
Abstract
Upadacitinib is a Janus kinase 1 inhibitor developed for treatment of moderate to severe rheumatoid arthritis (RA) and was recently approved by the US Food and Drug Administration for this indication in adults who have had an inadequate response or intolerance to methotrexate. Upadacitinib is currently under regulatory review by other agencies around the world. Ongoing trials are investigating the use of upadacitinib in other inflammatory autoimmune diseases. In this article, we review the clinical pharmacokinetic data available to date for upadacitinib that supported the clinical development program in RA and ultimately regulatory applications for upadacitinib in treatment of patients with moderate to severe RA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Upadacitinib exhibits dose-proportional pharmacokinetics and biphasic elimination with a terminal half-life of 9–14 h following administration of the extended-release formulation. |
Strong CYP3A4 inhibitors increase upadacitinib exposure by 75%. Strong inducers of CYP3A4 reduce upadacitinib exposure by approximately half. No dosage adjustment is required for concomitant medications when administered with upadacitinib based on drug-interaction studies. |
Mild, moderate, and severe renal impairment as well as mild and moderate hepatic impairment had no clinically relevant effect on upadacitinib systemic exposures (< 45% increase in AUC or Cmax relative to subjects with normal renal/hepatic function). |
Body weight, sex, race, ethnicity, and age did not have a clinically relevant effect on the upadacitinib AUC or Cmax. |
1 Introduction
The Janus kinase (JAK)/signal transducers and activators of transcription (STAT) pathway is well-known for initiating multiple signaling cascades for development and homeostasis in humans; specifically, this pathway is the principal signaling mechanism for cytokines and growth factors [1]. There are four JAK enzymes: JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). The role of JAK/STAT signaling in immune-mediated disease has been well-established, leading to new therapeutic targets and treatment options for rheumatoid arthritis (RA) and other immune inflammatory diseases [2, 3].
RA is characterized by chronic systemic inflammation with persistent polyarthritis of synovial joints, which can lead to bone erosion, deformity, and disability [4]. Treatments for RA have evolved over the last 20 years with the development of biologic and targeted synthetic disease-modifying antirheumatic drugs (DMARDs) (bDMARDs and tsDMARDs, respectively), meaning a number of patients can now be diagnosed early and treated promptly, leading to remission or very low disease activity [5]. However, there still remains a proportion of patients who do not respond to existing therapies and need alternative treatments to manage their condition and reduce disability associated with the disease [3, 5].
Upadacitinib (ABT-494) is a novel selective JAK1 inhibitor that was recently approved by the US Food and Drug Administration (FDA) for the treatment of adult patients with moderately to severely active RA who have had an inadequate response or intolerance to methotrexate [6]. Upadacitinib is currently under regulatory review by other agencies around the world and is being evaluated in clinical trials for the treatment of other inflammatory conditions [7,8,9,10]. Upadacitinib potently inhibits JAK1 and is less potent against the other isoforms, JAK2, JAK3, and TYK2 [11]. The hypothesis behind the development of upadacitinib is that higher potency against JAK1 has the potential to maximize efficacy in RA while limiting the effects on physiological functions that involve JAK enzymes (e.g., hematopoiesis and immune function) [11, 12].
Upadacitinib safety and efficacy were evaluated in two phase II dose-ranging studies in patients with RA who were inadequate responders to anti-tumor necrosis factor (TNF) agents (BALANCE I) [13] or to methotrexate (BALANCE II) [14]. A regional phase IIb/III study was conducted in Japanese patients who had inadequate response to conventional synthetic DMARDs (csDMARDs) (SELECT-SUNRISE) [15]. The efficacy and safety of upadacitinib has been confirmed in five global Phase 3 studies in patients with moderately to severely active RA who were inadequate responders to methotrexate (SELECT-COMPARE [16] and SELECT-MONOTHERAPY [17]), inadequate responders to csDMARDs (SELECT-NEXT [18]), inadequate responders to bDMARDs (SELECT-BEYOND [19]), or methotrexate-naïve (SELECT-EARLY [20]). Upadacitinib demonstrated superior efficacy to comparators in patients with RA who were inadequate responders to bDMARDs (vs. placebo) or methotrexate/csDMARDs (vs. placebo and/or active comparators) when it was used in combination with methotrexate/csDMARDs and as a monotherapy in patients who did not respond to methotrexate or in patients who were methotrexate-naïve [16,17,18,19,20].
Upadacitinib is also being investigated for treatment of other inflammatory disorders, including psoriatic arthritis, juvenile idiopathic arthritis, Crohn’s disease, ulcerative colitis, atopic dermatitis, ankylosing spondylitis, and giant cell arteritis. In phase IIb trials evaluating efficacy and safety of upadacitinib in Crohn’s disease, atopic dermatitis, and ulcerative colitis, clinical benefit has been demonstrated that supported evaluation of upadacitinib in phase III trials in these diseases [7, 10, 21].
In this article, we review the clinical pharmacokinetics data available to date for upadacitinib that supported the clinical development program in RA and regulatory submissions for upadacitinib in treatment of patients with moderate to severe RA.
Upadacitinib is highly permeable and highly soluble at clinically relevant doses across the pH range of 1–7.5 [22]. Upadacitinib is 52% bound to plasma proteins [23]; therefore, no relevant interactions are expected through displacement from plasma proteins. Upadacitinib is metabolized in vitro by cytochrome P450 (CYP) enzyme 3A with minor contribution from CYP2D6 [12]. Upadacitinib did not inhibit or induce the activity of CYP enzymes (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) and did not inhibit the transporters P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), organic anion transporting polypeptide (OATP) 1B1, OATP1B3, organic cation transporter (OCT) 1, OCT2, organic anion transporter (OAT) 1, OAT3, multidrug and toxin extrusion (MATE) 1, and MATE2K at clinically relevant concentrations [23]. Review of in vitro assessments of upadacitinib is beyond the scope of this article as we focus on the relevant clinical data.
