg-3 lines are used to study placental permeability to drugs [23], while BeWo cells are the most widely used to study nanoparticle translocation studies and syncytialization due to their ability to form a barrier with little permeability [20]. While the ACH-3P cell line was developed to better simulate first trimester trophoblastic cells, and as an alternative to study drug transport in vitro, ACH-3P cells do not form syncytiotrophoblast, which is vital for placental transport [23]. Other cell lines used for drug transport studies include cells derived from the nephron [24]. For example, Cerveny et al. successfully used Madin–Darby Canine Kidney Cells II (MDCKII), cells expressing human efflux transporters, to show that atazanavir is a substrate for p-glycoprotein 1 (P-gp or ABCB1), and confirmed its functionality in limiting fetal exposure using dual perfusion studies in a pregnant rat placenta model [25]. In vitro assays have also reliably predicted materno-fetal transport of both tenofovir (TFV) and tenofovir disoproxil fumarate (TDF) in studies assessing the role of these drug transporters in the transplacental PK of TFV and TDF [26]. In addition, in vitro assays reliably predicted etravirine/TDF drug–drug interactions during transplacental passage of TDF through interactions with ABCG2 [27].

Overview of innovative approaches to studying drug disposition in pregnancy and lactation. PK pharmacokinetic, PBPK physiologically based pharmacokinetic
Perfusion of an isolated human placental cotyledon/lobule (ex vivo studies) are also increasingly utilized in predicting drug disposition within the placenta [28]. The utility of ex vivo models have proved successful in predicting in vivo placental drug transfer. Placental ex vivo systems exist in two forms: closed (recirculating) and open (non-recirculating) ex vivo circuit systems [29]. The closed-circuit ex vivo systems are more physiologic since they mimic the maternal–fetal circulation (both maternal and fetal perfusate are recirculated), thus making it easy to understand mechanisms of drug transport and transplacental transfer for different drugs and molecules. Hence, this approach would be useful to provide a rational basis to study the extent of drug transfer to the developing fetus. For example, placental transfer of maraviroc was studied in an ex vivo human cotyledon perfusion model, and this model was used to inform dose selection and optimal timing of sample collection in human studies [30]. Novel techniques using ex vivo approaches may improve our understanding of placental drug transfer by studying preterm placentas (as human cord blood plasma drug concentrations can only be collected safely at the time of birth and not earlier during pregnancy), improving replication of placental studies through standardization of existing closed ex vivo experimental protocols, and maintaining viability and structure of the placenta over extended periods of time to extend the duration and number of placental experiments performed over several days. To address these challenges, nano technologically driven microengineered models of the human placenta (placental drug transport-on-a-chip models) are currently being utilized to simulate and investigate drug transfer across maternal–fetal circulation [31,32,33]. The placenta-on-a-chip promises to serve as a new screening tool to enable precise prediction of drug transport across the feto-placental unit [34, 35].
A recent review highlighted the limitations of animal models (e.g. mice or rat) on which preclinical information on breastmilk excretion in drug labels are often based, including interspecies differences in milk composition and variances in asymmetrical drug transport [36]. Substantial differences have been observed in the extent of drug excretion into breastmilk between human and mice; a geometric mean milk-to-plasma ratio between mice and humans of 2.03 (95% confidence interval 1.42–2.89) was reported in a study of 27 drugs [37]. Primary epithelial cell models derived directly from human mammary glands is limited by their short lifespan. Some studies have demonstrated that conditional reprogramming allows these cells to be cultured in vitro with an extended lifespan [38]. Importantly, the cells exhibit in vivo heterogeneous morphologic features and self-organize into distinct structures capable of differentiation into milk-producing cells when grown in 3D. However, their functionality in studying drug excretion into breastmilk has not been investigated. An in vitro mouse mammary epithelial cell culture model that recapitulates the secretory and tight-junction properties of human mammary epithelium has been described and has been used to study the directionality of passive drug transport [39] and milk-to-plasma ratio of drugs [40], a critical parameter in predicting breastfed infant drug exposure through breastmilk. In vitro assays may play a role in predicting drug exposure in breastmilk to feed into in silico models simulating infant exposure through breastmilk. The European public–private partner initiative ‘ConcePTION’, which is currently exploring several in vitro and in silico techniques for studying drug exposure into breastmilk (work package 3), is an excellent example of this approach [41].
