
Abstract
Background
Oral insulin 338 is a novel tablet formulation of a long-acting basal insulin. This randomised, open-label, four-period crossover trial investigated the effect of timing of food intake on the single-dose pharmacokinetic properties of oral insulin 338.
Methods
After an overnight fast, 44 healthy males received single fixed doses of oral insulin 338 administered 0, 30, 60 or 360 min before consuming a standardised meal (500 kcal, 57 energy percent [E%] carbohydrate, 13 E% fat, 30 E% protein). Blood samples for pharmacokinetic assessment were taken up to 288 h post-dose.
Results
Total exposure (area under the concentration-time curve from time zero to infinity [AUCIns338,0–∞]) and maximum concentration (Cmax,Ins338) of insulin 338 were both significantly lower for 0 versus 360 min post-dose fasting (ratio [95% confidence interval (CI)]: 0.36 [0.26–0.49], p < 0.001, and 0.35 [0.25–0.49], p < 0.001, respectively). There were no significant differences in AUCIns338,0–∞ and Cmax,Ins338 for 30 or 60 versus 360 min post-dose fasting (ratio [95% CI] 30 versus 360 min: 0.85 [0.61–1.21], p = 0.36, and 0.86 [0.59–1.26], p = 0.42; ratio [95% CI] 60 versus 360 min: 0.96 [0.72–1.28], p = 0.77, and 0.99 [0.75–1.31], p = 0.95). The mean half-life was ~ 55 h independent of the post-dose fasting period. Oral insulin 338 was well-tolerated with no safety issues identified during the trial.
Conclusions
Oral insulin 338 pharmacokinetics are not affected by food intake from 30 min after dosing, implying that patients with diabetes mellitus do not need to wait more than 30 min after a morning dose of oral insulin 338 before having their breakfast. This is considered important for convenience and treatment compliance.
ClinicalTrials.gov identifier
NCT02304627.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Oral insulin 338 is a long-acting, basal insulin formulated in a Gastro-Intestinal Permeation Enhancement Technology One (GIPET® I) tablet with the absorption enhancer sodium caprate. The effect of timing of food intake on the pharmacokinetics of oral insulin 338 was investigated in healthy males. |
Absorption of oral insulin 338 was reduced by ~ 65% when a meal was consumed immediately after dosing, while absorption of oral insulin 338 was comparable when the same meal was given 30, 60 or 360 min post-dose. Oral insulin 338 was well-tolerated in this trial. |
Patients with diabetes mellitus do not need to wait more than 30 min before eating their breakfast after a morning dose of oral insulin 338, which is considered relevant for patient convenience and treatment compliance. |
1 Introduction
Over nearly a century, several attempts have been made to develop insulin for oral administration to patients with diabetes mellitus. Potential advantages of oral insulin delivery, as compared to subcutaneous injection, include improved patient convenience and compliance, and a more physiological insulin profile through a higher portal-to-peripheral insulin gradient. The latter has the potential to reduce diabetes adverse effects such as hypoglycaemia, weight gain, neuropathy and retinopathy [1,2,3,4]. The main challenge to oral insulin delivery is that systemic absorption of orally administered protein-based drugs is hindered by their vulnerability to proteolytic degradation in the gastrointestinal tract and by their low intestinal permeability [5, 6].
Since oral insulin is absorbed relatively fast from the gastrointestinal tract, the rate of systemic appearance of orally administered insulin is high relative to subcutaneously administered insulin. This is probably the reason why oral insulin has always been considered a potential mealtime insulin [3]. However, specific considerations exist for an oral mealtime insulin, such as the variability of insulin exposure after oral administration and the potential effect of food on insulin absorption. These particular challenges may be less relevant for an oral basal insulin.
Oral insulin 338 is a novel tablet formulation of a long-acting basal insulin formulated in a Gastro-Intestinal Permeation Enhancement Technology One (GIPET® I) tablet with the absorption enhancer sodium caprate [7]. Insulin 338 has been modified compared to human insulin in order to make it more resistant to enzymatic degradation in the gastrointestinal tract, while acylation of insulin 338 causes reversible binding to albumin, resulting in a plasma half-life of up to 70 h at steady state [8]. Sodium caprate is approved by the US Food and Drug Administration (FDA) as a food additive for human consumption with no limit for daily intake set by the World Health Organization [9]. The absorption enhancement by sodium caprate is believed to occur via tight junction modulation and cell membrane fluidisation, thus acting on both paracellular and transcellular pathways [9].
