
Abstract
Nintedanib is an oral, small-molecule tyrosine kinase inhibitor approved for the treatment of idiopathic pulmonary fibrosis and patients with advanced non-small cell cancer of adenocarcinoma tumour histology. Nintedanib competitively binds to the kinase domains of vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF). Studies in healthy volunteers and in patients with advanced cancer have shown that nintedanib has time-independent pharmacokinetic characteristics. Maximum plasma concentrations of nintedanib are reached approximately 2–4 h after oral administration and thereafter decline at least bi-exponentially. Over the investigated dose range of 50–450 mg once daily and 150–300 mg twice daily, nintedanib exposure increases are dose proportional. Nintedanib is metabolised via hydrolytic ester cleavage, resulting in the formation of the free acid moiety that is subsequently glucuronidated and excreted in the faeces. Less than 1% of drug-related radioactivity is eliminated in urine. The terminal elimination half-life of nintedanib is about 10–15 h. Accumulation after repeated twice-daily dosing is negligible. Sex and renal function have no influence on nintedanib pharmacokinetics, while effects of ethnicity, low body weight, older age and smoking are within the inter-patient variability range of nintedanib exposure and no dose adjustments are required. Administration of nintedanib in patients with moderate or severe hepatic impairment is not recommended, and patients with mild hepatic impairment should be monitored closely and the dose adjusted accordingly. Nintedanib has a low potential for drug–drug interactions, especially with drugs metabolised by cytochrome P450 enzymes. Concomitant treatment with potent inhibitors or inducers of the P-glycoprotein transporter can affect the pharmacokinetics of nintedanib. At an investigated dose of 200 mg twice daily, nintedanib does not have proarrhythmic potential.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Nintedanib has a straightforward and time-independent pharmacokinetic profile that is consistent across a range of patient populations. |
Hepatic and intestinal metabolism as well as biliary excretion are the major routes of elimination for nintedanib. |
The intrinsic factors sex and renal function do not affect nintedanib pharmacokinetics. |
Nintedanib has a low potential for drug–drug interactions via cytochrome P450 but coadministration of drugs that are potent inhibitors or inducers of P-glycoprotein should be undertaken with care. |
1 Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrosing form of interstitial lung disease of unknown origin [1] characterised by excessive and disordered collagen deposition and scarring of the lung parenchyma, leading to irreversible loss of lung function [2]. Although the underlying pathogenetic mechanisms remain elusive multiple growth factors and signalling pathways appear to be involved in the pathogenesis of IPF offering various targets for therapeutic intervention [2, 3].
Receptors for pro fibrotic growth factors such as the members of the vascular endothelial growth factor (VEGF) receptor (VEGFR), platelet-derived growth factor (PDGF) receptor (PDGFR) and fibroblast growth factor (FGF) receptor (FGFR) families are essential regulators of fibroblast proliferation and migration in the lung and have been implicated in the pathogenesis of IPF [4,5,6]. Various IPF treatments have been used, although until recently none have proven to be effective or been approved for clinical use. In 2014, two compounds with pleiotropic mechanisms of action, pirfenidone and nintedanib, were approved after they were shown to be effective in slowing progressive functional decline and disease progression in IPF [7, 8].
Besides their role in fibrosis, VEGF, PDGF and FGF and their respective receptors (VEGFR, PDGFR and FGFR) are recognised as playing an important role in the formation and maintenance of tumour vasculature through angiogenesis, whereby new capillary blood vessels are created from pre-existing vessels, resulting in tumour growth, progression and metastasis [9, 10]. The angiogenic process is complex with multiple proangiogenic and antiangiogenic pathways involved. The best characterised proangiogenic regulator in tumour angiogenesis is the VEGF signalling pathway, which is released by tumour cells as a response to hypoxia and nutrient deficiency during tumour growth [10]. Compensatory signalling via FGF and PDGF pathways can result in the development of resistance to therapies that target VEGF alone [11,12,13].
Nintedanib is a potent, oral, small-molecule tyrosine kinase inhibitor (TKI) that blocks the kinase activity of a variety of receptors, including the receptor tyrosine kinases (RTKs) VEGFR 1–3; PDGFR-α and -β; and FGFR 1–3 [14, 15]. By competitively and reversibly inhibiting the adenosine triphosphate (ATP) binding pocket of VEGFR, PDGFR and FGFR, nintedanib blocks the intracellular signalling pathways that are crucial for the proliferation, migration and transformation of lung fibroblasts [6, 16], as well as the proliferation and survival of endothelial as well as perivascular cells (pericytes and vascular smooth muscle cells) in tumour tissues [14].
Nintedanib also blocks the activity of several non-RTKs (nRTKs) [17], including FLT-3 (Fms-like tyrosine-protein kinase-3), RET (ret proto-oncogene) and members of the Src-family, including Lck (lymphocyte-specific tyrosine-protein kinase), Lyn (tyrosine-protein kinase lyn) and Src (proto-oncogene tyrosine-protein kinase src) kinases [14, 15]. The Src-family kinases have been shown to play a role in the pathogenesis of pulmonary fibrosis [18].
Nintedanib was approved by the US Food and Drug Administration (FDA) in October 2014 and by the European Medicines Agency (EMA) in January 2015 for the treatment of patients with IPF [19, 20], based on results of one phase II dose-finding trial (TOMORROW) [21] and two replicate phase III trials (INPULSIS-1 and INPULSIS-2) [8] that demonstrated a significant reduction in the rate of decline in forced vital capacity (FVC) over a 52-week treatment period, consistent with slowing disease progression.