2.2 In Vivo Studies to Assess Drug Exposure during Pregnancy and Lactation
Opportunistic approaches to the study of pregnant women during clinical trials should be encouraged, meaning that women who become pregnant during a clinical trial should have the informed option to stay on the investigational drug throughout pregnancy [5]. These women can be enrolled into a specific substudy within the trial that incorporates pregnancy, delivery and postpartum PK sampling, fetal monitoring, and infant follow-up for safety. This opportunistic approach was used in the ARIA [42], ADVANCE [43], and WAVES [44] randomized clinical drug trials. Although the ARIA study evaluated the safety and efficacy of dolutegravir/abacavir/lamivudine in women living with HIV, women who became pregnant had the option of participating in a dedicated substudy [42]. Similarly, the ADVANCE trial evaluated the efficacy and safety of tenofovir alafenamide–emtricitabine–dolutegravir (TAF–FTC–DTG) and TDF–FTC–DTG, as compared with the first-line regimen of efavirenz (EFV)-based combination antiretroviral therapy (cART) used at the time of the trial in the majority of people living with HIV in low- and middle-income countries. Female patients who became pregnant had the option of remaining in the ADVANCE trial pregnancy PK substudy, with active follow-up of infants to 18 months [43]. WAVES was a randomized, controlled, double-blind, phase III study investigating an integrase strand inhibitor regimen (elvitegravir [EVG]–cobicistat [Cobi]–FTC-TDF) versus a protease inhibitor-based regimen (atazanavir/ritonavir [ATV/RTV]–FTC–TDF) for women with HIV [44]. Women who became pregnant during the study had the option to continue unblinded study ARVs after providing additional informed consent [44]. Furthermore, studies involving long-acting ARVs (e.g. cabotegravir, dapivirine [DPV] intravaginal rings) should study pregnancy PK and safety outcomes if women become pregnant during such trials, as exposure will be prolonged due to their long half-lives. An example of this is the HPTN and IMPAACT collaborative clinical trial design to evaluate the safety and PK of injectable long-acting cabotegravir administered for pre-exposure prophylaxis or treatment of adolescents living with HIV who become pregnant while on study [45]. In addition to adaptive, early-phase trial designs, it is pertinent to encourage phase III PK studies in pregnant and lactating women, including PK substudies in pregnant women within phase III clinical trials. DolPHIN 1 [46], DolPHIN-2 [47], and VESTED [48] are examples of dedicated clinical trials in pregnancy and lactation. While these trials were conducted after drug registration, they can serve as models for efficacy, PK, and safety trials in pregnancy conducted during drug development prior to licensure.
A novel approach that can provide early information regarding pregnancy effects on PK is microdosing studies during pregnancy and lactation. Microdosing studies are defined as exploratory acquisition of human PK data using subtherapeutic doses expected to be well tolerated (typically 0.01% of the expected pharmacological dose of a drug), but at a high enough dose to allow cellular responses to be studied [49, 50]. These studies have been suggested to acquire PK information in special populations such as children and pregnant women [51]. Subtherapeutic exposure to drugs in microdosing studies has been identified as no more than minimal risk [51, 52]. Since such low doses are unlikely to produce meaningful PD effects and would be too small to cause any major adverse effects after single doses, it is reasonable to encourage such studies in pregnant and lactating women. An approach for ARVs could be to perform a microdosing study in pregnant women living with HIV who are virologically suppressed under treatment. This microdosing study on top of optimized background therapy (addition of a microdose of a new compound to a current standard therapy) could be performed during phase III or earlier in drug development to identify the pregnancy effect on drug PK. Although microdosing provides no direct benefit to the individual patient, its characterization as involving only minimal risk may allow this approach to be an ethical option for studying drugs in pregnant and lactating women. Despite the lack of clinical benefit, microdosing studies have been successfully completed in pediatric populations [53, 54]. Microdosing studies in pediatric populations are considered acceptable under certain circumstances—in complementing and enhancing physiologically based PK studies in children [51, 55], and in preventing potential drug toxicity and unpredictability surrounding first-in-pediatric dosing [56]. As microdosing has been instrumental in accelerating pediatric-specific drug development programs, a similar strategy could be used for advancing drug development in pregnant women. Although microdosing has great potential in shortening the time taken for drug development in pregnant women, practical issues remain. For example, extrapolation based on results from microdosing to therapeutic dosing may not be reliable because the PK processes of absorption, distribution, metabolism and elimination in microdosing studies is assumed linear [55]. Therefore, microdosing might be a poor predictor of therapeutic dosing in drugs with non-linear PK, especially orally administered medications [57]. While it is difficult to delineate particular drug characteristics that impact the predictability of microdose PK, characterizing the dose–response curve of the particular drug from multiple microdoses, and determining how close the microdose is to the therapeutic dose, is usually the first step, taking the drawbacks into consideration [50, 58]. Microdosing studies in lactating women is even more challenging, as in most cases only a small fraction of the total dose transfers into breastmilk, which will compromise the analysis method, i.e. very low concentrations need to be assessed reliably. However, for drugs that are expected to transfer into breastmilk, this is a potential method to approximate the milk-to-plasma ratio and estimate potential infant exposure.