As already mentioned, absorption of orally administered drugs may be affected by food ingestion [10]. Therefore, it is crucial to evaluate the effect of food on the pharmacokinetics of drugs intended to be administered via the oral route [11, 12]. Accordingly, the rationale for the present trial was to investigate the effect of timing of post-dose food intake on the single-dose pharmacokinetic properties of oral insulin 338 in healthy subjects.
2 Methods
2.1 Trial Design and Subjects
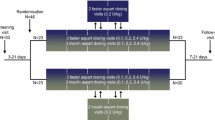
This was a randomised, single-centre (Profil, Neuss, Germany), open-label, four-period crossover trial (Electronic Supplementary Material Online Resource 1, Fig. S1). The trial protocol was reviewed and approved by the local health authorities (Bundesinstitut für Arzneimittel und Medizinprodukte, Bonn, Germany) and an independent ethics committee (Ethik-Kommission der Ärztekammer Nordrhein, Düsseldorf, Germany).
Eligible subjects were healthy men 18–64 years of age (both inclusive) with a body mass index (BMI) of 18.5–28.0 kg/m2 (both inclusive). Exclusion criteria included clinically significant concomitant diseases (including acute or chronic gastrointestinal symptoms), clinically significant abnormal values in clinical laboratory screening tests (including a fasting plasma glucose ≥ 5.6 mmol/L), previous clinically significant gastrointestinal surgery, smoking (defined as > 5 cigarettes or the equivalent per day), or use of prescription or non-prescription medication within 2 weeks prior to first dosing (occasional use of paracetamol and acetylsalicylic acid [aspirin] was permitted up to 48 h prior to dosing).
2.2 Trial Procedures and Assessments
The trial consisted of seven visits: an informed consent visit, a screening visit, four dosing visits separated by 7–15 days’ washout (from last pharmacokinetic sampling until next dosing) and a follow-up visit.
At each dosing visit, subjects attended the clinical site in the evening on the day prior to dosing. Subjects were asked not to smoke, perform strenuous physical exercise, take any prescription or non-prescription medication, including routine vitamins, or to consume alcohol, excessive amounts of coffee or tea (> 5 cups per day), other xanthine-containing beverages or chocolate during the last 48 h prior to dosing. Subjects received a standardised dinner and subsequently fasted overnight (from 22:00 h).
In the morning of the dosing day at approximately 08:00 h, subjects received a single fixed oral dose of insulin 338 (8100 nmol; Novo Nordisk, Bagsværd, Denmark) formulated in a film coated (Opadry® II Yellow) GIPET® I tablet with the absorption enhancer sodium caprate (550 mg). The tablet was administered with 100 mL still water. No other water consumption was allowed from 1 h before dosing until 1 h after dosing. Subjects were dosed in a semi-supine position and were not allowed to lie supine for the first 2 h post-dose except for trial procedures.
A test meal was served either at 0, 30, 60 or 360 min after dosing. The composition of the test meal was identical at all dosing visits. The post-dose test meal consisted of chicken with whole grain pasta and tomato sauce plus a dessert of apple–strawberry compote with cottage cheese. This mixed meal with fixed energy content (approximately 500 kcal) contained approximately 57 energy percent (E%) carbohydrate, 13 E% fat and 30 E% protein. For the 0 min post-dose fasting, ingestion of the post-dose test meal was initiated immediately after dosing. Independent of the post-dose fasting period, the entire meal had to be ingested within 15 min. The 0 and 360 min post-dose fasting periods corresponded to the fed and fasted conditions, respectively, and were hypothesised to represent the lower and upper limits of exposure. The 30 and 60 min post-dose fasting periods were included to evaluate two clinically feasible intervals in case of morning dose administration.