In November 2014, nintedanib was approved in Europe and subsequently in other countries for use in combination with docetaxel in patients with locally advanced, metastatic or recurrent non-small cell lung carcinoma (NSCLC) of adenocarcinoma tumour histology after first-line chemotherapy [22]. This was based on results of the phase III LUME-Lung 1 study that showed a significant improvement in overall survival (OS) with nintedanib plus docetaxel (median 12.6 vs. 10.3 months; hazard ratio [HR] 0.83; 95% confidence interval [CI], 0.70–0.99; p = 0.036) compared with docetaxel plus placebo in previously treated patients with advanced NSCLC of adenocarcinoma tumour histology [23]. In patients with aggressive tumour disease a pronounced significant OS benefit (median 10.9 vs. 7.9 months; HR 0.75; 95% CI 0.60–0.92; p = 0.0073) was seen as well.
This review discusses the pharmacokinetic and pharmacodynamic properties and drug–drug interaction profile of nintedanib.
2 Preclinical Pharmacology
2.1 Structure and Physicochemical Properties
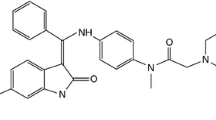
Nintedanib (Vargatef®, Ofev®), also known as BIBF 1120, has the chemical name 1H-indole-6-carboxylic acid, 2,3-dihydro-3-[[[4-[methyl[(4-methyl-1-piperazinyl)acetyl]-amino]phenyl]amino]phenylmethylene]-2-oxo-,methyl ester, (3Z)-,ethanesulfonate and a molecular weight of 539.62 as free base. The chemical structure is shown in Fig. 1. Structural modelling studies with recombinant VEGFR-2 kinase demonstrated that nintedanib binds to the ATP-binding site in the cleft between the NH2 and COOH terminal lobes of the kinase domain [14].

Metabolism and excretion of nintedanib. The total percentage of each metabolite excreted as a proportion of dose is shown in parentheses. The CYP isozymes and glucuronidation (UGT) enzymes involved in the metabolism of nintedanib are shown. CYP cytochrome P450, UGT uridine diphosphate glucuronosyltransferase
Nintedanib is dosed as a soft gelatine capsule. It displays a pH-dependent solubility profile with low aqueous solubility under neutral pH and increased solubility at acidic pH < 3 [24]. Nintedanib is considered to be a class 2 or 4 compound under the Biopharmaceutics Classification System (i.e. having low water solubility at neutral pH together with an ambiguous classification regarding permeability). Although nintedanib shows a high in vitro permeability, the absorption rate and bioavailability of nintedanib are decreased due to P-glycoprotein (P-gp) transporter effects [24].
2.2 Pharmacodynamic Properties
The competitive binding of nintedanib to the ATP-binding pocket of the intracellular receptor kinase domain of VEGFR 1–3, PDGFR-α and -β and FGFR 1–3 inhibits the tyrosine kinase activity of these receptors, thereby interfering with the cross-autophosphorylation of the receptor homodimers and blocking the signalling cascade [25]. Nintedanib has been shown to inhibit the FGFR, PDGFR and VEGFR signalling cascades mediating lung fibroblast proliferation and migration [6, 16]. It inhibits protein kinase signalling pathways in three cell types contributing to angiogenesis, endothelial cells, pericytes and smooth muscle cells, resulting in inhibition of cell proliferation and apoptosis [14].
In cell-free in vitro kinase assays, nintedanib showed nanomolar potency to inhibit all three VEGFR subtypes, PDGFR-α and -β and FGFR-1, -2 and -3 [14]. In cell-based assays, inhibition of VEGFR-2 phosphorylation was sustained for at least 32 h in VEGFR-2-transfected NIH3T3 cells exposed to nintedanib for 1 h [14], suggesting a sustained inhibitory effect.
In preclinical disease models nintedanib showed antifibrotic effects in animal models of pulmonary fibrosis [16], highlighted by significant reductions in total lung collagen and reduced fibrosis in histological analyses. Nintedanib exerted anti-inflammatory effects as shown by significant reductions in lymphocyte and neutrophil counts in bronchoalveolar lavage fluid, reductions in inflammatory cytokines, and reduced inflammation and granuloma formation in histological analysis of lung tissue [16]. Additional data show that nintedanib significantly reduces lung collagen levels in a mouse model of rheumatoid arthritis-associated interstitial lung disease [26].
Nintedanib has also demonstrated antifibrotic effects in preclinical models of systemic sclerosis (SSc), an immune-mediated rheumatic disease of unknown aetiology that is characterised by fibrosis of the skin and internal organs and vasculopathy [27]. In two preclinical mouse models of SSc, nintedanib effectively inhibited fibroblast activation and exerts potent antifibrotic effects [28]. In a further mouse model, nintedanib ameliorated histological features of pulmonary arterial hypertension, destructive microangiopathy, and pulmonary and dermal fibrosis [29].
Nintedanib also effectively interfered with the formation and maintenance of the tumour vascular system resulting in tumour growth inhibition and stasis [14, 30]. Treatment with nintedanib in mouse xenograft models led to a rapid reduction in tumour microvessel density, pericytes vessel coverage and tumour perfusion [14] and resulted in significant tumour growth inhibition in models of different human cancer types including NSCLC [14].