Another innovative approach is to assess drugs in pregnant and lactating women in a short-course (targeted) PK study. This approach is useful in assessing the PK properties of a drug administered in a continuous slow-release fashion (transdermally administered medications, depots, implants and intravaginal rings). Because the steady-state concentrations of medications administered through these routes are stable over an extended period of time, targeted PK evaluations (limited only to specific times during pregnancy or postpartum during lactation) could be performed. For example, breastmilk and maternal concentrations of DPV, a non-nucleoside reverse transcriptase inhibitor to be used for pre-exposure prophylaxis via a vaginal ring (25 mg), were assessed in a short course PK study in 16 healthy women [59]. Although breastmilk and maternal plasma were collected, the infants were not breastfed during this PK study. Such a study could also be performed around the weaning period, when breastmilk is being produced but infants are no longer being breastfed. Women who choose not to breast feed can also be enrolled in such studies. A disadvantage is that the plasma exposure of the infant to medications received through breastmilk cannot be determined using this study design since infants are not being breastfed, but the amount of drug ingested over 24 h can still be estimated from the breastmilk drug concentrations.
2.3 Population Pharmacokinetic (PK) Modeling to Assess Drug Exposure in Pregnancy and Breastmilk
Using maternal-to-fetal drug exposures as an index of placental drug transfer, population PK models allow the simultaneous use of sparse sampling methods and multiple covariates (for example, maternal body size, age, fetal weight, disease status, hematocrit, genetic polymorphisms and gestational age) to explain intrasubject, intersubject, and residual variabilities during drug development. Using this approach, for several ARVs, maternal, fetal and infant PK parameters have been reliably predicted during pregnancy and lactation [60, 61]. In addition, post-delivery neonatal raltegravir exposure as a result of transplacentally acquired raltegravir during pregnancy has been successfully predicted using population PK modeling, allowing for neonatal dose selection for treatment of the neonate, with raltegravir [62].
2.4 In Silico Approaches: Maternal and Fetal Physiologically Based PK (PBPK) Modeling
2.4.1 PBPK Models to Assess Maternal Drug Exposure during Pregnancy
PBPK models constitute another innovative mechanistic technique for investigating drug disposition during pregnancy. These models usually include physiologic maternal tissues such as brain, heart, kidney, muscle, skin, liver, lung, adipose tissue, feto-placental unit, and bone. These in silico approaches have the ability to generate reliable predictions of maternal drug exposure in clinically relevant scenarios, including gestation-specific changes, drug–drug interactions, food–drug interactions, as well as new knowledge on the mode of action of drugs, the mechanisms underlying their interactions, and adverse effects following maternal exposure [63,64,65]. Some examples include a maternal PBPK model used to predict darunavir/ritonavir [66] and rilpivirine [64] PK during pregnancy. A better understanding of the relationship between predicted drug exposure in maternal PBPK models and observed maternal drug exposures during pregnancy are avenues that should be the focus of research in the future.
2.5 PBPK Models to Assess Fetal Drug Exposure during Pregnancy and Breastfeeding
PBPK modeling based on ex vivo placental perfusion studies enable the more efficient estimation of fetal drug exposure. The most promising PBPK models for predicting drug disposition in the feto-placental interface were described by Zhang and Unadkat [67] and De Sousa Mendes et al. [28]. The fetal PBPK model of Zhang and Unadkat, published in 2017, was the first to extensively detail specific characteristics of the fetal circulation, including incorporating the amniotic fluid and placental compartments, fetal volume of distribution, and fetal hepatic metabolism and renal excretion. The PK of FTC, TFV, and nevirapine in pregnancy and fetal circulation were reliably predicted by the PBPK models of De Sousa Mendes et al. [28, 68], while Schalkwijk et al. recently used a similar model to reliably predict fetal darunavir exposure [69]. PBPK models have proved invaluable for predicting the PK of several ARVs [69], including drug–drug interactions [70, 71] and food–drug interactions [72]. For example, Atoyebi et al. applied a similar PBPK model as a case study to predict fetal exposure to EFV and found significantly lower prenatal exposure when compared with known teratogens such as thalidomide [73]. In silico generation of data on the extent of fetal exposure to drugs may give us more insight into fetal exposure–safety relationships without exposing pregnant women and their fetuses to these teratogenic drugs. These models could be applied for the prediction of drug–drug interactions in pregnancy.
Despite these advances of in silico pregnancy modeling, PBPK approaches can be improved and made more innovative by addressing the deficiencies of currently used PBPK models [74]. Innovative approaches with future PBPK models would include incorporating the effects of understudied phase I metabolic enzymes (e.g. alcohol dehydrogenase); phase II metabolic enzymes (e.g. UGT1A1); integrating other routes of drug elimination (e.g. biliary excretion); and accounting for earlier gestational ages, changes in protein binding, placental drug transporters, and placental drug metabolizing enzymes. In addition, since currently used in silico approaches were modeled using healthy pregnant women, newer PBPK models would need to incorporate the effects of maternal and fetal medical and surgical conditions (for example, HIV, pre-gestational diabetes, pre-eclampsia, cholestasis, women with Fontan cardiac repairs), and how these conditions affect maternal–fetal physiology. In addition, more research is needed to increase prediction of drug disposition in the feto-placental unit, as only very few currently used PBPK models predict fetal drug exposure within the feto-placental unit [75]. Use of in silico frameworks may leverage available information and ultimately help to improve knowledge about the adequacy and safety of pharmacotherapy in pregnant and lactating women and their fetuses [76].
