
Abstract
Introduction
As patients who receive cannabidiol (CBD) may have co-existing renal morbidities, it is important to understand whether dose adjustments are necessary to mitigate the risk of exposure-related toxicity. This study was conducted to evaluate the pharmacokinetics, safety, and tolerability of CBD in patients with renal impairment.
Methods
The pharmacokinetics and safety of a single oral 200 mg dose of a plant-derived pharmaceutical formulation of highly purified CBD in oral solution (Epidiolex® in the USA; 100 mg/mL) were assessed in subjects with mild, moderate, or severe renal impairment (n = 8/group) relative to matched subjects with normal renal function (n = 8). Blood samples were collected until 48 h post-dose and evaluated by liquid chromatography with tandem mass spectrometry. Analysis of variance was used to compare primary pharmacokinetic parameters (maximum measured plasma concentration [Cmax], oral clearance of drug from plasma [CL/F], renal clearance [CLR], area under the plasma concentration–time curve [AUC] from time zero to last measurable concentration [AUCt], and AUC from time zero to infinity [AUC∞]); descriptive analysis was used for secondary pharmacokinetic parameters (time to Cmax [tmax], terminal [elimination] half-life [t½], cumulative amount excreted from time zero to the last quantifiable sample [Aelast], and fraction of the systemically available drug excreted into the urine [fe]).
Results
No statistically significant differences were observed in Cmax, AUCt, AUC∞, or tmax values between subjects with mild, moderate, or severe renal impairment and subjects with normal renal function for CBD or its major metabolites, 7-carboxy-CBD (7-COOH-CBD) and 7-hydroxy-CBD (7-OH-CBD), and minor metabolite, 6-hydroxy-CBD (6-OH-CBD); geometric mean ratio for Cmax values ranged from 0.68 to 1.35. No differences were observed for other secondary parameters (Aelast and fe). CBD, 7-COOH-CBD, 7-OH-CBD, and 6-OH-CBD were highly protein bound (> 90%); binding was similar in all subject groups. Urine analysis for CBD recorded no appreciable amount, and thus no urinary pharmacokinetic parameters could be derived. Adverse events (AEs) affected two subjects; all five AEs were mild in severity and resolved during the trial. There were no serious AEs or discontinuations due to AEs. Laboratory, physical examination, vital sign, and 12-lead electrocardiogram findings were not clinically significant.
Conclusion
Renal impairment had no effect on the metabolism of CBD after a single oral 200 mg dose. CBD was generally well tolerated in subjects with varying degrees of renal function.
Registration
European Union Clinical Trials Register (EudraCT) no. 2015-002122-39.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Following a single oral 200 mg dose of cannabidiol (CBD), renal impairment status was found to have no effect on CBD or its biotransformation products. |
No statistically significant differences were observed in maximum measured plasma concentration (Cmax), area under the plasma concentration–time curve (AUC) from time zero to last measurable concentration (AUCt), AUC from time zero to infinity (AUC∞), or time to Cmax (tmax) values between subjects with renal impairment and subjects with normal renal function. |
A single dose of 200 mg CBD was well tolerated across all subject groups, and no safety concerns were observed. |
1 Introduction
Highly purified cannabidiol (CBD; Epidiolex®) is approved by the Food and Drug Administration (FDA) in the USA for seizures associated with Lennox–Gastaut syndrome (LGS) or Dravet syndrome (DS) in patients ≥ 2 years of age [1,2,3,4]. CBD likely exerts a cumulative anticonvulsant effect via several endogenous systems, including, but not limited to, modulation of intracellular calcium (Ca2+) and neuronal excitability [5]. Results from non-clinical studies and the scientific literature support at least three targets underlying the mechanism of anticonvulsant action of CBD: modulation of intracellular Ca2+ by antagonism of G protein–coupled receptor 55 (GPR55) and desensitization of transient receptor potential vanilloid type 1 (TRPV1) channels, and inhibition of adenosine reuptake via inhibition of the equilibrative nucleoside transporter 1 (ENT1) [5,6,7,8]. Importantly, and unlike Δ9-tetrahydrocannabinol (THC), CBD is unlikely to cause euphoric effects associated with propensity for abuse, due to its lack of orthosteric engagement of the cannabinoid receptor type 1 (CB1) at physiologically achievable concentrations [9].