For a more thorough discussion of the preclinical efficacy profile of nintedanib, the reader is referred to comprehensive reviews by Hilberg et al. [14] and Wollin et al. [6, 16].
2.3 Preclinical Pharmacokinetics Relevant to Human Pharmacokinetics
The pharmacokinetics and drug metabolism of nintedanib (dosed via intravenous [IV] infusion or oral gavage) were studied in several animal species.
Mean plasma protein binding of nintedanib was > 97% in mice and rats, 91–93% in monkeys, and 98% in humans, over a concentration range of 50–2000 ng/mL [13, 17, 31]. Albumin was the major binding protein.
Following administration of [14C]-radiolabelled nintedanib to rats, radioactivity was widely distributed into most of the tissues (except the central nervous system [CNS]). Repeated oral dosing ([14C]-radiolabelled nintedanib 30 mg/kg) for 13 days showed a slight accumulation in some tissues, although a similar accumulation in plasma concentrations was not apparent [32].
2.4 In Vitro Drug–Drug Interaction Victim and Perpetrator Properties
Several in vitro metabolism, transport and drug interaction studies were performed to quantitatively assess the drug–drug interaction potential of nintedanib.
In vitro studies with human hepatocytes and/or human liver microsomes showed that nintedanib is a minor substrate for cytochrome P450 (CYP) 3A4 isoenzyme and has a very low potential (along with its two major metabolites [BIBF 1202 and BIBF 1202 glucuronide]) to inhibit or induce CYP isoenzymes [24], including those that are most relevant or genetically polymorphic for drug metabolism in humans (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 and CYP3A4) [32]. In human liver microsomes, nintedanib was rapidly hydrolysed by esterases (major mechanism) and demethylated by CYP3A4 (minor mechanism) to form the metabolites BIBF 1202 and BIBF 1053, respectively [33]. CYP-dependent metabolism accounted for about 5% compared to about 25% ester cleavage [20]. Thus, drug–drug interactions with nintedanib as a victim of CYP enzyme-modulating agents (e.g. with co-medication with CYP inhibitors or inducers) are considered very unlikely. Furthermore, drug–drug interactions with nintedanib as a perpetrator of CYP enzymes (e.g. nintedanib acting as a CYP enzyme inhibitor or inducer) are also considered very unlikely.
Further in vitro data indicated that nintedanib, at clinically relevant concentrations, did not inhibit glucuronidation by uridine 5′-diphospho-glucuronosyltransferase (UDP-glucuronosyltransferase, UGT) 1A1 (UGT1A1) in human liver microsomes. UGT1A1 is responsible for the glucuronidation of the metabolite BIBF 1202 to BIBF 1202 glucuronide in human liver microsomes [33]. In addition, BIBF 1202 was glucuronidated by several intestinal UGTs (UGT1A7, UGT1A8, UGT1A10). A clinically relevant drug–drug interaction based on inhibition of UGT after oral administration of nintedanib is considered less likely as all half-maximal inhibitory concentration (IC50) values are substantially higher than the therapeutic plasma concentrations [32].
In vitro assays using transfected MDCK cells demonstrated that nintedanib is a substrate of the efflux transporter P-gp and weakly inhibits P-gp (Table 1) [24]. Studies using cell lines that express different drug transporters showed that nintedanib was not a substrate of organic anion-transporting polypeptide (OATP) 1B1, OATP1B3, OATP2B1, organic cation transporter (OCT) 2, multidrug resistance-associated protein 2 (MRP-2) or the efflux breast cancer resistance protein (BCRP), but was a weak substrate for OCT1 [31]. Nintedanib did not inhibit OATP1B1-, OATP1B3-, OATP2B1-, OCT1-, OCT2-, P-gp- or BRCP-mediated transport at clinically relevant concentrations [33].
Although the principal metabolite BIBF 1202 shows pharmacological activity at some of the target receptors, in vivo cellular activity indicates a substantially lower potency than nintedanib [32]. BIBF 1202 was not effective in mouse xenograft models [31], suggesting that BIBF 1202 plasma concentrations themselves are unlikely to contribute to the clinical effects of nintedanib in vivo. BIBF 1202 glucuronide did not show in vitro activity at the target receptors and is a non-reactive glucuronide [34].
In summary, in the clinical setting, nintedanib is unlikely to affect the pharmacokinetics of other drugs that are substrates of major drug-metabolising CYPs or uptake transporters, such as OATP1B1, OATP1B3, OAT1, OAT3 or OCT2. Drug–drug interactions with nintedanib as a victim of CYP enzyme-modulating agents (e.g. with co-medication with CYP inhibitors or inducers) are considered very unlikely. Nintedanib is a substrate of P-gp in vitro. The potential for a clinical drug–drug interaction between nintedanib and inhibitors or inducers of P-gp was addressed in dedicated phase I studies, as described in Sect. 5.