Several lactation PBPK models have been described, with most of the early models focusing on environmental risk assessment [77]. Observations of opioid toxicities in infants whose mothers received opioid prescriptions [78] prompted the development of PBPK models to study infant drug exposure to maternal therapeutic drugs. The model described by Willmann et al. included cytochrome P450 (CYP) 2D6 genotype status and confirmed comparable risks of opioid poisoning for neonates of both the ultra-rapid and extensive metabolizer mothers [79]. Several such models have been reported, with one of their major limitations being the absence of a milk-to-plasma ratio prediction component [36]. Hence, they often depend on the availability of published ratios from clinical studies, such as those described previously, to generate predictions of infant exposure. For new drugs with no breastmilk data, using a range of clinically feasible scenarios of milk-to-plasma ratios, and also taking the physicochemical properties of the compound into account, to simulate potential infant exposure could be a starting point to generate data for follow-up clinical lactation studies [80].
3 Discussion and conclusion
Information describing pregnancy-specific safety and PK data are critically important for all medications that could potentially be prescribed for use in pregnant women. In order for drugs to be used safely and effectively in pregnant women, studies are necessary to describe maternal drug exposure during pregnancy, identify the optimal dose for use in pregnancy, assess the magnitude of human placental transfer and resulting fetal drug exposure, and describe maternal and infant clinical outcomes. While some information on drug toxicity may be derived from adult data, i.e. which organs or functions are likely to affected, the extent to which drugs influence the development of organs in a human fetus remains unknown without experience from human dosing during pregnancy.
The new approaches described in this paper can facilitate and accelerate studies to define the optimal use of medications in pregnant women.
-
1.
Placental transfer of drugs can be predicted from placental cell lines, ex vivo placenta transfer studies, and/or placenta-on-a-chip experiments. These studies can inform pregnancy PBPK models, predict fetal exposure during pregnancy, and improve the placental unit in these models.
-
2.
Pregnancy PBPK models can predict the alterations in maternal drug exposure during pregnancy and suggest an initial dose for in vivo pregnancy studies, enhancing safety and accelerating the timeline of human pregnancy studies. Microdosing studies in pregnancy may also be used to describe the effect of pregnancy on PK, and can provide data to refine PBPK and population PK models to improve dose predictions. PK modeling can be used to optimally design pregnancy PK substudies, either as stand-alone studies or embedded as substudies in phase III clinical trials.
In the context of breastfeeding the following approaches can help to provide important information.
-
1.
Breastmilk transfer can be assessed using in vitro cell cultures, which can inform lactation PBPK models, predict milk concentrations, and estimate total daily intake by infants.
-
2.
Infant exposure can be predicted using the dose per feeding moment (derived from the lactation PBPK model), and different organ exposure can be predicted using pediatric PBPK models. These can generate some insight into potential developmental problems at the organ level, which then informs which studies should be conducted in breastfed infants.
An integrated approach incorporating a range of methodologies (in vitro, ex vivo, in silico and in vivo studies) (Fig. 1) is key to accelerating the availability of pharmacology data in pregnancy and lactation to allow the safe and effective use of medications during pregnancy and lactation.
References
Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernandez-Diaz S. Medication use during pregnancy, with particular focus on prescription drugs: 1976-2008. Am J Obstet Gynecol. 2011;205(1):51.e1–8.
Lupattelli A, Spigset O, Nordeng H. Adherence to medication for chronic disorders during pregnancy: results from a multinational study. Int J Clin Pharm. 2014;36(1):145–53.
Stock SJ, Norman JE. Medicines in pregnancy [version 1; peer review: 3 approved]. F1000Research 2019, 8 (F1000 Faculty Rev):911 [cited 20 Feb 2020]. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6587138/pdf/f1000research-8-19177.pdf. Accessed 20 Feb 2020.
Matsui D. Ethics of studies of drugs in pregnancy. Paediatr Drugs. 2015;17(1):31–5.
Eke AC, Dooley KE, Sheffield JS. Pharmacologic research in pregnant women—time to get it right. N Engl J Med. 2019;380(14):1293–5.
Pernia S, DeMaagd G. The new pregnancy and lactation labeling rule. P & T. 2016;41(11):713–5.
Eke AC, Chakhtoura N, Kashuba A, Best BM, Sykes C, Wang J, et al. Rilpivirine plasma and cervicovaginal concentrations in women during pregnancy and postpartum. J Acquir Immune Defic Syndr. 2018;78(3):308–13.