2 Clinical Pharmacokinetic Assessments
Table 1 summarizes 11 phase I studies of upadacitinib that were key in characterization of its clinical pharmacology attributes. These studies include characterization of the pharmacokinetics and relative bioavailability of the immediate-release (IR) and extended-release (ER) formulations, the potential for drug interactions with concomitant medications, and the effect of intrinsic factors on upadacitinib systemic exposures. In addition to these individual studies, population pharmacokinetic analyses were performed using combined data across multiple studies to support the overall clinical pharmacokinetic characterization for upadacitinib. Summary of phase II and III studies in patients with moderate to severe RA that included pharmacokinetic assessments are summarized in Table 2. No data are available at this time for upadacitinib pharmacokinetics in pediatric patients and a phase I study in subjects with polyarticular course juvenile arthritis is currently ongoing [24].
Upadacitinib was administered as an IR capsule formulation in early phase I and in RA phase II studies, and as an ER once-daily (QD) tablet formulation in later phase I studies, a Japan local phase IIb/III study, and global phase III studies as described in Table 1 (phase I studies) and Table 2 (phase II and III studies).
High-level pharmacokinetic results and conclusions from these studies and analyses are summarized in the following sections.
3 Clinical Pharmacokinetics of Upadacitinib
3.1 Single- and Multiple-Dose Pharmacokinetics of Upadacitinib Immediate-Release and Extended-Release Formulations
Over a single dose range of 1–48 mg of upadacitinib IR formulation in healthy volunteers, upadacitinib was rapidly absorbed, with a median time to maximum plasma concentration (tmax) of approximately 1 h under fasting conditions [12]. A summary of the single-dose pharmacokinetic parameters of upadacitinib is provided in Table 3. After reaching maximum plasma concentration (Cmax), upadacitinib plasma concentrations declined in a biphasic fashion with an apparent terminal elimination half-life (t½) of approximately 6–15 h for most doses. The percentage of upadacitinib excreted as unchanged parent drug ranged from approximately 16–21% [12]. The upadacitinib Cmax was dose proportional in the 1–48 mg dose range, and there was no trend for change in upadacitinib dose-normalized area under the plasma concentration–time curve (AUC) with increasing dose over the 3–36 mg single-dose range [12].
Twice-daily (BID) doses of up to 24 mg of upadacitinib IR were also evaluated in healthy subjects. A summary of the multiple-dose pharmacokinetic parameters for the IR formulation is provided in Table 4 and for the ER formulation in Table 5. At steady state, upadacitinib showed no accumulation with BID dosing. The upadacitinib mean tmax was approximately 2 h under non-fasting conditions and the harmonic mean terminal t½ ranged from approximately 8–16 h [12]. The upadacitinib functional t½, estimated from the Cmax to trough concentration (Ctrough) ratio at steady state, was approximately 3 h. Upadacitinib exposure (Cmax and AUC) was approximately dose proportional across the 3–24 mg BID dose range. At steady state, approximately 20% of the upadacitinib dose was excreted unchanged in the urine [12].
Because QD dosing would be more ideal for patients, an ER formulation for upadacitinib was developed. Following administration of the ER formulation, the upadacitinib median tmax was 2–3 h under fasting conditions and 4 h under non-fasting conditions (Table 3) [25]. Steady state was achieved by Day 4 and there was no accumulation of upadacitinib in plasma with multiple QD dosing of the ER formulation (Table 5). The upadacitinib harmonic mean terminal t½ ranged from 9 to 14 h after administration of the ER formulation. The 15 and 30 mg QD doses of the ER formulation provided equivalent AUC from time zero to 24 h (AUC24) and comparable Cmax and minimum plasma concentration (Cmin) values with the 6 and 12 mg BID doses of the IR formulation under fasting conditions, respectively [25]. The fluctuation index over a 24-h period was similar between the BID dosing of the IR formulation and the QD dosing of the ER formulation [25]. Based on non-compartmental analyses of phase I studies, the upadacitinib ER formulation was estimated to have bioavailability of 76% relative to the same dose of the IR formulation (Table 6).
An in vitro–in vivo correlation (IVIVC) was established between the upadacitinib in vitro dissolution and in vivo pharmacokinetics profile using data from ER formulations with different in vitro release rates [22]. The established IVIVC demonstrated that the upadacitinib in vivo pharmacokinetic profile can be adequately predicted using in vitro dissolution data at pH 6.8 for within the evaluated formulation design boundaries.
3.2 Population Pharmacokinetic Analyses
Following conclusion of the RA phase II studies, population pharmacokinetic analyses using data from three phase I studies (Studies 1, 2, and 3) and two phase II studies (BALANCE I and II) were conducted [26]. The analyses, including data from 573 subjects (81% with RA and 19% healthy), characterized upadacitinib population pharmacokinetics in healthy volunteers as well as in patients with RA and informed the comparability of upadacitinib plasma exposures for the doses selected in phase III studies across different patient subgroups.
Upadacitinib pharmacokinetics were adequately characterized using a two-compartment model with a linear elimination process and a first-order absorption process with absorption lag time for the IR formulation. Upadacitinib typical oral clearance in RA patients was 24% lower (leading to 32% higher estimated AUC) than in healthy subjects [26]. Females and mild or moderate renal impairment had a statistically significant, but non-clinically relevant, effect on the upadacitinib AUC (16% higher estimated AUC in females than in males and 16% and 32% higher AUC in mild and moderate renal impairment, respectively, relative to normal renal function). The upadacitinib typical apparent central volume of distribution (Vc/F) for females was estimated to be 75% of the corresponding values for males. Total bodyweight was a significant covariate on the upadacitinib Vc/F but did not reach statistical significance for the apparent oral clearance (CL/F) in this analysis. CYP2D6 metabolic phenotype had no effect on upadacitinib CL/F. There was no correlation between the upadacitinib IR dose and its oral bioavailability, CL/F, or Vc/F [26].