3 Clinical Pharmacokinetics
The clinical pharmacokinetics of nintedanib monotherapy were investigated in healthy subjects [34,35,36,37,38], volunteers with hepatic impairment [39,40,41], and in patients with IPF [8, 21, 42] or various advanced types of cancer [25, 38, 43,44,45,46,47,48]. In healthy volunteers, only single-dose administration was performed. Key pharmacokinetic parameters following single and steady-state twice-daily dosing of nintedanib in patients with advanced cancer are presented in Table 2. The pharmacokinetics of nintedanib were further characterised by two successive population pharmacokinetic (PopPK) analyses, the first based on combined pharmacokinetic data from NSCLC (n = 849) and IPF (n = 342) patients [49] and the second in IPF patients only (n = 933) [50] enrolled in the phase II and III trials. For comparison, Table 3 gives key pharmacokinetic parameters after multiple dosing of nintedanib to typical patients with IPF or NSCLC based on the PopPK analyses. These results show that the key pharmacokinetic parameters are consistent across the two patient populations.
3.1 Absorption and Distribution
Following oral administration of soft capsules under fed conditions, nintedanib is absorbed promptly, with maximum concentration (Cmax) being reached at 2–4 h (range 0.5–8 h) [25, 34, 35]. A representative plasma concentration–time profile of nintedanib after single 100 mg doses is shown in in Fig. 2. After reaching Cmax nintedanib follows at least bi-phasic disposition kinetics.

Geometric mean plasma concentration–time profile of nintedanib after single-dose administration of nintedanib (100 mg) to healthy volunteers (n = 14) (main figure, semi-log scale; insert figure, linear scale) [39]
The absolute bioavailability of the oral capsule formulation of nintedanib (100 mg) relative to IV dosing (4-h infusion, 6 mg) is around 5% (95% CI 3.6–6.1) [38]. Absorption and bioavailability are thought to be decreased by transporter effects and substantial first-pass metabolism [20].
After a high-fat, high-calorie meal [35] nintedanib exposure appears to be slightly increased, with Cmax and area under the drug plasma concentration–time curve (AUC) from time zero to infinity (AUC∞) increased by ~ 15% (geometric mean [gMean] ratio point estimate 115.3%, 90% CI 84.6–157.2) and 20% (gMean ratio 120.6%, 90% CI 95.3–152.5), respectively [24]. The 90% CIs of the AUC∞ and Cmax ratios under fed versus fasted conditions were not entirely within the range of 80–125% but include 100% in both cases. Absorption is delayed by about 2 h (from 2 to 4 h) in the fed state and inter-patient variability is increased. Nintedanib is recommended to be taken with food to improve gastrointestinal tolerability [19, 22].
Visual inspection of dose-normalised pharmacokinetic parameter estimates (Cmax and AUC) show no relevant deviation from dose proportionality across the dose range of 50–450 mg once daily and 150–300 mg twice daily (Fig. 3) [38]. In line with this, the pharmacokinetics of nintedanib 150–250 mg twice daily in a PopPK analysis of patients with NSCLC or IPF were found to be dose proportional [49]. Clearance of nintedanib does not change after multiple dosing compared with single administration, meaning that the pharmacokinetics of nintedanib can be considered to be time independent. Thus, the pharmacokinetics of nintedanib are linear with respect to dose and time (i.e. single-dose data can be extrapolated to multiple-dose data) [20]. Based on trough concentrations, steady state is reached by 7 days at the latest (no sampling at earlier timepoints) [38, 42]. Accumulation after repeated twice-daily dosing is negligible (ratio of about 1.4- to 1.7-fold based on the AUC from time zero to 12 h [AUC12]) [38] (Tables 2, 3). The pharmacokinetics of nintedanib do not differ substantially between healthy volunteers and patients with IPF or cancer.

Comparison of dose-normalised nintedanib Cmax,ss (a) and AUC0-12h,ss (b) after multiple twice-daily oral doses (150–300 mg) in patients with advanced solid tumours or multiple myeloma [25, 44, 45, 48] enrolled in four phase I trials (n = 53). Results are based on a meta-analysis of data from these four trials [38]. The box represents the 25th–75th percentiles of the distribution, the whiskers extend to 10th and 90th percentiles, and the central line is the median. The dots show individual datapoints outside the 10th–90th percentile ranges. AUC0-12h,ss,norm dose-normalised area under the drug plasma concentration–time curve from time zero to 12 h at steady state, bid twice daily, Cmax,ss,norm dose-normalised maximum concentration at steady state, gMean geometric mean, Max maximum, Min minimum, n maximum number of patients contributing to the geometric means of each timepoint at steady state
Pharmacokinetic parameters show moderate to high inter-individual variability, as demonstrated by coefficients of variation (CVs) between 46 and 83% [25, 34, 44, 47] (see Table 2). This variability seems to be caused by differences in absorption rather than disposition and elimination. The low bioavailability (< 5%) and high first-pass metabolism of the drug may contribute to this observation [38]. Intra-individual variability is low to moderate (geometric CV [gCV] < 40%).
Nintedanib is ~ 98% plasma protein bound, with serum albumin considered to be the major binding protein [20]. After IV administration of nintedanib (6 mg), a high volume of distribution (gMean 1050 L, gCV 45%) is observed [38], indicating extensive distribution into peripheral tissues. This observation is in accordance with the high in vitro permeability of nintedanib through biomembranes in cellular assays and the results of a quantitative whole-body autoradiography study in rats, in which a rapid and relatively homogenous tissue distribution apart from the CNS and thus a pronounced blood–brain barrier was observed [17, 24]. Nintedanib is preferentially distributed in plasma with a blood to plasma ratio of 0.87 [20].