Eke AC, McCormack SA, Best BM, Stek AM, Wang J, Kreitchmann R, et al. Pharmacokinetics of increased nelfinavir plasma concentrations in women during pregnancy and postpartum. J Clin Pharmacol. 2019;59(3):386–93.
Eke AC, Stek AM, Wang J, Kreitchmann R, Shapiro DE, Smith E, et al. Darunavir pharmacokinetics with an increased dose during pregnancy. J Acquir Immune Defic Syndr. 2020;83(4):373–80.
Eke AC, Wang J, Amin K, Shapiro DE, Stek A, Smith E, et al. Fosamprenavir with ritonavir pharmacokinetics during pregnancy. Antimicrob Agents Chemother. 2020;64(4):e02260–022619.
Boyd SD, Sampson MR, Viswanathan P, Struble KA, Arya V, Sherwat AI. Cobicistat-containing antiretroviral regimens are not recommended during pregnancy: viewpoint. AIDS. 2019;33(6):1089–93.
Eke AC, Mirochnick MH. Cobicistat as a pharmacoenhancer in pregnancy and postpartum: progress to date and next steps. J Clin Pharmacol. 2019;59(6):779–83.
Eke AC, Brooks KM, Gebreyohannes RD, Sheffield JS, Dooley KE, Mirochnick M. Tenofovir alafenamide use in pregnant and lactating women living with HIV. Expert Opin Drug Metab Toxicol. 2020;16(4):333–42.
Momper JD, Best BM, Wang J, Capparelli EV, Stek A, Barr E, et al. Elvitegravir/cobicistat pharmacokinetics in pregnant and postpartum women with HIV. AIDS. 2018;32(17):2507–16.
Crauwels HM, Osiyemi O, Zorrilla C, Bicer C, Brown K. Reduced exposure to darunavir and cobicistat in HIV-1-infected pregnant women receiving a darunavir/cobicistat-based regimen. HIV Med. 2019;20(5):337–43.
Momper JS, Stek A, Wang J, Shapiro DE, Smith E, Chakhtoura N, et al. Pharmacokinetics of atazanavir boosted with cobicistat during pregnancy and postpartum. In: 20th International Workshop on Clinical Pharmacology of HIV, Hepatitis, and Other Antiviral Drugs. 14–16 May 2019; Noordwijk, The Netherlands.
Colbers A, Mirochnick M, Schalkwijk S, Penazzato M, Townsend C, Burger D. Importance of prospective studies in pregnant and breastfeeding women living with human immunodeficiency virus. Clin Infect Dis. 2019;69(7):1254–8.
Syme MR, Paxton JW, Keelan JA. Drug transfer and metabolism by the human placenta. Clin Pharmacokinet. 2004;43(8):487–514.
Cerveny L, Ptackova Z, Ceckova M, Karahoda R, Karbanova S, Jiraskova L, et al. Equilibrative nucleoside transporter 1 (ENT1, SLC29A1) facilitates transfer of the antiretroviral drug abacavir across the placenta. Drug Metab Dispos. 2018;46(11):1817–26.
Rothbauer M, Patel N, Gondola H, Siwetz M, Huppertz B, Ertl P. A comparative study of five physiological key parameters between four different human trophoblast-derived cell lines. Sci Rep. 2017;7(1):5892.
Kitano T, Iizasa H, Hwang IW, Hirose Y, Morita T, Maeda T, et al. Conditionally immortalized syncytiotrophoblast cell lines as new tools for study of the blood-placenta barrier. Biol Pharm Bull. 2004;27(6):753–9.
Vahakangas K, Myllynen P. Experimental methods to study human transplacental exposure to genotoxic agents. Mutat Res. 2006;608(2):129–35.
Lacconi V, Massimiani M, Magrini A, Pietroiusti A. In vitro experimental models to study the efficiency of the placental barrier for environmental toxicants: tumor cell lines versus trophoblast primary cells. Biomed Prev issues. 2018;1:157.
Karlgren M, Simoff I, Backlund M, Wegler C, Keiser M, Handin N, et al. A CRISPR-Cas9 generated MDCK cell line expressing human MDR1 without endogenous canine MDR1 (cABCB1): an improved tool for drug efflux studies. J Pharm Sci. 2017;106(9):2909–13.
Cerveny L, Ptackova Z, Durisova M, Staud F. Interactions of protease inhibitors atazanavir and ritonavir with ABCB1, ABCG2, and ABCC2 transporters: effect on transplacental disposition in rats. Reprod Toxicol. 2018;79:57–65.
Neumanova Z, Cerveny L, Ceckova M, Staud F. Interactions of tenofovir and tenofovir disoproxil fumarate with drug efflux transporters ABCB1, ABCG2, and ABCC2; role in transport across the placenta. AIDS. 2014;28(1):9–17.