CBD is highly lipophilic and is extensively metabolized by the liver to form monohydroxylated metabolites. The major circulating metabolite in vivo in humans is 7-carboxy-CBD (7-COOH-CBD), followed by parent CBD, 7-hydroxy-CBD (7-OH-CBD; an active metabolite), and 6-hydroxy-CBD (6-OH-CBD; a relatively minor metabolite) [10]. CBD is also subject to a large first-pass effect. Wall et al. [11] found that at 72 h after dosing, approximately 33% of CBD and its metabolites were excreted in the feces (of which a large amount was intact) and 16% were excreted in the urine. It is possible that the patients who receive CBD will have co-existing morbidities, which can lead to drug accumulation. There are no previously published studies investigating the pharmacokinetics of GW Research Ltd’s formulation of CBD in renally impaired subjects. This trial aimed to assess the effect of renal impairment on systemic exposure to a single dose of CBD.
2 Methods
2.1 Trial Design
All relevant trial-related documents were reviewed by independent ethics committees, and approval for the trial was granted on 11 September 2015. All subjects provided written informed consent for participation in the trial, which was performed in full conformity with the current Declaration of Helsinki [12], the International Council for Harmonisation guidelines for Good Clinical Practice [13], and all other applicable regulations. The trial was performed between 14 September 2015 and 10 February 2016 at three Pharmaceutical Research Associates (PRA) sites specializing in clinical pharmacology trials (one each in Hungary, the Czech Republic, and Slovakia). The trial was performed considering the FDA [14] and European Medicines Agency (EMA) [15] recommendations for the evaluation of pharmacokinetics in subjects with impaired renal function.
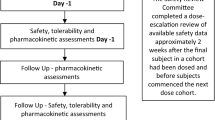
The trial consisted of a screening period (Days − 28 to − 2), a treatment period (hospitalization from Day − 1 until Day 3), and a follow-up visit (Day 14 [± 2 days]). Renal function was assessed by the estimated creatinine clearance (CLcr) using the Cockcroft–Gault equation at screening.
During the in-house treatment period, baseline assessments were performed on Day − 1 (after an overnight fast of at least 8 h). On the morning of Day 1, subjects received a standardized low-protein breakfast 2 h prior to dosing with a single oral 200 mg dose of a pharmaceutical formulation of highly purified CBD derived from Cannabis sativa L. plant in oral solution (100 mg/mL; Epidiolex® in the USA; GW Research Ltd, Cambridge, UK). Results from this study cannot be extrapolated to other CBD-containing products.
Observations were made until release following 48-h post-dose assessments on Day 3. A follow-up visit was performed on Day 14 (± 2 days). Fluid intake except water was prohibited during fasting (from 2 h pre-dose to 4 h post-dose).
2.2 Inclusion and Exclusion Criteria
2.2.1 Trial Population
The inclusion criteria specified that the trial population should consist of male and female subjects (age 18–75 years; body mass index [BMI] 18–35 kg/m2) with mild, moderate, or severe renal impairment, as defined by estimated CLcr, and subjects with normal renal function (matched to renally impaired subjects with respect to age and BMI).
The renal function categories were classified as follows:
Group 1: mild renal impairment (CLcr 50–80 mL/min).
Group 2: moderate renal impairment (CLcr 30 to < 50 mL/min).
Group 3: severe renal impairment (CLcr < 30 mL/min).Footnote 1
Group 4: normal renal function (CLcr > 80 mL/min).
Female subjects were non-pregnant and non-lactating at screening. Male and female subjects agreed to use effective contraception for the duration of the trial and for 3 months and 30 days thereafter, respectively.
Subjects with impaired renal function were included only if deemed to have stable disease status and no history of kidney transplant. In addition, serum albumin concentrations must not have been < 25 g/L and hemoglobin concentrations not < 95 g/L (< 100 g/L in the Czech Republic) at screening and baseline.
2.3 Trial Assessments
2.3.1 Pharmacokinetic Assessments
At the times specified later in this section, 6 mL blood samples were taken from subjects via an indwelling intravenous catheter or direct venepuncture into lithium heparin vacutainers; blood samples were then centrifuged for 10 min at 2600g at 18 °C. The resultant plasma was stored upright in a freezer at − 70 °C.