3.2 Metabolism and Elimination
Nintedanib is predominantly metabolised via hydrolytic ester cleavage, with subsequent glucuronidation and excretion via the liver. The disposition and mass balance of nintedanib was investigated in a dedicated absorption, distribution, metabolism, and elimination (ADME) study in eight male subjects who received a single oral dose of 100 mg [14C-]-radiolabelled nintedanib solution [34]. The major route of elimination of drug-related radioactivity following a single oral dose of [14C]-radiolabelled nintedanib solution was via faecal/biliary excretion (93.4% in faeces within 120 h after dosing) with negligible clearance in the urine (0.65% of the dose within 72 h after dosing). Renal excretion of unchanged nintedanib was 0.05% within 48 h of dosing. The combined faecal and urinary recovery after 4 days accounted for 92.4% of the administered [14C]-radiolabelled dose, indicative of an essentially complete mass balance. Most of the recovery occurred within 24–48 h of dosing.
Metabolite profiling showed that metabolism is the major pathway of clearance for nintedanib, primarily through rapid hydrolytic ester cleavage of the methyl ester moiety, resulting in the formation of BIBF 1202, which is subsequently glucuronidated by UGT enzymes (UGT1A1, UGT1A7, UGT1A8 and UGT1A10) in the intestine and by UGT1A1 in the liver to form BIBF 1202 glucuronide [34] (Fig. 1). Most of the drug-related radioactivity determined in plasma was accounted for by nintedanib (24%), BIBF 1202 (32%) and BIBF 1202 glucuronide (30%). Other metabolites were only observed in low amounts in plasma or excreta. One of the minor metabolic pathways occurred via CYP enzymes, predominantly CYP3A4, and yielded a demethylated metabolite (designated BIBF 1053) that could not be detected in plasma but only in excreta, predominantly faeces (4% of dose) [19, 34].
These findings indicate that oxidative metabolism mediated via CYPs is of negligible importance for the metabolism and elimination of nintedanib, suggesting that the risk of drug–drug interactions between nintedanib and other therapies modulating CYP enzymes (i.e. CYP inhibitors and CYP inducers) is minimal.
In addition, nintedanib has no in vitro liability to inhibit or induce CYP enzymes (see Sect. 5), which may facilitate its combination with treatments for IPF (e.g. pirfenidone) and cytotoxic chemotherapies (e.g. docetaxel and paclitaxel) that utilise CYP enzymes.
The disposition and metabolism of nintedanib were also investigated following IV infusion (6 mg) [38]. Nintedanib showed a high total plasma clearance (gMean 1390 mL/min, gCV 28.8%), which classifies it as a high clearance drug. Comparison of exploratory metabolite pharmacokinetic data after IV and oral administration suggests that BIBF 1202 and its glucuronide are formed faster and to a larger extent after oral administration. The urinary excretion of unchanged drug within 48 h after IV administration was about 1% of dose, indicating that non-renal mechanisms are responsible for the clearance of nintedanib. The renal clearance was about 20 mL/min. Thus, renal excretion plays a minor role in the elimination process of nintedanib, both after IV and oral administration.
Elimination of nintedanib from plasma occurs with a terminal half-life of approximately 10–15 h (gCV approximately 50%) [20, 38]. The effective half-life of nintedanib in patients with IPF was estimated to be about 9.5 h (gCV 31.9%) based on PopPK model-predicted accumulation ratios (Table 3) [19].
4 Pharmacokinetics in Selected Special Populations
4.1 Renal Impairment
Nintedanib is not being excreted via the kidneys to a relevant extent. The contribution of renal excretion after oral administration of nintedanib, both as unchanged drug (about 0.05% of dose) and as drug-related radioactivity (about 0.6% of dose), is minor.
In line with this, results from the PopPK analyses based on phase II and III data indicate that exposure to nintedanib was not influenced by mild (classified as creatinine clearance [CLCR] 60–90 mL/min) or moderate (CLCR 30–60 mL/min) renal impairment [49, 50]. Therefore, adjustment of the starting dose in patients with mild to moderate renal impairment is not required [19, 22] and no dedicated study in renally impaired patients has been performed. The safety and efficacy of nintedanib have not been thoroughly studied in patients with severe renal impairment (CLCR < 30 mL/min) and therefore treatment in these patients is not recommended.
4.2 Hepatic Impairment
Administration of nintedanib in patients with moderate or severe hepatic impairment is not recommended, and patients with mild hepatic impairment should be monitored closely and the dose adjusted accordingly. Several studies have evaluated the effect of hepatic impairment on the pharmacokinetics of nintedanib. In a dedicated single-dose study of nintedanib 100 mg in subjects with mild (n = 8) or moderate (n = 8) hepatic impairment (Child-Pugh A and B grades [51], respectively) and matched healthy control subjects (n = 17) [39], exposure to nintedanib based on Cmax and AUC∞ was approximately two-fold higher in subjects with mild hepatic impairment and approximately eight-fold higher in subjects with moderate hepatic impairment than in matched controls (Table 4). Subjects with severe hepatic impairment (Child-Pugh C) have not been studied.