From dosing until 360 min post-dose, subjects were not allowed to eat any foods except for the standardised post-dose test meal. Subjects were allowed to drink up to 500 mL of water from 60 to 360 min post-dose. For the 0, 30 and 60 min post-dose fasting, subjects were served standardised meals at 360 min (identical to the post-dose test meal) and 12 h (standardised dinner). With respect to 360 min post-dose fasting, subjects were only served the post-dose test meal at 360 min and a standardised dinner at 12 h. Subjects stayed in-house at the clinical site until 48 h after dosing for safety observation and pharmacokinetic blood sampling. From 12 to 48 h after dosing, subjects were served meals (breakfast, lunch, dinner and snacks) at appropriate timepoints during the in-house stay and no other food intake was allowed. The subjects attended the clinical site for short visits at 72, 96, 120, 168, 216 and 288 h post-dose for pharmacokinetic blood sampling (Online Resource 1, Table S1).
Serum insulin 338 concentrations were measured by a validated insulin 338-specific luminescent oxygen channelling immunoassay (LOCI) with a lower limit of quantification (LLOQ) of 200 pmol/L. Serum capric acid concentrations were measured by a validated chromatographic assay with an LLOQ of 100 ng/mL.
Safety assessments included adverse events (AEs), hypoglycaemic episodes (classified according to the American Diabetes Association [13]), laboratory safety parameters, physical examination, vital signs and electrocardiogram.
2.3 Trial Endpoints
Insulin 338 pharmacokinetic endpoints were total exposure (area under the concentration–time curve [AUC] from time zero to infinity [AUCIns338,0–∞]; primary endpoint), maximum concentration (Cmax,Ins338), time from dosing until the first time that serum insulin 338 concentration was above LLOQ (onset of appearanceIns338), time to maximum concentration (tmax,Ins338) and terminal half-life (t½,Ins338).
AUCIns338,0–∞ was calculated as the sum of the AUC from dosing until the time of last quantifiable insulin 338 concentration (using the linear trapezoidal technique) and the AUC from the time of last quantifiable insulin 338 concentration derived by extrapolation until infinity based on the terminal slope. t½,Ins338 was calculated as log(2)/λz, where λz is the terminal rate constant determined by linear regression on the terminal part of the serum insulin 338 concentration–time profiles.
Capric acid pharmacokinetic endpoints were the exposure from 0 to 360 min (AUCCapric acid,0–360min; calculated by the linear trapezoidal technique), maximum concentration (Cmax,Capric acid) and time to maximum concentration (tmax,Capric acid).
2.4 Statistical Analyses
The sample size calculation was based on the precision of the ratio of AUCIns338,0–∞ between any two post-dose fasting periods. Based on a previous trial with oral insulin 338, it was assumed that the within-subject standard deviation for AUCIns338,0–∞ was 1.01 on a log scale. To obtain a probability of at least 80% that the 95% confidence interval (CI) for the true ratio of AUCIns338,0–∞ between two post-dose fasting periods ranged from 0.62 to 1.61 times the observed estimate of the ratio, a total of 40 evaluable pharmacokinetic profiles for each of the four post-dose fasting periods were needed. In order to account for dropouts, it was planned to randomise 45 subjects.
Statistical analyses were performed using SAS® version 9.4 (SAS Institute, Cary, NC, USA) at a 5% significance level based on all randomised subjects receiving at least one dose of trial product.
In order to compare the effect of post-dose fasting duration on the pharmacokinetics of insulin 338, AUCIns338,0–∞ and Cmax,Ins338 were analysed in a linear mixed model with the log-transformed endpoint as dependent variable, post-dose fasting period and trial period as fixed effects and subject as a random effect. The model also included residual variance depending on the post-dose fasting period. From the model, mean differences between post-dose fasting periods in the log-transformed endpoints were estimated and back-transformed to the original scale and presented as ratios together with corresponding two-sided 95% CIs. Specifically, the three shorter post-dose fasting periods were compared with 360 min of post-dose fasting, since 360 min of post-dose fasting was expected to result in the greatest insulin 338 absorption due to expected complete or near complete insulin 338 absorption within 360 min after oral dosing.
In order to estimate and compare the total pharmacokinetic variability between each of the four post-dose fasting periods, the total variability in AUCIns338,0–∞ and Cmax,Ins338 was estimated for each of the four post-dose fasting periods in the model described. Total variability consisted of both the within-subject day-to-day variability and the between-subject variability. It was tested by a likelihood ratio test whether the model with different variances for the four post-dose fasting periods could be reduced to a model with a common variance for all four post-dose fasting periods, suggesting no difference in variance among the four post-dose fasting periods.