Validated liquid chromatographic–tandem mass spectrometric bioanalytical methods were used to quantify plasma concentrations of CBD, 6-OH-CBD, 7-OH-CBD, and 7-COOH-CBD. For analysis of CBD and metabolites, samples were stored for a maximum of 227 days at − 80 °C, and stability in plasma was determined for at least 309 days for all analytes. As THC is present as a trace impurity (≤ 0.1% w/w active pharmaceutical ingredient) in the CBD used in this formulation, plasma concentrations of THC and its metabolites 11-hydroxy-Δ9-tetrahydrocannabinol (11-OH-THC) and 11-nor-9-carboxy-Δ9-tetrahydrocannabinol (11-COOH-THC) were also determined.
For analysis of THC and metabolites, samples were stored for a maximum of 244 days at − 80 °C, and stability in plasma was determined for at least 356 days for all analytes.
Blood samples for pharmacokinetic analysis were taken at the following time points:
Pre-dose, then at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 7, 8, 9, 10, 12, 24, 36, and 48 h post-dose.
Urine samples for pharmacokinetic analysis were collected pre-dose and then at intervals of 0–4, 4–8, 8–12, 12–24, and 24–48 h post-dose.
Pharmacokinetic parameters were determined by non-compartmental analysis using Phoenix® WinNonlin® (Pharsight Inc., Princeton, NJ, USA) version 6.3. Pharmacokinetic parameters evaluated included maximum measured plasma concentration (Cmax), area under the plasma concentration–time curve (AUC) from time zero to infinity (AUC∞), AUC from time zero to last measurable concentration (AUCt), terminal (elimination) half-life (t½), time to Cmax (tmax), cumulative amount excreted from time zero to the last quantifiable sample (Aelast), fraction of the systemically available drug excreted into the urine (fe), oral clearance of drug from plasma (CL/F), renal clearance (CLR), and percentage of estimated extrapolated part for the calculation of AUC∞ ([AUC∞ − AUCt]/AUC∞) × 100 (%AUCextra).
2.3.2 Chromatographic Conditions and Specifications
High-performance liquid chromatography (HPLC) with tandem mass spectrometry was performed on a Waters Acquity (Waters Corporation, Milford, MA, USA) HPLC system. Chromatographic separations were performed on an Acquity BEH Phenyl Column (internal diameter 1.7 μm, 2.1 × 100 mm).
The selectivity of the HPLC method was checked by comparing chromatograms from blank plasma with the corresponding spiked peaks. Each blank plasma sample was tested to ensure that there were no significant interfering peaks with any analyte under investigation. The assay ranges were between 2.00 and 10,000 ng/mL for CBD, 0.250 and 250 ng/mL for 6-OH-CBD, 0.250 and 1250 ng/mL for 7-OH-CBD, and 0.250 and 20,000 ng/mL for 7-COOH-CBD. Calibration standards were between 0.125 and 62.5 ng/mL for THC and 0.250 and 125 ng/mL for 11-OH-THC and 11-COOH-THC. All analyte calibration curves were created using weighted least-squares linear/quadratic regression. There were no significant interfering peaks observed at the retention times for any of the analytes, indicating adequate selectivity of the methods.
The precision (coefficient of variation [CV]) and accuracy (relative error [RE]/mean % different [Bias]) of the HPLC method were determined by analysis of the plasma samples and were acceptable for all analytes (≤ 15% [20% at the lower limit of quantification (LLOQ)]). The recovery of CBD, 6-OH-CBD, 7-OH-CBD, and 7-COOH-CBD from human plasma was between 92 and 100% at three concentrations tested (low, medium, and high) and was considered acceptable. The recovery of THC, 11-OH-THC, and 11-COOH-THC from human plasma was between 56.3% and 124% at three concentrations tested (low, medium, and high). Although the recovery for THC and its metabolites varied between quality control (QC) levels, this did not affect the linearity of the assay and therefore was not considered to impact the validity of the data.
Most analytes passed an assessment of matrix effect in population plasma for renally impaired subjects; however, 11-COOH-THC failed this assessment at the low QC level (suggesting the method did not compensate appropriately for matrix effects). As such, 11-COOH-THC data in subjects with renal impairment should be regarded with a degree of caution.