In addition, supportive data on the pharmacokinetics of nintedanib in patients with hepatic impairment have been collected as part of the clinical development programmes in IPF and cancer patients. Despite some differences in the classification of hepatic impairment (Child-Pugh classification not used), the pharmacokinetic data of nintedanib in individuals with hepatic impairment were aligned across datasets, with an increase in nintedanib exposure observed in patients with impaired liver function [39]. The supportive data comprise two phase I dose-escalation studies of open-label nintedanib in Asian and European patients with impaired hepatic function and advanced hepatocellular carcinoma (HCC) [40, 41] as well as the two PopPK analyses in patients with NSCLC and IPF [49, 50]. In both analyses, patients were defined as having mild hepatic impairment if their aspartate aminotransferase (AST) or alanine aminotransferase (ALT) or bilirubin levels were greater than the upper limit of normal (ULN), but AST and ALT were ≤ 10 × ULN and bilirubin was ≤ 1.5 × ULN at start of treatment. The number of patients with mild hepatic impairment at the start of treatment was 116 (of 849 patients with NSCLC and 342 patients with IPF) in the first analysis [49] and 44 (of 933 patients with IPF) in the second [50]. A trend toward elevated nintedanib exposure of up to 1.4-fold was observed in patients with mild hepatic impairment compared with patients with normal hepatic function. Because of missing information about underlying hepatic disease, a robust assessment of the effect of hepatic impairment defined by elevation of transaminases or bilirubin on nintedanib was not possible. Pharmacokinetic observations for nintedanib and its major metabolites in patients with hepatic impairment point towards an increase in the bioavailable fraction of nintedanib in these subjects (i.e. higher exposure in subjects with hepatic impairment than in healthy subjects, with comparable plasma concentration–time profiles). This is in line with nintedanib being a high clearance drug with high first-pass metabolism. Renal clearance of nintedanib is not influenced by impaired hepatic elimination.
Based on these findings, treatment with nintedanib is not recommended for patients with moderate or severe hepatic impairment (Child-Pugh B or C) [19, 20, 22]. Patients with mild hepatic impairment (Child-Pugh A) should be monitored closely and the dose adjusted accordingly. Prescribing guidelines in the USA and Europe recommend a dose reduction to 100 mg twice daily for patients with IPF and associated mild hepatic impairment [19, 20], while patients with NSCLC and associated mild hepatic impairment should be closely monitored and the dose adapted based on tolerability [22].
5 Drug–Drug Interactions
Nintedanib has limited drug–drug interaction potential, based on the minor role of CYP pathways in its metabolism and the lack of induction or inhibition of CYP enzymes (IC50> 50 μmol/L) and drug transporters by nintedanib and its metabolites in vitro [34].
Nintedanib displays a pH-dependent solubility profile with increased solubility at acidic pH < 3. However, in clinical trials, coadministration with proton pump inhibitors or histamine H2 antagonists did not influence trough concentrations of nintedanib [19].
Since nintedanib is a substrate of P-gp in vitro, coadministration of potent inhibitors and inducers of this efflux transporter can potentially modify its exposure (AUC and Cmax). Two phase I drug–drug interaction studies were performed to investigate the interaction potential with potent P-gp modulators [52].
5.1 Ketoconazole
A drug–drug interaction study in healthy male subjects evaluated the effect of the potent P-gp inhibitor ketoconazole on nintedanib exposure [52]. Administration of ketoconazole increased exposure to nintedanib by 61% (AUC∞) and 83% (Cmax) (Fig. 4). Therefore, patients should be monitored closely for tolerability of nintedanib if a potent P-gp inhibitor (e.g. ketoconazole, erythromycin or cyclosporine [ciclosporin]) is being coadministered as exposure to nintedanib may be increased [19, 22]. Management of adverse effects may require interruption, dose reduction or discontinuation of nintedanib therapy.

Effect of ketoconazole [52], rifampicin [52], pirfenidone [62] and bosentan [66] coadministration on nintedanib exposure. The shaded area illustrates the typical bioequivalence limits (80–125%) used in the assessment of drug interactions. AUC area under the drug plasma concentration–time curve, CI confidence interval, Cmax maximum concentration
5.2 Rifampicin
In healthy male subjects [52], coadministration with the potent P-gp inducer rifampicin (rifampin) decreased plasma exposure of nintedanib to 50.3% (AUC∞) and 60.3% (Cmax) compared to administration of nintedanib alone (Fig. 4). Therefore, coadministration with P-gp inducers (e.g. rifampicin, carbamazepine, phenytoin and St. John’s wort) should be avoided (US label [19]) or carefully considered (EU label [22]) as this may result in decreased exposure to nintedanib.
5.3 Anticancer Therapy
The pharmacokinetics of nintedanib have been studied in combination with standard chemotherapy agents, including pemetrexed [53, 54], docetaxel [55, 56], afatinib [57, 58], carboplatin plus paclitaxel [59], cisplatin plus gemcitabine [60] and mFOLFOX-6 (leucovorin calcium [folinic acid], 5-fluorouracil, and oxaliplatin) [61]. None of the studies demonstrated a significant pharmacokinetic drug–drug interaction between nintedanib and the other therapies.