All other pharmacokinetic endpoints for insulin 338 and capric acid as well as all safety endpoints were summarised by descriptive statistics based on all subjects receiving at least one dose of trial product.
3 Results
3.1 Subjects
Of 56 subjects screened, 45 were randomised, 44 were treated and 42 completed the trial. One subject withdrew consent before dosing at the first dosing visit, and one subject withdrew consent after completion of one trial period (60 min post-dose fasting). Furthermore, one subject was withdrawn due to an AE of increased blood creatine kinase-MB (CK-MB) with onset 12 days after dosing in the first trial period (30 min post-dose fasting), indicating non-compliance with the requirement to avoid strenuous physical exercise.
The mean (± standard deviation) age of the 44 exposed subjects was 34.7 (± 8.2) years. All subjects were white, mean body weight was 79.1 (± 8.8) kg and mean BMI was 24.2 (± 1.9) kg/m2.
3.2 Pharmacokinetics of Insulin 338
Insulin 338 pharmacokinetic mean profiles were comparable for post-dose fasting periods of 30, 60 and 360 min (Fig. 1 and Online Resource 1, Fig. S2) and there were no significant differences in AUCIns338,0–∞ and Cmax,Ins338 for 30 and 60 min post-dose fasting compared with 360 min post-dose fasting (Fig. 2). The 0 min post-dose fasting resulted in generally lower mean exposure of insulin 338 (Fig. 1 and Online Resource 1, Fig. S2), with a significant reduction of ~ 65% in AUCIns338,0–∞ and Cmax,Ins338 compared with 360 min post-dose fasting (Fig. 2).

Mean serum insulin 338 concentration–time profiles after oral administration and varying duration of post-dose fasting in healthy males. Error bars show standard error of the mean

Effect of post-dose fasting period on (a) AUCIns338,0–∞ and (b) Cmax,Ins338 after oral administration of insulin 338 in healthy males. Bars are estimated means and 95% CIs. Treatment comparisons show estimated treatment ratios [95% CI] and p value for the pairwise comparisons of 0, 30 and 60 min post-dose fasting with 360 min post-dose fasting. AUCIns338,0–∞ area under the concentration–time curve for insulin 338 from time zero to infinity, CI confidence interval, Cmax,Ins338 maximum concentration of insulin 338
The median onset of appearanceIns338 was 10 min independent of post-dose fasting period, while tmax,Ins338 appeared to be shorter with a median of 25 min for the 0 min post-dose fasting compared with a median of 40–60 min for the 30, 60 and 360 min post-dose fasting periods (Table 1). Harmonic mean t½,Ins338 ranged between 54.5 and 55.5 h for the four different post-dose fasting periods.
For AUCIns338,0–∞, the coefficient of variation for the total variability ranged from 82.4% to 131.5% for the four post-dose fasting periods (Online Resource 1, Table S2). The model with different variance for each post-dose fasting period could be reduced to a model with a common variance for the four post-dose fasting periods (p = 0.112), suggesting that the total variability in AUCIns338,0–∞ did not differ between post-dose fasting periods. For Cmax,Ins338, the coefficient of variation for the total variability was 122.2%, 163.2%, 90.0% and 84.5% for 0, 30, 60 and 360 min post-dose fasting, respectively (Online Resource 1, Table S2). The model with different variance for each post-dose fasting period could not be reduced (p = 0.006), suggesting that the total variability in Cmax,Ins338 differed between the four post-dose fasting periods.
3.3 Pharmacokinetics of Capric Acid
Capric acid pharmacokinetic profiles are shown for the four different post-dose fasting periods in Online Resource 1, Fig. S3. Compared to insulin 338, capric acid was rapidly eliminated. Mean serum capric acid concentrations were at the usual baseline level from ~ 2 h after dosing. The capric acid pharmacokinetic profile was slightly left-shifted for 0 min post-dose fasting compared with 30, 60 and 360 min post-dose fasting. Accordingly, Cmax,Capric acid appeared to be higher and tmax,Capric acid appeared to be shorter with 0 min post-dose fasting than with longer post-dose fasting periods. There were only minor differences in the AUC for capric acid from time zero to 6 h (AUCCapric acid,0–6h) between the four post-dose fasting periods (Online Resource 1, Table S3).