These analyses were later updated with inclusion of data from the five phase III studies in RA, which provide characterization of upadacitinib pharmacokinetics from both the IR and ER formulations and assessment of impact of intrinsic and extrinsic factors from a much larger sample size. These population pharmacokinetic analyses were conducted using data from four phase I studies that enrolled healthy volunteers and RA patients (Studies 1, 2, 3, and 4), two phase II studies in RA patients (BALANCE I and II), five phase III studies in RA patients (SELECT-COMPARE, SELECT-NEXT, SELECT-BEYOND, SELECT-MONOTHERAPY, and SELECT-EARLY), and one regional phase IIb/III study in Japanese RA patients (SELECT-SUNRISE). The analysis dataset included a total of 4170 subjects (96% with RA and 4% healthy) [27].
A two-compartment model with first-order absorption with lag time for the IR formulation, mixed zero and first-order absorption with lag time for the ER formulation, and linear elimination adequately described upadacitinib pharmacokinetics. Statistically significant covariates were patient population (RA vs. healthy), creatinine clearance (CrCL), and baseline bodyweight on CL/F; and body weight on Vc/F [27].
For a typical RA patient and reference body weight of 74 kg and CrCL of 109 mL/min, upadacitinib CL/F was estimated to be 40.5 L/h and the apparent volume of distribution at steady state (Vss/F) was estimated to be 294 L following administration of the ER formulation. The inter-subject variability for upadacitinib CL/F and Vc/F were estimated to be 21% and 24%, respectively, in the phase I studies, and 37% and 53%, respectively, in the phase II/III studies. The oral bioavailability of the ER formulation relative to the IR formulation was estimated to be 76% [27].
3.3 Effect of Intrinsic Factors on Upadacitinib Pharmacokinetics
3.3.1 Race/Asian Ethnicity
Upadacitinib pharmacokinetics were characterized in Japanese and Chinese healthy subjects (Study 2) following administration of the IR formulation [28]. Additionally, the effect of race on upadacitinib pharmacokinetics was assessed as a covariate in the population pharmacokinetic analyses. Dose-normalized Cmax and AUC values in Japanese and Chinese healthy subjects were comparable (within ~ 20%) to exposures in Western/non-Japanese subjects (Table 7). Similarly, upadacitinib plasma exposures in Japanese patients with RA were consistent (within 20%) overall with exposures in non-Japanese patients with RA based on the population pharmacokinetic analyses [26, 27, 29] and race was not a significant covariate on upadacitinib clearance in population pharmacokinetic analyses. Therefore, current data indicate that upadacitinib pharmacokinetics are not sensitive to differences between races or ethnicities. No dose adjustment for upadacitinib is warranted based on patients’ race or ethnicity [23].
3.3.2 Hepatic Impairment
A dedicated open-label, single-dose study was conducted to assess the effect of mild and moderate hepatic impairment (based on the Child-Pugh classification) on upadacitinib pharmacokinetics (Study 5) [30]. Subjects were assigned to one of three groups, with the mild hepatic impairment group consisting of those individuals that met Child-Pugh category A and the moderate hepatic impairment group aligning with Child-Pugh category B. The third group was subjects with normal hepatic function. Demographic characteristics were similar between subjects with normal and impaired hepatic function. Treatment with upadacitinib was anticipated to be not recommended in subjects with severe hepatic impairment; therefore, the impact of severe hepatic impairment on upadacitinib pharmacokinetics was not evaluated in this study [30]. There was no statistically significant difference in upadacitinib Cmax or AUC values in subjects with mild and moderate hepatic impairment compared with subjects with normal hepatic function. The upadacitinib AUC central value was 28% and 24% higher in subjects with mild and moderate hepatic impairment, respectively, than in subjects with normal hepatic function (based on a conservative analysis excluding one outlier with low AUC in the moderate hepatic impairment group; Fig. 1) [30]. The upadacitinib Cmax central value was similar in subjects with mild hepatic impairment and 43% higher in subjects with moderate hepatic impairment than in subjects with normal hepatic function. These results indicate that mild and moderate hepatic impairment have no clinically relevant effect on upadacitinib exposures (AUC or Cmax) [30].

Effect of intrinsic factors on upadacitinib exposures and dosing recommendations for upadacitinib per the US prescribing information. AUC area under the plasma concentration–time curve, CI confidence interval, Cmax maximum plasma concentration, PK pharmacokinetics
3.3.3 Renal Impairment
A single-dose, open-label study was conducted to evaluate the pharmacokinetics and safety of upadacitinib in subjects with mild, moderate, and severe renal impairment (based on estimated glomerular filtration rate [eGFR]) relative to subjects with normal renal function (Study 6) [31]. The eGFR was calculated by the Modification of Diet in Renal Disease (MDRD) equation and groups were assigned as follows: normal renal function (eGFR ≥ 90 mL/min/1.73 m2), mild renal impairment (eGFR 60–89 mL/min/1.73 m2), moderate renal impairment (eGFR 30–59 mL/min/1.73 m2), and severe renal impairment (eGFR 15–29 mL/min/1.73 m2) [32]. The pharmacokinetics of upadacitinib have not been studied in subjects with end-stage renal disease (eGFR < 15 mL/min/1.73 m2). Upadacitinib AUC from time zero to infinity (AUC∞) central values were 18%, 33%, and 44% higher in subjects with mild, moderate, and severe renal impairment, respectively, than in subjects with normal renal function (based on a conservative analysis excluding one outlier with a low AUC∞ in the moderate renal impairment group) (Fig. 1) [31]. Upadacitinib Cmax central values were similar in subjects with mild, moderate, and severe renal impairment compared with subjects with normal renal function. These results indicate that mild to severe renal impairment has only a limited and clinically non-relevant effect on the upadacitinib plasma AUC∞. Also, the estimates from the population pharmacokinetic analyses including subjects with RA were consistent with the dedicated renal impairment study in non-RA patients; this confirms the lack of need for dose adjustments in RA patients with renal impairment [31]. The results from this dedicated renal impairment study were consistent whether the eGFR was normalized based on the body surface area or not.