Protein binding was determined by extracting and analyzing pre-dose plasma that had been spiked with CBD, 6-OH-CBD, 7-OH-CBD, 7-COOH-CBD, THC, 11-OH-THC, and 11-COOH-THC and equilibrated for 20–24 h at 37 °C to provide a concentration of total analyte content: protein bound + unbound analyte. A supernatant fraction (ultracentrifuged at 45,000 rpm for 20 h at 37 °C) from aliquots of spiked plasma was extracted and analyzed for each subject to provide a concentration of unbound fraction of each analyte.
The bioanalytical methods used in this trial were validated according to guidelines from the EMA [16] and FDA [17]; these methods are further described in the Electronic Supplementary Material.
Various bioanalytical limitations were observed during the protein-binding validation process. During validation of the method for separation of unbound analytes by ultracentrifugation, potential bias due to non-specific binding and analyte instability was observed; as such, total CBD (bound and unbound) and metabolite pharmacokinetic data were used for the primary assessment and are presented in this article.
2.3.3 Safety Assessments
The safety and tolerability of CBD were evaluated by recording the incidence of adverse events (AEs) throughout the trial, clinical laboratory tests, vital signs, 12-lead electrocardiography (ECG), and physical examinations.
2.4 Statistical Analysis
The primary objective of the trial was to assess the effects of a single dose of CBD on the pharmacokinetic parameters of CBD and its major metabolites in subjects with impaired renal function compared with subjects with normal renal function. Secondary objectives were to assess the safety and tolerability of CBD in the same population. Descriptive statistics of subject demographics and safety outcomes were based on the safety analysis set (all subjects who received CBD).
The pharmacokinetic parameters of CBD, THC, and their metabolites were calculated for the pharmacokinetic analysis set (all subjects who received CBD and had evaluable pharmacokinetic data) using Phoenix® WinNonlin® version 6.3. Pharmacokinetic parameters for all analytes with sufficient data above LLOQ were estimated from the concentration–time profiles for individuals in the pharmacokinetic analysis set. At least three data points (not including Cmax) were required to calculate kel (elimination rate constant [from the central compartment]), and percentage extrapolation of ≤ 30% was required to retain AUC∞ and t½; subjects who did not satisfy this criterion were excluded from the analysis. Analysis of variance (ANOVA) was used to compare primary pharmacokinetic parameters (Cmax, CL/F, CLR, AUC∞, and AUCt) between the control group of healthy subjects and each of the groups with renal impairment. Pharmacokinetic values were log-transformed prior to analysis. Covariates included sex, age, and BMI, if significant. Geometric least-squares means were used to calculate the ratios of primary pharmacokinetic parameters in each renal impairment group to those in the control group, together with 90% confidence intervals (CIs). A Wilcoxon rank-sum test with Hodges–Lehmann estimator was used for comparison of the tmax values between the control and each renal impairment group. Estimates of the median differences between groups were determined along with 90% CIs.
The relationship between log-transformed primary pharmacokinetic parameters and estimated CLcr at screening was explored by a linear regression approach that included sex, age, and BMI, if significant. Secondary pharmacokinetic parameters and safety data were analyzed descriptively.
2.4.1 Sample Size
The planned sample size of eight participants per group was based on practical considerations and guidance from the FDA [14] and EMA [15].
3 Results
3.1 Subject Demographics
A total of 32 subjects were enrolled into one of four subject groups (mild [n = 8], moderate [n = 8], or severe [n = 8] renal impairment or normal renal function [n = 8]). All 32 subjects completed the trial without any major protocol deviations and were included in the safety and pharmacokinetic analysis sets.
All subjects enrolled were white. The ratio of male to female subjects was 5:3 in the mild and moderate renal impairment groups and 3:5 in the severe renal impairment and normal renal function groups. Mean age across the groups ranged from 58.8 to 64.6 years, and mean BMI ranged from 27.6 to 29.9 kg/m2 (Table 1).
3.2 Concomitant Medication
Twenty-six (81.3%) subjects took at least one concomitant medication during the trial. The most common concomitant medication classes reported were blood pressure-regulating agents (β-blockers, xanthine oxidase inhibitors, calcium channel blockers), thyroid hormones, and diuretics. None were considered to impact the safety or interpretation of the trial data.