Reznicek J, Ceckova M, Tupova L, Staud F. Etravirine inhibits ABCG2 drug transporter and affects transplacental passage of tenofovir disoproxil fumarate. Placenta. 2016;47:124–9.
De Sousa Mendes M, Hirt D, Vinot C, Valade E, Lui G, Pressiat C, et al. Prediction of human fetal pharmacokinetics using ex vivo human placenta perfusion studies and physiologically based models. Br J Clin Pharmacol. 2016;81(4):646–57.
Kovo M, Golan A. In vitro models using the human placenta to study fetal exposure to drugs. Clin Med Reprodu Health. 2008;2:15–24.
Vinot C, Gavard L, Treluyer JM, Manceau S, Courbon E, Scherrmann JM, et al. Placental transfer of maraviroc in an ex vivo human cotyledon perfusion model and influence of ABC transporter expression. Antimicrob Agents Chemother. 2013;57(3):1415–20.
Blundell C, Yi YS, Ma L, Tess ER, Farrell MJ, Georgescu A, et al. Placental drug transport-on-a-chip: a microengineered in vitro model of transporter-mediated drug efflux in the human placental barrier. Adv Healthc Mater. 2018. https://doi.org/10.1002/adhm.201700786.
Lee JS, Romero R, Han YM, Kim HC, Kim CJ, Hong JS, et al. Placenta-on-a-chip: a novel platform to study the biology of the human placenta. J Matern Fetal Neonatal Med. 2016;29(7):1046–54.
Yin F, Zhu Y, Zhang M, Yu H, Chen W, Qin J. A 3D human placenta-on-a-chip model to probe nanoparticle exposure at the placental barrier. Toxicol In Vitro. 2019;54:105–13.
Pemathilaka RL, Reynolds DE, Hashemi NN. Drug transport across the human placenta: review of placenta-on-a-chip and previous approaches. Interface Focus. 2019;9(5):20190031.
Grafmuller S, Manser P, Krug HF, Wick P, von Mandach U. Determination of the transport rate of xenobiotics and nanomaterials across the placenta using the ex vivo human placental perfusion model. J Vis Exp. 2013;76:50401.
Anderson PO. Drugs in lactation. Pharm Res. 2018;35(3):45.
Ito N, Ito K, Koshimichi H, Hisaka A, Honma M, Igarashi T, et al. Contribution of protein binding, lipid partitioning, and asymmetrical transport to drug transfer into milk in mouse versus human. Pharm Res. 2013;30(9):2410–22.
Jin L, Qu Y, Gomez LJ, Chung S, Han B, Gao B, et al. Characterization of primary human mammary epithelial cells isolated and propagated by conditional reprogrammed cell culture. Oncotarget. 2018;9(14):11503–14.
Gerk PM, Moscow JA, McNamara PJ. Basolateral active uptake of nitrofurantoin in the CIT3 cell culture model of lactation. Drug Metab Dispos. 2003;31(6):691–3.
Athavale MA, Maitra A, Patel S, Bhate VR, Toddywalla VS. Development of an in vitro cell culture model to study milk to plasma ratios of therapeutic drugs. Indian J Pharmacol. 2013;45(4):325–9.
ConcePTION. The Work Packages—determination of drug transfer and infant drug exposure during lactation: generation of quantitative and translatable data. https://www.imi-conception.eu/work/. Accessed 22 Mar 2020.
Orrell C, Hagins DP, Belonosova E, Porteiro N, Walmsley S, Falco V, et al. Fixed-dose combination dolutegravir, abacavir, and lamivudine versus ritonavir-boosted atazanavir plus tenofovir disoproxil fumarate and emtricitabine in previously untreated women with HIV-1 infection (ARIA): week 48 results from a randomised, open-label, non-inferiority, phase 3b study. Lancet HIV. 2017;4(12):e536–46.
Venter WDF, Moorhouse M, Sokhela S, Fairlie L, Mashabane N, Masenya M, et al. Dolutegravir plus two different prodrugs of tenofovir to treat HIV. N Engl J Med. 2019;381(9):803–15.
Squires K, Kityo C, Hodder S, Johnson M, Voronin E, Hagins D, et al. Integrase inhibitor versus protease inhibitor based regimen for HIV-1 infected women (WAVES): a randomised, controlled, double-blind, phase 3 study. Lancet HIV. 2016;3(9):e410–20.
Aherfi S, Solas C, Motte A, Moreau J, Borentain P, Mokhtari S, et al. Hepatitis C virus NS3 protease genotyping and drug concentration determination during triple therapy with telaprevir or boceprevir for chronic infection with genotype 1 viruses, southeastern France. J Med Virol. 2014;86(11):1868–76.
Waitt C, Orrell C, Walimbwa S, Singh Y, Kintu K, Simmons B, et al. Safety and pharmacokinetics of dolutegravir in pregnant mothers with HIV infection and their neonates: a randomised trial (DolPHIN-1 study). PLoS Med. 2019;16(9):e1002895.