3.4 Safety
Oral administration of an insulin 338 tablet was well-tolerated with no safety issues identified during the trial. A total of 42 AEs were reported in 25 subjects (57%). No apparent differences in number of AEs were observed between the four post-dose fasting periods (8, 10, 11 and 13 events with 0, 30, 60 and 360 min post-dose fasting, respectively). All AEs were non-serious, and all AEs were either mild (6, 7, 9 and 12 events) or moderate (2, 3, 2 and 1 events) in severity and either unlikely related (6, 7, 7 and 9 events) or possibly related (2, 3, 4 and 4 events) to trial product. The most frequently reported AEs were headache (2, 2, 5 and 4 events) and nasopharyngitis (4, 0, 3 and 2 events). All subjects recovered from the AEs. One AE (increased blood CK-MB with onset 12 days after last dosing) led to withdrawal of the subject.
A total of 90 hypoglycaemic episodes were reported in 25 subjects (56.8%). Fewer hypoglycaemic episodes were reported for 0 min post-dose fasting (13 episodes) than for longer post-dose fasting periods (26, 25 and 26 episodes for 30, 60 and 360 min post-dose fasting). During the first 360 min after dosing, 2, 9, 14 and 9 hypoglycaemic episodes were reported for 0, 30, 60 and 360 min post-dose fasting, respectively. The majority of hypoglycaemic episodes were asymptomatic (85 episodes), while 4 episodes were documented symptomatic and 1 episode was probably symptomatic. No severe hypoglycaemic episodes were reported.
There were no clinically significant findings in safety laboratory parameters, vital signs, physical examination or electrocardiogram.
4 Discussion
The main finding of the current trial was that the pharmacokinetic properties of insulin 338 were comparable for post-dose fasting periods of 30, 60 and 360 min, while a marked reduction in insulin 338 exposure of ~ 65% was seen with food ingestion immediately after dosing compared with 360 min after dosing. A previous trial investigating the effect of post-dose fasting period on the pharmacokinetics of orally administered mealtime insulin showed no difference in insulin exposure between post-dose fasting periods of 10, 45 or 90 min [14]. On the other hand, studies with oral administration of other peptide drugs have shown that no or limited post-dose fasting reduced the systemic drug absorption [15,16,17]. Meal ingestion 10 min after oral dosing of salmon calcitonin co-formulated with 5-CNAC [8-(N-2-hydroxy-5-chlorobenzoyl)-amino-caprylic acid] reduced the exposure of salmon calcitonin by up to 30% versus 60 min post-dose fasting and by 41% versus 4 h post-dose fasting [15, 16]. Increasing post-dose fasting periods of 15, 30, 60 and 120 min following oral administration of semaglutide co-formulated with SNAC {sodium N-(8-[2-hydroxybenzoyl] amino) caprylate} were associated with increasing semaglutide exposure [17]. In contrast to mealtime insulin, the therapeutic effect of basal insulin is not highly dependent on the timing of dosing relative to meal ingestion. Therefore, it is also less relevant for an oral basal insulin such as oral insulin 338, as compared with an oral mealtime insulin, to avoid any effect of food intake immediately after dosing on the insulin absorption from the gastrointestinal tract. In the current trial, meal ingestion immediately after oral dosing resulted in shorter tmax,Ins338, suggesting that insulin 338 absorption was affected by the post-dose meal. Thus, the current results indicate that food intake shortly after oral insulin 338 administration should be avoided in order not to compromise insulin 338 serum exposure levels. Importantly, t½,Ins338 was unaffected by the post-dose fasting period, suggesting that the metabolism and elimination of insulin 338 are not affected by post-dose food intake.