3.4 Effect of Extrinsic Factors on Upadacitinib Pharmacokinetics
3.4.1 Drug–Drug Interactions
In vitro studies demonstrate that upadacitinib is a substrate for metabolism by CYP3A4 and to a minor extent by CYP2D6 [11]. Therefore, assessments of the in vivo effect of modulation of CYP3A4 and CYP2D6 on upadacitinib pharmacokinetics were conducted. Additionally, assessments of the effect of coadministration of methotrexate, statin (HMG-CoA reductase inhibitor) drugs, OATP1B inhibitors, or pH-modifying medications on upadacitinib pharmacokinetics were conducted [27, 33, 34].
Upadacitinib is also a substrate for P-gp and BCRP efflux transporters. However, inhibition of P-gp or BCRP transporters is not expected to have a relevant effect in vivo on the upadacitinib AUC value due to the high solubility and high permeability of upadacitinib. The lack of a relevant role of efflux transporters on upadacitinib AUC is also supported by linearity of the upadacitinib plasma AUC value over a wide range of doses [12, 25, 27].
3.4.2 Effect of Strong Cytochrome P450 (CYP) 3A4 Inhibitors
In vitro metabolism studies suggested that upadacitinib is a non-sensitive substrate for CYP3A4. A study that evaluated the in vivo impact of potent CYP3A4 inhibition by ketoconazole was conducted according to a randomized crossover design in 12 healthy subjects (Study 7). Administration of upadacitinib IR 3 mg on Day 4 of a 6-day regimen of ketoconazole 400 mg QD resulted in an increased upadacitinib Cmax and AUC∞ by 70% and 75%, respectively, compared with upadacitinib administered alone (Fig. 2) [33]. These results indicate that administration of upadacitinib with repeated doses of a strong CYP3A inhibitor results in a weak increase (< 2-fold) in the upadacitinib AUC∞ [35].

Effect of concomitant medications on upadacitinib exposures and dosing recommendations for upadacitinib per the US prescribing information. AUC area under the plasma concentration–time curve, CI confidence interval, Cmax maximum plasma concentration, CYP cytochrome P450, OATP organic anion transporting polypeptide, PK pharmacokinetics
3.4.3 Effect of Strong CYP3A4 Inducers
Study 8 was a phase I open-label study conducted according to a two-period sequential design in 12 healthy subjects that examined the effect of rifampin (rifampicin), a strong CYP3A4 inducer, on the metabolism and pharmacokinetics of upadacitinib. A single oral 12 mg dose of upadacitinib IR was administered under non-fasting conditions at three different timepoints: in the morning on Day 1, Period 1 (without rifampin); and in the morning on Days 1 and 8 of a 9-day regimen of rifampin 600 mg QD in Period 2 (both rifampin and upadacitinib were administered at the same time on study Days 1 and 8). Concomitant administration of upadacitinib IR 12 mg formulation on Day 8 of a 9-day regimen of rifampin 600 mg QD decreased the central values of the upadacitinib Cmax and AUC by approximately 50% and 60%, respectively, compared with administration of upadacitinib alone (Fig. 2) [33].
The clinical drug interaction assessments for the effect of ketoconazole and rifampin on upadacitinib pharmacokinetics were conducted using the IR formulation, prior to the availability of the ER formulation. The magnitude of effect of strong CYP3A4 inhibition or broad CYP induction was expected to be consistent for the IR and the ER formulations because upadacitinib is not expected to undergo an extensive first-pass metabolism (based on the minor fraction of dose eliminated as metabolites and the < 2-fold increase in exposure with strong CYP3A4 inhibition) [23, 30, 33].
3.4.4 Effect of CYP2D6 Genotype or CYP2D6 Inhibitors
The population pharmacokinetic analysis of phase I and II trials evaluated the effect of CYP2D6 phenotype (based on CYP2D6 genotype) on upadacitinib CL/F and showed that CYP2D6 metabolic phenotype has no correlation with upadacitinib CL/F [26]. For CYP2D6, subjects with the poor metabolizer phenotype have no functional copies of CYP2D6; therefore, the poor metabolizer phenotype represents a worst-case scenario for CYP2D6 inhibition. Given the comparability of upadacitinib CL/F between subjects with extensive and poor metabolizer phenotypes for CYP2D6, concomitant medications that are strong inhibitors of CYP2D6 are expected to have no effect on upadacitinib plasma AUC values and this covariate was not considered clinically relevant for the remainder of the upadacitinib clinical program [26].
3.4.5 Effect of Organic Anion Transporting Polypeptide (OATP) 1B Inhibitors
In Study 8 (as described in Sect. 3.4.3), the rifampin effect was assessed after the first dose to evaluate the effect of inhibition of hepatic uptake OATP1B transporters on upadacitinib pharmacokinetics. Concomitant administration of upadacitinib IR 12 mg QD with rifampin 600 mg QD on Day 1 of Period 2 had no effect on the upadacitinib plasma AUC (Fig. 2) [33].
3.4.6 Effect of pH-Modifying Medications
In the population pharmacokinetic analyses of phase I–III studies, concomitant use of pH-modifying drugs (histamine H2 antagonists, proton pump inhibitors, or antacids) had no effect on upadacitinib CL/F or the bioavailability of the upadacitinib ER formulation relative to the IR formulation [27, 36]. This was also consistent with the similarity of the upadacitinib ER formulation dissolution under different pH conditions. Therefore, coadministration of upadacitinib with pH-modifying medications is not expected to have a relevant effect on the upadacitinib plasma AUC.