3.3 Pharmacokinetics
Systemic exposure to CBD and its metabolites was unaffected by renal impairment (any severity; Fig. 1).

Geometric mean plasma concentration–time profiles for a cannabidiol (CBD), b 6-hydroxy-cannabidiol (6-OH-CBD), c 7-hydroxy-cannabidiol (7-OH-CBD), and d 7-carboxy-cannabidiol (7-COOH-CBD) after a single oral 200 mg CBD dose, by renal function group (semi-logarithmic) (pharmacokinetics analysis set)
For CBD and its metabolites, there were no significant differences in Cmax values between subjects with normal renal function and subjects with renal impairment; geometric mean ratios ranged from 0.68 to 1.35 (Table 2). AUCt and AUC∞ (not calculable for 7-COOH-CBD due to a long t½) were unaffected by renal impairment when subjects with normal renal function were compared with subjects with renal impairment; geometric mean ratios ranged from 0.95 to 1.74 for AUCt and from 1.05 to 2.01 for AUC∞, with no systematic trend to any decrease in CL/F with increasing severity of renal impairment (Table 2).
Regression analysis showed no apparent relationship between the log-transformed primary pharmacokinetic parameters for CBD (Cmax, AUCt, and AUC∞) and estimated CLcr (at screening) in all subject groups (p values > 0.05 [range 0.1–1]).
The tmax values for CBD and the metabolites 6-OH-CBD and 7-OH-CBD were independent of renal impairment status, with geometric mean plasma tmax reached between 2 and 3 h post-dose for all subject groups. The tmax for 7-COOH-CBD appeared slightly later but was also independent of renal impairment status, with geometric mean plasma tmax reached between approximately 3 and 4 h (range 2–9 h) post-dose for all subject groups. Statistical analysis showed that renal impairment had no effect on tmax values for CBD or any of its metabolites (Table 2). 7-COOH-CBD was the most abundant circulating product in plasma, followed by CBD, 7-OH-CBD, and then 6-OH-CBD (Fig. 1).
The t½ values for CBD, 6-OH-CBD, and 7-OH-CBD ranged from 11 to 22 h in all subject groups. Although statistical analyses were not performed for t½ values, there were no clear trends between subjects with normal renal function and subjects with renal impairment (Table 3). For 7-COOH-CBD, t½ was longer than the 48-h sampling time for all renal function groups. As such, t½ values for 7-COOH-CBD were not calculated (Table 3).
For CBD, CL/F ranged from 351 to 510 L/h and the apparent volume of distribution (Vz/F) ranged from 5800 to 7778 L. No statistically significant differences in CL/F were observed between renal impairment groups (mild to severe) and subjects with normal renal function; geometric mean ratios ranged from 0.83 to 0.96 and all 90% CIs of ratios contained 1 (Table 2).
Urine analysis for CBD recorded no appreciable amount of CBD. Concentrations were below the LLOQ (< 0.125 ng/mL) for most subjects at most time points and thus the primary parameter CLR and the secondary parameters Aelast and fe were not reported.
THC and its major metabolites were either not detected or detected only in trace concentrations (≤ 1.93 ng/mL) in plasma and were independent of renal impairment status.
3.4 Plasma Protein Binding of Cannabidiol (CBD)
There was no trend observed between the degree of renal impairment and plasma protein binding. CBD and its major metabolites were all highly bound to plasma proteins. The extent of protein binding ranged from 86.7 to 92.2% bound for CBD and from 96.8 to 99.0% bound for its major metabolites. Data could only be considered qualitative due to bioanalytical issues (the process and stability of the free fraction was not supported during bioanalytical method validation). The results from the free unbound data were consistent with the plasma data; however, only total drug (bound and unbound) pharmacokinetic parameters are presented in this article, as they can be supported by validation.
3.5 Safety
A single oral 200 mg dose of CBD was well tolerated in all subject groups; all AEs were mild in severity and resolved during the trial. There were no serious AEs, deaths, AEs of special interest, pregnancies, or early withdrawals due to AEs.
There were only five AEs reported throughout the trial, and no AEs were reported in the moderate or severe renal impairment groups. One subject in the mild renal impairment group reported three AEs: one case each of visual disturbance, nausea, and drowsiness. One subject in the normal renal function group reported two AEs: one case each of back pain and pain in hip.