Reduced exposure of oral insulin when administered close to a meal is one of the main reasons why several previous investigations of various oral insulin formulations have been conducted in a fasting condition [18,19,20]. However, studies of oral insulin formulations have also been performed in which insulin was administered together with a meal or only 10–20 min prior to meal ingestion, both with detectable exposure and glucose-lowering effect [14, 19, 21]. Based on the current pharmacokinetic results, it would be recommended that patients with diabetes mellitus treated with oral insulin 338 as a basal insulin can have their breakfast 30 min after taking their once-daily dose of oral insulin 338 each morning. Such a dosing recommendation is considered to be feasible with respect to patient convenience and treatment compliance. Interestingly, another widely used drug, levothyroxine for the treatment of hypothyroidism, is also usually dosed in a fasting state ≥ 30 min prior to breakfast to ensure that food ingestion does not impair its absorption [22].
The total variability in AUCIns338,0–∞ and Cmax,Ins338 after oral insulin 338 administration was found to be within the range of 82–132% and 84–163%, respectively, for the coefficient of variation. This may seem to be high, and is indeed higher than seen for an oral human insulin product in subjects with type 2 diabetes with a variability in absorption of 60–70% for the coefficient of variation [23]. However, in the previous study, pharmacokinetic measures included both exogenous human insulin and endogenous insulin, which may have reduced the measured pharmacokinetic variability [23]. It is also important to note that the current trial was a single-dose trial, while the intended dosing regimen for oral insulin 338 is regular once-daily dosing. Due to its extended half-life, once-daily dosing of oral insulin 338 implies that the exposure from sequential daily doses will overlap. This will in turn reduce the day-to-day variability in exposure and, therefore, also the total variability. Accordingly, a phase II trial in subjects with type 2 diabetes showed that once-daily oral insulin 338 treatment for 8 weeks improved glycaemic control to an extent not different from that seen with 8 weeks of once-daily insulin glargine [8]. The total variability in insulin 338 exposure increased with a decreasing post-dose fasting period, although it was only significant for Cmax,Ins338. The total variability in exposure associated with administering oral insulin 338 within 30 min prior to food intake does not, however, seem to be extreme and should therefore not exclude the dosing recommendation of taking the daily dose of oral insulin 338 at least 30 min prior to breakfast.
There were no unexpected safety findings in the present trial, and orally administered insulin 338 appeared to be safe in healthy male subjects. The observations of a numerically lower number of AEs and fewer hypoglycaemic episodes reported for 0 min post-dose fasting than for longer post-dose fasting periods were consistent with the lower level of insulin 338 exposure when food was ingested immediately after dosing.
Two important conclusions can be made based on the pharmacokinetic results for capric acid in the current trial. Firstly, elimination of capric acid occurred rapidly as mean serum capric acid concentrations had returned to the baseline level at approximately 2 h post-dose. Thus, once-daily dosing of oral insulin 338 with sodium caprate as an absorption enhancer will not lead to any build-up of circulating capric acid. Secondly, Cmax,Capric acid was slightly higher and tmax,Capric acid was slightly shorter when subjects had their meal immediately after dosing of oral insulin 338 than after post-dose fasting periods of 30–360 min. This finding suggests that food can at least to some extent increase capric acid absorption from the gastrointestinal tract. It is tempting to speculate that due to increased capric acid absorption into the circulation, the smaller amounts of absorption enhancer left in the gastrointestinal tract might explain part of the reduced absorption of insulin 338 seen when food intake occurred immediately after oral insulin 338 administration. However, several in situ studies with sodium caprate on animal intestinal preparations suggest that close proximity as well as contemporaneous presence of promoter and candidate drug are more important for successful absorption enhancement than the exact local concentration of promoter [9]. Therefore, an alternative explanation could be that presence of food in the gastrointestinal tract interferes with the close interaction of insulin 338 and sodium caprate and thereby leads to the large decrease seen in insulin 338 absorption following immediate food ingestion.
The current trial was conducted in accordance with relevant regulatory guidelines on food and drug interaction and bioequivalence [11, 12, 24]. Thus, the trial was a single-dose trial. Due to the long half-life of oral insulin 338, the pharmacokinetic assessments after a single dose in the current trial may not be entirely reflective of the steady-state situation achieved with repeated once-daily administration in clinical practice. Still, differences or similarities in exposure of oral insulin 338 with varying post-dose fasting periods shown after a single dose should, at least in theory, be able to translate to the steady-state situation. A strength of the present trial was the specific pharmacokinetic measurement of exogenous insulin 338. Thus, current results on the absorption of insulin 338 after oral administration were not biased by any endogenous insulin secretion in the trial population of healthy subjects.