3.4.7 Effect of Methotrexate or Other Conventional Synthetic Disease-Modifying Antirheumatic Drugs (csDMARDs)
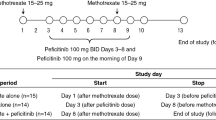
In Study 3, the effect of stable methotrexate treatment on upadacitinib pharmacokinetics was evaluated in patients with RA. The study included patients with mild to moderate RA who were on methotrexate for at least 3 months and receiving a stable dose of 10–25 mg/week for at least 4 weeks before the first dose of upadacitinib, and continued their weekly stable dose of methotrexate on Days 1, 8, 15, 22, and 29. Upadacitinib was administered BID at three doses (6, 12, and 24 mg and placebo in parallel groups) for 26 days (on Day 3 through 28) and a single morning dose on Day 29. Coadministration of upadacitinib with methotrexate had no effect on the upadacitinib plasma AUC relative to administration of upadacitinib alone in patients with RA who were on background treatment with methotrexate (Fig. 2) [12].
The most commonly used csDMARDs in combination with upadacitinib across phase III studies were methotrexate, hydroxychloroquine, sulfasalazine, and leflunomide. Use of csDMARDs did not have a significant effect on upadacitinib CL/F in population pharmacokinetics analyses (extension of analyses reported in Klunder et al. [27]; data on file, AbbVie). Additionally, upadacitinib estimated plasma AUC values were comparable between patients with RA who were administered upadacitinib on background treatment of csDMARDs and patients who received upadacitinib as monotherapy. Therefore, use of csDMARDs has no effect on upadacitinib pharmacokinetics. This was consistent with the lack of expected interactions with upadacitinib based on the elimination pathways and what is known about the drug–drug interaction potential of each of the aforementioned csDMARDS.
3.4.8 Evaluation of the Effect of Select Statins
RA patients have a higher risk of cardiovascular disease than the general population. Therefore, lipid-lowering medications, such as statins, are expected to be used concomitantly with upadacitinib in some patients. Study 9 was a phase 1, two-part, two-period, open-label drug–drug interaction study in 36 healthy subjects. A single dose of rosuvastatin (5 mg) or atorvastatin (10 mg) was administered on Day 1, Period 1, followed by a washout interval of 5 days. Doses of upadacitinib (30 mg QD) were administered on Days 1–10, Period 2 and a single dose of rosuvastatin (5 mg) or atorvastatin (10 mg) was administered 1 h after the upadacitinib dose on Day 7, Period 2. Upadacitinib pharmacokinetics were assessed at steady state when upadacitinib was administered alone (on Day 6, Period 2) and with rosuvastatin or atorvastatin (on Day 7, Period 2). Administration of a single 5 mg dose of rosuvastatin or a single 10 mg dose of atorvastatin had no relevant effect on upadacitinib Cmax or AUC values [34]. Thus, concomitant administration of rosuvastatin or atorvastatin had no effect on upadacitinib steady-state AUC and Cmax in healthy subjects [34].
3.4.9 Effect of Food on Upadacitinib Exposure
The effect of food on upadacitinib exposures from the IR and ER formulations was determined by comparing upadacitinib pharmacokinetic parameters under fasting conditions and after a high-fat breakfast. For the IR formulation, the upadacitinib tmax was delayed by approximately 2 h under the high-fat/high-calorie meal condition that also resulted in a 23% decrease in Cmax and no effect on AUC∞ [33]. As a result of the lack of a clinically relevant effect of food on upadacitinib exposure with the IR formulation, upadacitinib was administered without regard to food in phase II trials.
Administration of upadacitinib after a high-fat/high-calorie meal also had a limited and clinically non-relevant effect (~ 20% increase) on upadacitinib AUC and Cmax values compared with administration under fasting conditions [25]. Additionally, upadacitinib steady-state AUC24, Cmax, and Cmin values were comparable when 30 mg QD was administered under fasting and non-fasting conditions [25]. Based on results from this study, the upadacitinib ER formulation was administered without regard to food in phase III studies [25].
3.5 Effect of Upadacitinib on Pharmacokinetics of Concomitant Medications
Clinical phase I studies were conducted to evaluate the effect of coadministration of multiple doses of upadacitinib on the pharmacokinetics of specific probe substrates for different CYP enzymes, specifically on oral contraceptives (levonorgestrel and ethinylestradiol), rosuvastatin and atorvastatin, and methotrexate. The studies were conducted using a 30 mg QD dose of the upadacitinib ER formulation (a dose that is twice the FDA-approved dose of upadacitinib in RA) because these studies were conducted prior to completion of phase III studies and 30 mg QD was the highest upadacitinib dose evaluated in phase III RA studies.
A summary of the results for the effect of upadacitinib on plasma exposures of concomitant medications is provided in Fig. 3.

Effect of upadacitinib on exposures of co-administered drugs and associated dosing recommendations per the US prescribing information. 5-OH 5-hydroxy, AUC area under the plasma concentration–time curve, CI confidence interval, Cmax maximum plasma concentration, CYP cytochrome P450, PK pharmacokinetics
3.5.1 CYP-Sensitive Substrates
Typically, a drug is classified as a weak inhibitor if it increases the AUC of a sensitive index CYP substrate by ≥ 1.25- to ≤ 2-fold and a drug is classified as a weak inducer if it decreases the AUC of a sensitive index CYP substrate by ≥ 20 to < 50% [35, 37]. Therefore, an effect that is less than a 25% increase or 20% decrease in the AUC of an index CYP substrate is not considered to be a relevant inhibition or induction, respectively, particularly for drugs without a narrow therapeutic index.