Kintu K, Malaba T, Nakibuka J, Papamichael, Colbers A, Byrne K. Dolutegravir versus efavirenz in women starting HIV therapy in late pregnancy (DolPHIN-2): an open-label, randomised controlled trial. Lancet HIV. 2020;7:332–9.
Sadler BM, Stein DS. Clinical pharmacology and pharmacokinetics of amprenavir. Ann Pharmacother. 2002;36(1):102–18.
Tewari T, Mukherjee S. Microdosing: concept, application and relevance. Perspect Clin Res. 2010;1(2):61–3.
Lappin G, Noveck R, Burt T. Microdosing and drug development: past, present and future. Expert Opin Drug Metab Toxicol. 2013;9(7):817–34.
Burt T, Yoshida K, Lappin G, Vuong L, John C, de Wildt SN, et al. Microdosing and other phase 0 clinical trials: facilitating translation in drug development. Clin Transl Sci. 2016;9(2):74–88.
Roth-Cline M, Nelson RM. Microdosing studies in children: a US regulatory perspective. Clin Pharmacol Ther. 2015;98(3):232–3.
Mooij MG, van Duijn E, Knibbe CA, Windhorst AD, Hendrikse NH, Vaes WH, et al. Pediatric microdose study of [(14)C]paracetamol to study drug metabolism using accelerated mass spectrometry: proof of concept. Clin Pharmacokinet. 2014;53(11):1045–51.
Turner MA, Mooij MG, Vaes WH, Windhorst AD, Hendrikse NH, Knibbe CA, et al. Pediatric microdose and microtracer studies using 14C in Europe. Clin Pharmacol Ther. 2015;98(3):234–7.
Burt T, Vuong LT, Baker E, Young GC, McCartt AD, Bergstrom M, et al. Phase 0, including microdosing approaches: applying the Three Rs and increasing the efficiency of human drug development. Alternat Lab Anim. 2018;46(6):335–46.
Barker CIS, Standing JF, Kelly LE, Hanly Faught L, Needham AC, Rieder MJ, et al. Pharmacokinetic studies in children: recommendations for practice and research. Arch Dis Child. 2018;103(7):695–702.
van Nuland M, Rosing H, Huitema ADR, Beijnen JH. Predictive value of microdose pharmacokinetics. Clin Pharmacokinet. 2019;58(10):1221–36.
Lappin G, Kuhnz W, Jochemsen R, Kneer J, Chaudhary A, Oosterhuis B, et al. Use of microdosing to predict pharmacokinetics at the therapeutic dose: experience with 5 drugs. Clin Pharmacol Ther. 2006;80(3):203–15.
Noguchi L, Beigi R, Biggio J, Marzinke M, Kelly C, Bunge K, et al. Breast milk dapivirine pharmacokinetics, estimated infant exposure, and safety during dapivirine intravaginal ring use among lactating women. Am J Obstet Gynecol. 2017;217(6):717.
Benaboud S, Ekouevi DK, Urien S, Rey E, Arrive E, Blanche S, et al. Population pharmacokinetics of nevirapine in HIV-1-infected pregnant women and their neonates. Antimicrob Agents Chemother. 2011;55(1):331–7.
Hirt D, Urien S, Jullien V, Firtion G, Chappuy H, Rey E, et al. Pharmacokinetic modelling of the placental transfer of nelfinavir and its M8 metabolite: a population study using 75 maternal-cord plasma samples. Br J Clin Pharmacol. 2007;64(5):634–44.
Lommerse J, Clarke D, Kerbusch T, Merdjan H, Witjes H, Teppler H, et al. Maternal-neonatal raltegravir population pharmacokinetics modeling: implications for initial neonatal dosing. CPT Pharmacometr Syst Pharmacol. 2019;8(9):643–53.
Darakjian LI, Kaddoumi A. Physiologically based pharmacokinetic/pharmacodynamic model for caffeine disposition in pregnancy. Mol Pharm. 2019;16(3):1340–9.
Gockenbach M, Grimstein M, Momper J, Mirochnick M, Capparelli E, Struble K. Physiologically-based pharmacokinetic modeling of rilpivirine during pregnancy [oral abstract 16]. 20th International Workshop on Clinical Pharmacology of HIV, Hepatitis, and Other Antiviral Drugs. 14–16 May 2019; Noordwijk, The Netherlands.
Alsmadi MM, Idkaidek N. Optimization of drugs pharmacotherapy during pregnancy using physiologically based pharmacokinetic models—an update. Curr Drug Metab. 2018;19(12):972–8.
Colbers A, Greupink R, Litjens C, Burger D, Russel FG. Physiologically based modelling of darunavir/ritonavir pharmacokinetics during pregnancy. Clin Pharmacokinet. 2016;55(3):381–96.
Zhang Z, Unadkat JD. Development of a novel maternal-fetal physiologically based pharmacokinetic model II: verification of the model for passive placental permeability drugs. Drug Metab Dispos. 2017;45(8):939–46.