5.4 Pirfenidone
Pirfenidone is another medication approved for patients with IPF. A dedicated pharmacokinetic drug–drug interaction study investigated the combination of nintedanib and pirfenidone in two groups of patients with IPF using an intra-individual comparison of the pharmacokinetic data [62]. In a group of patients naïve to nintedanib or pirfenidone (n = 20), nintedanib plasma exposure was similar when coadministered with pirfenidone (titrated to 801 mg three times daily) in comparison to administration of nintedanib alone (Fig. 4). In a second group of patients treated with pirfenidone (n = 17), treatment with nintedanib (150 mg twice daily) for 7 days had no effect on the pharmacokinetics of pirfenidone. Further studies with this combination therapy in patients with IPF [63, 64] support the findings of this study that there is no evidence of a relevant pharmacokinetic drug–drug interaction between nintedanib and pirfenidone when administered in combination [20]. Only an exploratory safety study in Japanese patients with IPF showed a more pronounced trend towards decreased nintedanib exposure (by 32% for AUC and by 41% for Cmax) [42], but these data should be treated with caution as exposure to nintedanib could only be compared between patient groups (inter-individually) and the number of patients included in the pharmacokinetic evaluation was low (< 12 per treatment group).
5.5 Bosentan
Nintedanib is also being investigated as a treatment for patients with SSc-associated interstitial lung disease (SSc-ILD) [65], which may be associated with pulmonary arterial hypertension. Bosentan, a dual endothelin receptor antagonist, is used in SSc-ILD patients to treat pulmonary hypertension and to prevent digital ulcers. As such, it might be frequently combined with nintedanib. Since bosentan is an inducer of CYP3A4 and CYP2C9, the potential for induction of P-gp can be assumed. The pharmacokinetics of nintedanib were studied in combination with bosentan [66]. In healthy subjects, pre-treatment with bosentan (125 mg twice daily) for 7 days had no effect on the single-dose pharmacokinetics of nintedanib 150 mg (Fig. 4).
5.6 Medications that Alter Haemostasis
In view of its mechanism of action (VEGFR inhibition), nintedanib has the potential for an increased risk of bleeding when used with medications that can alter haemostasis (e.g. antiplatelet therapy, anticoagulants). In the INPULSIS trials, low-dose antiplatelet therapy and prophylactic anticoagulation was permitted [8]. Patients at known risk for bleeding or treated with full-dose therapeutic anticoagulation were excluded. The frequency of patients who experienced bleeding was slightly higher with nintedanib (10.3%) than with placebo (7.8%); non-serious epistaxis and contusion represented the most frequent bleeding events [19, 20, 67]. Serious bleeding events occurred with low and similar frequencies in both groups (placebo group 1.4%, nintedanib group 1.3%). Based on these results, the prescribing information for nintedanib states that patients with a known risk of bleeding should be treated with nintedanib only if the anticipated benefit outweighs the potential risk, i.e. an individual risk assessment should be performed for each patient [19, 20]. Patients on full anticoagulation should be monitored for bleeding and have their anticoagulant treatment adjusted as necessary [19].
6 Effect of Other Intrinsic and Extrinsic Factors on Nintedanib Pharmacokinetics
The potential effects of various intrinsic and extrinsic factors on the pharmacokinetics of nintedanib were investigated by two successive PopPK analyses, one that included a combined population of patients with NSCLC and IPF [49], and a second that evaluated IPF patients only [50].
In each PopPK model, age, body weight, smoking and Asian race (with different effect sizes in different subpopulations) were statistically significant covariates influencing nintedanib exposure. In the IPF analysis, serum lactate dehydrogenase (LDH) levels were identified as an additional factor significantly influencing nintedanib plasma concentrations. The effects of these covariates on nintedanib exposure (AUC during dosing interval τ at steady state [AUCτ,ss]) in the two PopPK analyses are depicted in Fig. 5. Exposure to nintedanib increases linearly with age and decreases with increasing body weight and in current smokers. It also linearly increases with increasing LDH levels in IPF patients only. What physiological processes trigger the LDH effect and whether the relationship can be transferred to other populations is not known. LDH levels should therefore be regarded as a rather non-specific marker with questionable predictive power. Each of the covariates had a small to moderate influence on nintedanib exposure. Based on the estimates from both PopPK analyses, the population mean exposure to nintedanib was 33–50% higher in Chinese, Indian and Taiwanese patients and 16% higher in Japanese patients, while it was 16–22% lower in Koreans than in Caucasians (body weight corrected). For current smokers, the mean exposure was 21% lower than in ex- or non-smokers. Other than ethnic origin, the ratios of AUCτ,ss for nintedanib for each factor were generally within the 80–125% range when varying those covariates individually within the observed extreme values (5th and 95th percentiles of baseline values for continuous covariates) as compared to a typical patient (defined by the mode/median of the baseline covariate values).

Effects of the covariates age, body weight, smoking status, lactate dehydrogenase and ethnic origin on nintedanib exposure (AUCτ,ss) based on two PopPK analyses in NSCLC and IPF patients [49, 50]. Ratios (point estimates and 95% CIs based on bootstrap analysis) of nintedanib population mean exposure (AUCτ,ss) predicted by final PopPK models for different covariates compared with a typical patient receiving nintedanib treatment are shown. The solid vertical line indicates the population mean for the typical patient, and the shaded area is the 90% prediction interval reflecting inter-patient variability. The vertical dotted lines indicate the bioequivalence limits (80–125%). 5th and 95th percentiles of the baseline values observed in the analysed population are shown for age and body weight. AUCτ,ss area under the drug plasma concentration–time curve at steady-state over a uniform dosing interval τ, CI confidence interval, IPF idiopathic pulmonary fibrosis, NSCLC non-small cell lung carcinoma, PopPK population pharmacokinetic
Simulations revealed that none of the individual covariate effects alone identified in the PopPK analyses caused a change in exposure that exceeded the observed inter-patient variability range of nintedanib or resulted in exposure changes of more than 50% relative to a typical patient. More pronounced increases in simulated exposure were only found when considering more than one covariate. Overall, these changes in exposure are considered not sufficient to warrant a priori dose adjustment on the basis of ethnic origin, body weight, age, smoking or a combination of these factors [19].