The effects of upadacitinib on in vivo activity of different CYP enzymes was evaluated using a cocktail approach. In Study 10, healthy subjects (n = 20) received single oral doses of the modified Cooperstown 5 + 1 cocktail drugs (midazolam [CYP3A], caffeine [CYP1A2], warfarin + vitamin K [CYP2C9], omeprazole [CYP2C19], and dextromethorphan [CYP2D6]) without upadacitinib and on Day 11 (midazolam) or 12 (all other probes) of a 15-day regimen of upadacitinib ER 30 mg QD [36]. Of all the sensitive probes and markers evaluated, only the effect of upadacitinib 30 mg QD on midazolam slightly exceeded the established thresholds (26% decrease in midazolam exposures) (Fig. 3) [36]. This small effect is within the expected variability in plasma exposures of most drugs. Given that midazolam is a sensitive CYP3A substrate and the effect of upadacitinib on midazolam exposures is relatively small, it is not expected that upadacitinib will have clinically relevant effects on plasma exposures of drugs metabolized by CYP3A. The small effect of upadacitinib 30 mg QD on midazolam exposures is not expected to be clinically relevant and does not necessitate dose adjustment for concomitant medications that are substrates for CYP3A when coadministered with upadacitinib [23, 36].
Additionally, a drug–drug interaction study with bupropion, a probe substrate for CYP2B6 metabolism, evaluated the effects of multiple doses of upadacitinib on the pharmacokinetics of bupropion, where subjects received a single dose of the bupropion ER 150 mg formulation administered alone (Period 1) and on Day 12 of a 16-day regimen of upadacitinib 30 mg QD (Period 2). Coadministration of upadacitinib 30 mg QD and bupropion 150 mg on Day 12 had no clinically relevant impact on bupropion exposure (Cmax or AUC) [38]. The central values for bupropion Cmax and AUC ratios when administered with upadacitinib relative to when administered without upadacitinib were 0.87 and 0.92, respectively, with the 90% confidence interval for bupropion AUC ratio falling within the default 0.8–1.25 equivalence boundaries. These results confirmed that upadacitinib at clinically relevant exposures has no effect on the pharmacokinetics of substrates metabolized by CYP2B6 [38].
3.5.1.1 Oral Contraceptives
Another common concomitant medication of interest in the target patient population is oral contraceptives, which are partially metabolized by CYP3A [39]. Study 11 was designed to evaluate the effect of multiple doses of upadacitinib ER 30 mg QD on the pharmacokinetics of a single oral dose of ethinylestradiol/levonorgestrel (0.03/0.15 mg) administered alone in Period 1 and on Day 12 of a 14-day regimen of upadacitinib in Period 2 in healthy female subjects. Ethinylestradiol/levonorgestrel Cmax and AUC values were bioequivalent when administered with upadacitinib compared with when administered in the absence of upadacitinib (Fig. 3) [40]. These findings further support the lack of a relevant effect of upadacitinib 30 mg QD (a dose that is double the approved dose for the RA indication) on the AUC and Cmax of CYP3A substrates [40].
3.5.2 Select Statins
Study 9, as described in Sect. 3.4.9, evaluated the pharmacokinetic effect of upadacitinib on rosuvastatin or atorvastatin, substrates for OATP1B1/3 transporters. With multiple upadacitinib 30 mg QD dose administrations, there was no increase in rosuvastatin or atorvastatin systemic exposures, indicating lack of inhibition of OATP1B in vivo by upadacitinib. Rosuvastatin Cmax and AUC∞ values were 23% and 33% lower, respectively, and the atorvastatin AUC∞ was 23% lower when they were coadministered with upadacitinib relative to when administered alone (Fig. 3). AUC and Cmax values for ortho hydroxyatorvastatin (the major active metabolite for atorvastatin) remained unchanged for atorvastatin concomitant administration with upadacitinib relative to atorvastatin administration alone. This apparent small decrease in the AUC of statins after multiple dose administration of upadacitinib 30 mg QD is comparable with the intra-subject variability in AUC and Cmax values of statins; therefore, it is not expected to be clinically relevant.
3.5.3 Methotrexate
As described in Sect. 3.4.8, Study 3 evaluated the effect of coadministration of methotrexate on upadacitinib pharmacokinetics. During the same study, the effect of upadacitinib on methotrexate pharmacokinetics was also evaluated. The central values for the methotrexate AUC∞ and Cmax after multiple doses of upadacitinib (Day 29) were equivalent to those of administration of methotrexate without upadacitinib (Day 1) (within the bioequivalence thresholds; Fig. 3) [12]. No dosage adjustments are recommended for coadministration with methotrexate.
4 Conclusions
The clinical pharmacokinetics of upadacitinib have been well-characterized using data from phase I, II, and III studies in healthy subjects and RA patients following single- and multiple-dose administrations with both IR and ER formulations. These studies and analyses enabled robust characterization of the effects of different intrinsic and extrinsic factors on upadacitinib pharmacokinetics as well as the potential for upadacitinib to influence the pharmacokinetics of other medications. None of the intrinsic factors evaluated are expected to have clinically relevant effects on upadacitinib systemic exposures. Upadacitinib AUC and Cmax values are significantly influenced by strong CYP3A4 inhibitors and strong CYP3A4 inducers. Therefore, upadacitinib should be administrated with caution in patients receiving long-term treatment with strong CYP3A4 inhibitors (e.g., ketoconazole) and coadministration with strong CYP3A4 inducers (e.g., rifampin) is not recommended. No dose adjustment is required when upadacitinib is administered in subjects with mild, moderate, or severe renal impairment or in subjects with mild or moderate hepatic impairment. Similarly, the results suggest that upadacitinib has low drug interaction potential and that no dose adjustment is required for concomitant medications when coadministered with upadacitinib. These data supported FDA approval and dosing recommendations across patient subpopulations and with concomitant medications for upadacitinib 15 mg QD in patients with moderate to severe RA [23].