De Sousa Mendes M, Lui G, Zheng Y, Pressiat C, Hirt D, Valade E, et al. A physiologically-based pharmacokinetic model to predict human fetal exposure for a drug metabolized by several CYP450 pathways. Clin Pharmacokinet. 2017;56(5):537–50.
Schalkwijk S, Buaben AO, Freriksen JJM, Colbers AP, Burger DM, Greupink R, et al. Prediction of fetal darunavir exposure by integrating human ex-vivo placental transfer and physiologically based pharmacokinetic modeling. Clin Pharmacokinet. 2018;57(6):705–16.
Roberts O, Rajoli RKR, Back DJ, Owen A, Darin KM, Fletcher CV, et al. Physiologically based pharmacokinetic modelling prediction of the effects of dose adjustment in drug-drug interactions between levonorgestrel contraceptive implants and efavirenz-based ART. J Antimicrob Chemother. 2018;73(4):1004–12.
Marzolini C, Rajoli R, Battegay M, Elzi L, Back D, Siccardi M. Physiologically based pharmacokinetic modeling to predict drug-drug interactions with efavirenz involving simultaneous inducing and inhibitory effects on cytochromes. Clin Pharmacokinet. 2017;56(4):409–20.
Rose RH, Turner DB, Neuhoff S, Jamei M. Incorporation of the time-varying postprandial increase in splanchnic blood flow into a PBPK model to predict the effect of food on the pharmacokinetics of orally administered high-extraction drugs. AAPS J. 2017;19(4):1205–17.
Atoyebi SA, Rajoli RKR, Adejuyigbe E, Owen A, Bolaji O, Siccardi M, et al. Using mechanistic physiologically-based pharmacokinetic models to assess prenatal drug exposure: thalidomide versus efavirenz as case studies. Eur J Pharm Sci. 2019;140:105068.
Eke AC, Gebreyohannes RD. Physiologically based pharmacokinetic modeling (PBPK’s) prediction potential in clinical pharmacology decision making during pregnancy. Int J Gynaecol Obstet. Epub 4 Apr 2020. https://doi.org/10.1002/ijgo.13150.
Dallmann A, Pfister M, van den Anker J, Eissing T. Physiologically based pharmacokinetic modeling in pregnancy: a systematic review of published models. Clin Pharmacol Ther. 2018;104(6):1110–24.
Challa A, Beam A, Shen M, Peryea T, Lavieri R, Lippmann E, et al. Machine learning on drug-specific data to predict small molecule teratogenicity. 2019 [cited 20 Feb 2020]. https://www.biorxiv.org/content/10.1101/860627v1.
Corley RA, Mast TJ, Carney EW, Rogers JM, Daston GP. Evaluation of physiologically based models of pregnancy and lactation for their application in children’s health risk assessments. Crit Rev Toxicol. 2003;33(2):137–211.
Ito S, Blajchman A, Stephenson M, Eliopoulos C, Koren G. Prospective follow-up of adverse reactions in breast-fed infants exposed to maternal medication. Am J Obstet Gynecol. 1993;168(5):1393–9.
Willmann S, Edginton AN, Coboeken K, Ahr G, Lippert J. Risk to the breast-fed neonate from codeine treatment to the mother: a quantitative mechanistic modeling study. Clin Pharmacol Ther. 2009;86(6):634–43.
Olagunju A, Rajoli RK, Atoyebi SA, Khoo S, Owen A, Siccardi M. Physiologically-based pharmacokinetic modelling of infant exposure to efavirenz through breastfeeding [version 1; peer review: 2 approved with reservations]. AAS Open Res. 2018;1:16.
Acknowledgements
The findings and conclusions of this manuscript are those of the authors and do not necessarily represent the official position of the WHO.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were used to assist in the preparation of this article.
Conflict of interest
Angela Colbers has received research grant support from ViiV Healthcare, Gilead Sciences, Janssen Research, and Merck, paid to the institution; Mark Mirochnick has received research grant support from ViiV Healthcare, Gilead Sciences and Merck & Co.; and Jeremiah D. Momper has received research grant support from Gilead Sciences and Veloxis Pharmaceuticals. Brookie M. Best, Ahizechukwu C. Eke, Martina Penazzato, Tim R. Cressey, Adeniyi Olagunju, and Elaine Abrams have no conflicts of interest to declare.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors' contributions
AE, AC, and TC wrote the paper, and AO, BB, MM, JM, EA, MP, and TC reviewed the paper and provided input.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Eke, A.C., Olagunju, A., Best, B.M. et al. Innovative Approaches for Pharmacology Studies in Pregnant and Lactating Women: A Viewpoint and Lessons from HIV. Clin Pharmacokinet 59, 1185–1194 (2020). https://doi.org/10.1007/s40262-020-00915-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00915-w