There were no clinically significant changes for any laboratory parameter, and no laboratory abnormalities were considered AEs. There were no clinically significant physical examination, vital sign, or ECG findings.
4 Discussion
This trial is the first to investigate the pharmacokinetics of this oral formulation of CBD in subjects with renal impairment. As patients who receive CBD may have co-existing renal morbidities, it is important to understand whether dose adjustments are necessary to mitigate the risk of exposure-related toxicity [14, 15].
4.1 CBD and Metabolite Pharmacokinetics
Following a single oral 200 mg dose of CBD, renal impairment status was found to have no effect on CBD or its biotransformation products. No statistically significant differences were observed in Cmax, AUCt, AUC∞, or tmax values between subjects with renal impairment and subjects with normal renal function. CBD and its major metabolites were not detected (below LLOQ of assay) in urine, and thus this likely represents a minor route of elimination of intact drug. No differences were observed for any other secondary parameters.
Exposure to 7-COOH-CBD was much greater than that to the parent drug; however, in contrast to CBD, exposure to 7-COOH-CBD was lowest in subjects with severe renal impairment (compared with the other impairment groups and the normal renal function group). A possible explanation for this is the potential for a different biotransformation pathway for 7-COOH-CBD in severely renally impaired subjects [18].
4.2 Safety
A single dose of 200 mg CBD was well tolerated across all subject groups, and no safety concerns were observed. Only five mild AEs were reported by two subjects during the trial: one in the mild renal impairment group and one in the normal renal function group. There was no increase in AE frequency or severity with increasing degree of renal impairment. There were no moderate or severe AEs, deaths, serious AEs, or any other significant events. There were no clinically significant safety findings for laboratory parameters, physical examinations, vital signs, ECG, or body weight.
4.3 Trial Limitations
This trial was conducted prior to the availability of data from the multiple-dose pharmacokinetic trials that defined the t½ for CBD. Subsequent trials, now that pharmacokinetic data are available, have been designed to evaluate pharmacokinetic effects over longer time frames. Although data are limited to 48 h post-dose, the pharmacokinetic results during the first 48 h are consistent with pharmacokinetic results reported in other single-dose studies with CBD [10].
For 7-COOH-CBD, the t½ was longer than the 48-h sampling time for all renal function groups. As such, t½ values for 7-COOH-CBD were not calculated. However, Cmax and AUClast were the primary parameters for the evaluation of renal insufficiency and had no impact on the study results or conclusions. In addition, exposure data from plasma total drug (bound and unbound) pharmacokinetic parameters are presented here, as they can be supported by validation. It should be noted that although data generated for the unbound (free) drug during this study were only considered qualitative, the results were consistent with the total plasma data.
Results from this trial suggest that intact CBD parent drug is not significantly cleared by renal elimination; however, this trial was not designed to evaluate conjugated metabolites in the urine and therefore does not rule out the possibility that CBD metabolites are cleared by conjugation. CBD has not been tested in patients with end-stage renal disease, and it is not known if CBD and its metabolites are dialyzable.
Validation studies have been conducted for potential interference with concomitant medications commonly used to treat the approved indications for CBD; however, because this trial evaluated a special population, renal impairment, it was not possible to conduct validation studies for all possible concomitant medications within this rare subgroup.
5 Conclusion
Renal impairment status had no effect on CBD pharmacokinetics following a single oral 200 mg dose, with no statistically significant effects on Cmax, AUCt, AUC∞, or tmax. CBD was generally well tolerated; there were no serious or severe AEs, and no new safety concerns were identified.
Notes
Subjects requiring dialysis were not enrolled in this trial.
References
Devinsky O, Cross JH, Laux L, Cannabidiol in Dravet Syndrome Study Group, et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376(21):2011–20.
Devinsky O, Patel AD, Thiele EA, et al. Randomized, dose-ranging safety trial of cannabidiol in Dravet syndrome. Neurology. 2018;90(14):e1204–11.
Devinsky O, Patel AD, Cross JH, et al. Effect of cannabidiol on drop seizures in the Lennox–Gastaut syndrome. N Engl J Med. 2018;378(20):1888–97.
Thiele EA, Marsh ED, French JA, et al. Cannabidiol in patients with seizures associated with Lennox–Gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2018;391(10125):1085–96.