Finally, only males were recruited in the present trial as the preclinical reproduction toxicity studies with insulin 338 were not finalised at the time of trial conduct. It is, however, anticipated that the pharmacokinetic properties of oral insulin 338 in males do not differ from those in females, and furthermore that the pharmacokinetics of oral insulin 338 in healthy subjects are not markedly different from those in the target population of subjects with type 2 diabetes.
5 Conclusion
The present trial shows that absorption of oral insulin 338 administered in the fasting state is not affected by food intake from 30 min after dosing. This finding is important for patient convenience and treatment compliance, as it suggests that patients with diabetes mellitus can have their breakfast 30 min after their morning dose of basal oral insulin 338.
References
Heinemann L, Jacques Y. Oral insulin and buccal insulin: a critical reappraisal. J Diabetes Sci Technol. 2009;3:568–84.
Iyer H, Khedkar A, Verma M. Oral insulin—a review of current status. Diabetes Obes Metab. 2010;12:179–85.
Zijlstra E, Heinemann L, Plum-Mörschel L. Oral insulin reloaded: a structured approach. J Diabetes Sci Technol. 2014;8:458–65.
Wong CY, Martinez J, Dass CR. Oral delivery of insulin for treatment of diabetes: status quo, challenges and opportunities. J Pharm Pharmacol. 2016;68:1093–108.
Goldberg M, Gomez-Orellana I. Challenges for the oral delivery of macromolecules. Nat Rev Drug Discov. 2003;2:289–95.
Aguirre TA, Teijeiro-Osorio D, Rosa M, Coulter IS, Alonso MJ, Brayden DJ. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv Drug Deliv Rev. 2016;106:223–41.
Walsh EG, Adamczyk BE, Chalasani KB, Maher S, O’Toole EB, Fox JS, et al. Oral delivery of macromolecules: rationale underpinning Gastrointestinal Permeation Enhancement Technology (GIPET). Ther Deliv. 2011;2:1595–610.
Halberg IB, Lyby K, Wassermann K, Heise T, Zijlstra E, Plum-Mörschel L. Efficacy and safety of oral basal insulin versus subcutaneous insulin glargine in subjects with type 2 diabetes: an 8-week, randomised, double-blind phase 2 trial. Lancet Diabetes Endocrinol. 2019;7:179–88.
Maher S, Leonard TW, Jacobsen J, Brayden DJ. Safety and efficacy of sodium caprate in promoting oral drug absorption: from in vitro to the clinic. Adv Drug Deliv Rev. 2009;61:1427–49.
Schmidt LE, Dalhoff K. Food–drug interactions. Drugs. 2002;62:1481–502.
European Medicines Agency. Committee for Human Medicinal Products. Guideline on the investigation of drug interactions. 2012. https://www.ema.europa.eu/documents/scientific-guideline/guideline-investigation-drug-interactions_en.pdf. Accessed 16 Apr 2019.
U.S. Food and Drug Administration. Guidance for Industry. Food-effect bioavailability and fed bioequivalence studies. 2002. https://www.fda.gov/media/70945/download. Accessed 16 Apr 2019.
Seaquist ER, Anderson J, Childs B, Cryer P, Dagogo-Jack S, Fish L, et al. Hypoglycemia and diabetes: a report of a workgroup of the American Diabetes Association and the Endocrine Society. Diabetes Care. 2013;36:1384–95.
Eldor R, Arbit E, Miteva Y, Freier R, Kidron M. Oral insulin: type I diabetes (T1DM) patient response upon preprandial administration. Diabetes. 2010;59(Suppl 1):A141 (Abstract).
Karsdal MA, Byrjalsen I, Riis BJ, Christiansen C. Optimizing bioavailability of oral administration of small peptides through pharmacokinetic and pharmacodynamic parameters: the effect of water and timing of meal intake on oral delivery of salmon calcitonin. BMC Clin Pharmacol. 2008;8:5.
Karsdal MA, Byrjalsen I, Azria M, Arnold M, Choi L, Riis BJ, et al. Influence of food intake on the bioavailability and efficacy of oral calcitonin. Br J Clin Pharmacol. 2009;67:413–20.