References
Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117(Pt 8):1281–3. https://doi.org/10.1242/jcs.00963.
O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36(4):542–50. https://doi.org/10.1016/j.immuni.2012.03.014.
Fragoulis GE, McInnes IB, Siebert S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology (Oxford). 2019;58(Suppl_1):i43–54.
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–38. https://doi.org/10.1016/S0140-6736(16)30173-8.
Burmester GR, Pope JE. Novel treatment strategies in rheumatoid arthritis. Lancet. 2017;389(10086):2338–48. https://doi.org/10.1016/S0140-6736(17)31491-5.
AbbVie. AbbVie receives FDA approval of RINVOQ™ (upadacitinib), an oral JAK inhibitor for the treatment of moderate to severe rheumatoid arthritis. 2019. https://news.abbvie.com/news/press-releases/abbvie-receives-fda-approval-rinvoq-upadacitinib-an-oral-jak-inhibitor-for-treatment-moderate-to-severe-rheumatoid-arthritis.htm. Accessed 28 Nov 2019.
Guttman-Yassky E, Silverberg JI, Thaci D, Hong C-H, Mohamed M-EF, Othman AA, et al. Primary results from a phase 2b, randomized, placebo-controlled trial of upadacitinib for patients with atopic dermatitis [abstract no. #6533]. In: American Academy of Dermatology Annual Meeting; 16–20 Feb 2018; San Diego.
Mohamed MF, Klunder B, Lacerda AP, Othman AA. Exposure-response analyses for upadacitinib efficacy and safety in the Crohn’s disease CELEST study and bridging to the extended-release formulation. Clin Pharmacol Ther. 2019. https://doi.org/10.1002/cpt.1668(epub 2019 Oct 8).
Panaccione R, D’Haens G, Sandborn W, Ghosh S, Schreiber S, Tanida S, et al. Efficacy of upadacitinib as an induction therapy for patients with moderately to severely active ulcerative colitis, with or without previous treatment failure of biologic therapy: data from the dose-ranging phase 2B study U-Achieve [abstract no. 799]. Gastroenterology. 2019;156(6):S-170.
Sandborn W, Feagan B, Panes J, D’Haens G, Colombel JF, Zhou Q, et al. Safety and efficacy of ABT-494 (upadacitinib), an oral Jak1 inhibitor, as induction therapy in patients with Crohn’s disease: results from Celest. Gastroenterology. 2017;152(5):S1308–9. https://doi.org/10.1016/S0016-5085(17)34357-3.
Parmentier JM, Voss J, Graff C, Schwartz A, Argiriadi M, Friedman M, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. 2018;2:23. https://doi.org/10.1186/s41927-018-0031-x.
Mohamed MF, Camp HS, Jiang P, Padley RJ, Asatryan A, Othman AA. Pharmacokinetics, safety and tolerability of ABT-494, a novel selective JAK 1 inhibitor, in healthy volunteers and subjects with rheumatoid arthritis. Clin Pharmacokinet. 2016;55(12):1547–58. https://doi.org/10.1007/s40262-016-0419-y.
Kremer JM, Emery P, Camp HS, Friedman A, Wang L, Othman AA, et al. A phase IIb study of ABT-494, a selective JAK-1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti-tumor necrosis factor therapy. Arthritis Rheumatol. 2016;68(12):2867–77. https://doi.org/10.1002/art.39801.
Genovese MC, Smolen JS, Weinblatt ME, Burmester GR, Meerwein S, Camp HS, et al. Efficacy and safety of ABT-494, a selective JAK-1 inhibitor, in a phase IIb study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. 2016;68(12):2857–66. https://doi.org/10.1002/art.39808.
Tanaka Y, Takeuchi T, Yamaoka K, Oribe M, Kawano M, Zhou Y, Othman AA, Pangan AL, Kitamura S, Matsuda N, Meerwein S. Kameda H. SAT0257 A phase 2b/3 randomised, placebo-controlled, double-blind study of upadacitinib, a selective jak1 inhibitor, in japanese patients with active rheumatoid arthritis and inadequate response to conventional synthetic DMARDs. Ann Rheum Dis. 2018;77(Suppl 2):991–2.
Fleischmann R, Pangan AL, Song IH, Mysler E, Bessette L, Peterfy C, et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III, double-blind, randomized controlled trial. Arthritis Rheumatol. 2019;71(11):1788–800. https://doi.org/10.1002/art.41032.
Smolen JS, Pangan AL, Emery P, Rigby W, Tanaka Y, Vargas JI, et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet. 2019;393(10188):2303–11. https://doi.org/10.1016/S0140-6736(19)30419-2.
Burmester GR, Kremer JM, Van den Bosch F, Kivitz A, Bessette L, Li Y, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2018;391(10139):2503–12. https://doi.org/10.1016/S0140-6736(18)31115-2.
Genovese MC, Fleischmann R, Combe B, Hall S, Rubbert-Roth A, Zhang Y, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet. 2018;391(10139):2513–24. https://doi.org/10.1016/S0140-6736(18)31116-4.
Van Vollenhoven R, Pangan AL, Friedman A, Mohamed MF, Chen S, Rischmueller M, et al. A phase 3, randomized, controlled trial comparing upadacitinib monotherapy to MTX monotherapy in MTX-naïve patients with active rheumatoid arthritis [abstract no. 891]. Arthritis Rheumatol. 2018;70(suppl 10).
Sandborn W, Ghosh S, Panes J, et al. Efficacy and safety of upadacitinib as an induction therapy for patients with moderately-to-severely active ulcerative colitis: data from the phase 2b study u-achieve [oral presentation no. 195]. United European Gastroenterology Week; 20–24 Oct 2018; Vienna.
Mohamed MF, Trueman S, Othman AA, Han JH, Ju TR, Marroum P. Development of in vitro-in vivo correlation for upadacitinib extended-release tablet formulation. AAPS J. 2019;21(6):108.