Ibeas Bih C, Chen T, Nunn AV, Bazelot M, Dallas M, Whalley BJ. Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics. 2015;12(4):699–730.
Iannotti FA, Hill CL, Leo A, et al. Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: potential for the treatment of neuronal hyperexcitability. ACS Chem Neurosci. 2014;5(11):1131–41.
Yang H, Zhou J, Lehmann C. GPR55—a putative “type 3” cannabinoid receptor in inflammation. J Basic Clin Physiol Pharmacol. 2016;27(3):297–302.
Carrier EJ, Auchampach JA, Hillard CJ. Inhibition of an equilibrative nucleoside transporter by cannabidiol: a mechanism of cannabinoid immunosuppression. Proc Natl Acad Sci USA. 2006;103(20):7895–900.
Schoedel KA, Szeto I, Setnik B, et al. Abuse potential assessment of cannabidiol (CBD) in recreational polydrug users: a randomized, double-blind, controlled trial. Epilepsy Behav. 2018;88:162–71.
Taylor L, Gidal B, Blakey G, Tayo B, Morrison G. A phase I, randomized, double-blind, placebo-controlled, single ascending dose, multiple dose, and food effect trial of the safety, tolerability and pharmacokinetics of highly purified cannabidiol in healthy subjects. CNS Drugs. 2018;32(11):1053–67.
Wall ME, Brine DR, Perez-Reyes M. Metabolism of cannabinoids in man. In: Braude MC, Szara S, editors. The pharmacology of marihuana. New York: Raven Press; 1976. p. 93–113.
World Medical Association Declaration of Helsinki. Ethical principles for medical research involving human subjects. Edinburgh: World Medical Association; 2000 Oct, including the footnote of October 2002.
ICH harmonised tripartite guideline: guideline for good clinical practice E6(R1). Geneva: International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; 1996 Jun.
US Food and Drug Administration (FDA). Guidance for industry: pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling. 1998. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072127.pdf. Accessed 31 Aug 2018.
Committee for Medicinal Products for Human Use (CHMP). Note for guidance on the evaluation of the pharmacokinetics of medicinal products in patients with impaired renal function. 2004. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003123.pdf. Accessed 31 Aug 2018.
European Medicines Agency. Guideline on bioanalytical method validation. 2011 Jul 21. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf. Accessed 30 Aug 2019.
US Department of Health and Human Services, Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM). Guidance for industry, bioanalytical method validation. Rockville: US Department of Health and Human Services, FDA, CDER; 2001.
Tarantino G, Di Minno MN, Capone D. Drug-induced liver injury: is it somehow foreseeable? World J Gastroenterol. 2009;15(23):2817–33.
Acknowledgements
The authors would like to thank the volunteers who took part in the trial, as well as the staff that assisted with the trial at each site. The authors would also like to acknowledge and thank Nateisha Williams, BS, employee of Greenwich Biosciences, Inc., for providing medical writing support, and Laura Riordan, MS, for providing editorial support funded by Greenwich Biosciences, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This trial was sponsored by GW Research Ltd.
Conflict of interest
Bola Tayo, MD, and Gilmour Morrison, HND, are employees of GW Research Ltd and own shares in GW Pharmaceuticals Plc. Lesley Taylor, PhD, was employed at GW Research Ltd at the time this work was completed. Farhad Sahebkar, MD, is an employee of Greenwich Biosciences, Inc.
Data Availability Statement
The sponsor is adhering to current US and EU requirements so will not make individual de-identified participant data available; however, the protocol and statistical analysis plan will be made available upon request to the corresponding author.
Ethical Standards
All procedures performed in studies involving human participants were in accordance with the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. For information on local ethics committees involved in conducting the trial, refer to EudraCT no. 2015-002122-39.
Informed Consent
Informed consent was obtained from all individual participants included in the studies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tayo, B., Taylor, L., Sahebkar, F. et al. A Phase I, Open-Label, Parallel-Group, Single-Dose Trial of the Pharmacokinetics, Safety, and Tolerability of Cannabidiol in Subjects with Mild to Severe Renal Impairment. Clin Pharmacokinet 59, 747–755 (2020). https://doi.org/10.1007/s40262-019-00841-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00841-6