Bækdal TA, Borregaard J, Donsmark M, Breitschaft A, Søndergaard FL. Evaluation of the effects of water volume with dosing and post-dose fasting period on pharmacokinetics of oral semaglutide. Diabetes. 2017;66(Suppl 1):A315 (Abstract).
Kidron M, Dinh S, Menachem Y, Abbas R, Variano B, Goldberg M, et al. A novel per-oral insulin formulation: proof of concept study in non-diabetic subjects. Diabet Med. 2004;21:354–7.
Heise T, Nosek L, Arbit E, et al. Reduction of postprandial blood glucose excursions by an optimized formulation of oral insulin. Diabetes. 2005;54(Suppl 1):A103 (Abstract).
Luzio SD, Dunseath G, Lockett A, Broke-Smith TP, New RR, Owens DR. The glucose lowering effect of an oral insulin (Capsulin) during an isoglycaemic clamp study in persons with type 2 diabetes. Diabetes Obes Metab. 2010;12:82–7.
Khedkar A, Iyer H, Anand A, Verma M, Krishnamurthy S, Savale S, et al. A dose range finding study of novel oral insulin (IN-105) under fed conditions in type 2 diabetes mellitus subjects. Diabetes Obes Metab. 2010;12:659–64.
Chakera AJ, Pearce SH, Vaidya B. Treatment for primary hypothyroidism: current approaches and future possibilities. Drug Des Devel Ther. 2012;6:1–11.
Kapitza C, Zijlstra E, Heinemann L, Castelli MC, Riley G, Heise T. Oral insulin: a comparison with subcutaneous regular human insulin in patients with type 2 diabetes. Diabetes Care. 2010;33:1288–90.
European Medicines Agency. Committee for Medicinal Products for Human Use. Guideline on the investigation of bioequivalence. 2010. https://www.ema.europa.eu/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf. Accessed 16 Apr 2019.
Acknowledgements
The authors would like to thank Carsten Roepstorff, PhD, CR Pharma Consult, Copenhagen, Denmark for providing medical writing support, which was funded by Novo Nordisk A/S.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval and informed consent
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Funding
This trial was funded by Novo Nordisk.
Conflict of interest
Inge B. Halberg, Karsten Lyby and Karsten Wassermann are employees and shareholders of Novo Nordisk. Tim Heise is a shareholder of Profil, which has received research funds from Adocia, Boehringer Ingelheim, Dance Pharmaceuticals, Eli Lilly, Johnson & Johnson, MedImmune, Merck Sharp and Dohme, Mylan, Nordic Bioscience, Novo Nordisk, Poxel, Roche Diagnostics, Saniona, Sanofi, Senseonics and Zealand Pharma. In addition, Tim Heise is member of advisory panels for Novo Nordisk and Mylan and received speaker honoraria and travel grants from Dexcom, Eli Lilly, Mylan, Novo Nordisk, Sanofi and Zealand Pharma. Leona Plum-Mörschel has received speaker honoraria and travel grants from Eli Lilly and Novo Nordisk. Eric Zijlstra has received speaker honoraria and travel grants from Novo Nordisk, Roche Diabetes Care and Senseonics.
Trial registration
The trial was registered at ClinicalTrials.gov (trial identifier: NCT02304627).
Data availability
Individual participant data will be shared in datasets in a de-identified/anonymised format with bona fide researchers submitting a research proposal requesting access to data for use as approved by the Independent Review Board (IRB) according to the IRB Charter (see http://www.novonordisk-trials.com). Datasets from Novo Nordisk-sponsored clinical research completed after 2001 for product indications approved in both the EU and USA and study protocol and redacted Clinical Study Report (CSR) will be available according to Novo Nordisk data-sharing commitments. The data will be available permanently after research completion and approval of product and product use in both the EU and USA (no end date). The access request proposal form and the access criteria can be found at http://www.novonordisk-trials.com. The data will be made available on a specialised SAS® data platform.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Halberg, I.B., Lyby, K., Wassermann, K. et al. The Effect of Food Intake on the Pharmacokinetics of Oral Basal Insulin: A Randomised Crossover Trial in Healthy Male Subjects. Clin Pharmacokinet 58, 1497–1504 (2019). https://doi.org/10.1007/s40262-019-00772-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00772-2