AbbVie Inc. RINVOQ™ (upadacitinib extended-release tablets) [US package insert]. North Chicago: AbbVie Inc.; 2019.
AbbVie Inc. A study to evaluate the pharmacokinetics, safety, tolerability of upadacitinib in pediatric subjects with polyarticular course juvenile idiopathic arthritis [ClinicalTrials.gov identifier NCT03725007]. National Institutes of Health, ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/nct03725007. Accessed 4 Nov 2019.
Mohamed MF, Zeng J, Marroum PJ, Song IH, Othman AA. Pharmacokinetics of upadacitinib with the clinical regimens of the extended-release formulation utilized in rheumatoid arthritis phase 3 trials. Clin Pharmacol Drug Dev. 2019;8(2):208–16. https://doi.org/10.1002/cpdd.462.
Klunder B, Mohamed MF, Othman AA. Population pharmacokinetics of upadacitinib in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I and II clinical trials. Clin Pharmacokinet. 2018;57(8):977–88. https://doi.org/10.1007/s40262-017-0605-6.
Klunder B, Mittapalli RK, Mohamed MF, Friedel A, Noertersheuser P, Othman AA. Population pharmacokinetics of upadacitinib using the immediate-release and extended-release formulations in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I-III clinical trials. Clin Pharmacokinet. 2019;58(8):1045–58. https://doi.org/10.1007/s40262-019-00739-3.
Mohamed MF, Camp HS, Jungerwirth S, Othman AA. Pharmacokinetics of upadacitinib in healthy Japanese and Chinese subjects and comparability to western subjects [abstract]. Clin Pharmacol Ther. 2018;103(S1):S80.
Mohamed MF, Klunder B, Meerwein S, Othman A. Upadacitinib pharmacokinetics in Japanese subjects with rheumatoid arthritis with the extended release formulation and comparability to non-Japanese subjects [poster no. THU0183]. Ann Rheum Dis. 2019;78:367.
Trueman S, Mohamed MF, Feng T, Lacerda AP, Marbury T, Othman AA. Characterization of the effect of hepatic impairment on upadacitinib pharmacokinetics. J Clin Pharmacol. 2019;59(9):1188–94. https://doi.org/10.1002/jcph.1414.
Mohamed MF, Trueman S, Feng T, Anderson J, Marbury TC, Othman AA. Characterization of the effect of renal impairment on upadacitinib pharmacokinetics. J Clin Pharmacol. 2019;59(6):856–62. https://doi.org/10.1002/jcph.1375.
Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130(6):461–70. https://doi.org/10.7326/0003-4819-130-6-199903160-00002.
Mohamed MF, Jungerwirth S, Asatryan A, Jiang P, Othman AA. Assessment of effect of CYP3A inhibition, CYP induction, OATP1B inhibition, and high-fat meal on pharmacokinetics of the JAK1 inhibitor upadacitinib. Br J Clin Pharmacol. 2017;83(10):2242–8. https://doi.org/10.1111/bcp.13329.
Mohamed MF, Coppola S, Feng T, Camp H, Othman AA. Lack of clinically- relevant effect of upadacitinib on plasma exposures of rosuvastatin and atorvastatin. Clin Pharmacol Drug Dev. 2019; 8(S1)
U.S. Department of Health and Human Services. Guidance for industry: Clinical drug interaction studies—study design, data analysis, and clinical implications. October 2017. https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf. Accessed 6 Nov 2019
Mohamed MF, Feng T, Enejosa JV, Fisniku O, Othman AA. Effects of upadacitinib coadministration on the pharmacokinetics of sensitive cytochrome P450 probe substrates: a study with the Modified Cooperstown 5 + 1 Cocktail. J Clin Pharmacol. 2019. https://doi.org/10.1002/jcph.1496(epub 2019 Aug 5).
EMA. Guideline on the investigation of drug interactions. London: European Medicines Agency (EMA); 2012. p. 1–59.
Minocha M, Trueman S, Mohamed M-EF, Feng T, Enejosa J, Othman AA. Chronic dosing of upadacitinib, an oral selective JAK-1 inhibitor, does not impact exposures of bupropion, a probe substrate for cytochrome P450 2B6 metabolism. Clin Pharmacol Ther. 2019;105(Suppl 1):S63.
Zhang H, Cui D, Wang B, Han YH, Balimane P, Yang Z, et al. Pharmacokinetic drug interactions involving 17alpha-ethinylestradiol: a new look at an old drug. Clin Pharmacokinet. 2007;46(2):133–57. https://doi.org/10.2165/00003088-200746020-00003.
Mohamed MF, Trueman S, Feng T, Friedman A, Othman AA. The JAK1 inhibitor upadacitinib has no effect on the pharmacokinetics of levonorgestrel and ethinylestradiol: a study in healthy female subjects. J Clin Pharmacol. 2019;59(4):510–6. https://doi.org/10.1002/jcph.1350.
Acknowledgements
The authors thank Mia DeFino, MS, ELS, a freelance medical writer under contract with AbbVie, for medical writing support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The studies summarized in this review were supported by AbbVie Inc. AbbVie contributed to the study designs, research, and interpretation of data, and the writing, review, and approval of this review article.
Conflict of interest
Mohamed-Eslam F. Mohamed and Ben Klüender are employees of AbbVie and may hold AbbVie stock or stock options. Ahmed A. Othman is a former employee of AbbVie and may hold AbbVie stocks or stock options.
Additional information
AbbVie was Dr. Othman’s affiliation at time of conducting this research.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mohamed, ME.F., Klünder, B. & Othman, A.A. Clinical Pharmacokinetics of Upadacitinib: Review of Data Relevant to the Rheumatoid Arthritis Indication. Clin Pharmacokinet 59, 531–544 (2020). https://doi.org/10.1007/s40262-019-00855-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00855-0