Patient sex (body weight corrected), patient population (NSCLC vs. IPF) and mild or moderate renal impairment (based on CLCR) had no obvious effect on the pharmacokinetics of nintedanib.
7 Clinical Pharmacodynamics
7.1 Effect on Cardiac Repolarisation
The effect of a single 200 mg dose of nintedanib and multiple 200 mg doses of nintedanib (administered twice daily for 15 days) on cardiac repolarisation was studied in 64 patients with advanced renal cell carcinoma (RCC) [47]. The upper limits of the two-sided 90% CIs for the adjusted mean time-matched QT interval using the Fridericia correction method (QTcF) from baseline to days 1 and 15 (primary electrocardiogram [ECG] endpoint) were well below the regulatory threshold of 10 ms at all times. The largest mean time-matched increase of QTcF at steady state was 3.1 ms (two-sided 90% CI − 0.2 to 6.4). No changes in the mean corrected QT (QTc) interval of > 60 ms were detected on days 1 or 15 and no patient had new-onset prolongation of the QTcF interval or an uncorrected QT interval of > 500 ms on days 1 or 15. There was no correlation between pharmacokinetic parameters and ECG variables and no treatment-related morphological ECG abnormalities were detected. Thus, the data indicate that nintedanib does not affect the QTc interval and, therefore, no specific ECG monitoring is indicated clinically.
7.2 Dynamic Contrast-Enhanced Magnetic Resonance Imaging
Dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) is an established method for assessing the antivascular effects of antiangiogenic inhibitors [68]. Two studies used DCE-MRI to assess the size (bi-dimensional diameter) and vascular properties (vascular permeability, vascular density, regional flow) of suitable target lesions under nintedanib therapy in patients with advanced cancer [25, 48]. In both studies, changes consistent with an antivascular pharmacodynamic effect (decreased vascular permeability and tumour blood flow) were observed after nintedanib treatment. These included changes in key kinetic parameters such as Ktrans (transfer constant), kep (reflux constant) or iAUC60 (initial AUC for the first 60 s) of a gadolinium chelate complex. The effects were not clearly dose dependent, but most responses were seen at doses of ≥ 200 mg. Effects were already seen 24–48 h after the first drug intake and were preserved or even increased after continuous treatment with nintedanib over several weeks. DCE-MRI response was associated with disease stabilisation.
7.3 Exposure–Response Analyses
Exposure–response modelling of nintedanib in the treatment of patients with IPF was performed [69, 70] using data from 1403 patients who participated in the phase II TOMORROW and the phase III INPULSIS trials [8, 21] and received nintedanib doses of 50–150 mg twice daily or placebo. The association between nintedanib exposure (based on trough concentrations) and efficacy (based on the annual FVC decline in patients over time) and the relationship between nintedanib exposure and safety (based on the probability of experiencing diarrhoea or transaminase elevations ≥ 3 × ULN) was investigated.
The exposure–efficacy relationship was described by a linear disease progression model with a disease-modifying drug effect on the rate of FVC decline, indicating a slowdown of FVC decline with nintedanib treatment. A maximum effect (Emax) relationship between nintedanib exposure and FVC decline was established using observed and PopPK model predicted trough concentrations as exposure metrics. The trough concentration producing 80% of the Emax (EC80) on FVC decline was estimated to be 10–13 ng/mL, which was similar to the median trough concentration achieved with the therapeutic dose of nintedanib 150 mg twice daily (10 ng/mL) during the phase III trials (Fig. 6). A stepwise covariate selection approach was applied to explore factors influencing the baseline FVC, natural disease progression or the drug-dependent treatment effect. None of the tested patient characteristics (age, height, sex, smoking status, Asian subpopulations [including Chinese, Taiwanese, Korean, Indian and Japanese ethnicity], presence of honeycombing, diarrhoea or FVC percentage predicted at baseline) was a distinct predictor of the treatment effect of nintedanib.

Nintedanib exposure (trough concentrations [Cpre,ss]) versus predicted annual rate of FVC decline derived from a linear disease progression model before covariate analysis (with 90% CIs from bootstrap analysis) based on model predicted Cpre,ss levels [69]. For comparison, the horizontal line shows predicted Cpre,ss values (median, 5th and 95th percentiles) for patients with idiopathic pulmonary fibrosis starting with a dose of 150 mg bid in the phase II/III trials [8, 21]. bid twice daily, CI confidence interval, Cpre,ss pre-dose drug concentration in plasma at steady state, EC50 concentration of drug producing 50% of maximum effect, EC80 concentration of drug producing 80% of maximum effect, FVC forced vital capacity
The safety data were described by parametric time-to-first event models [70] using the same exposure metrics as for exposure–efficacy correlations. A reliable association between nintedanib exposure and the risk of developing diarrhoea (any severity grade) could not be established, with nintedanib dose shown to be a better predictor for diarrhoea than exposure. This conclusion was based on several findings. First, an exposure–diarrhoea model (using a sigmoidal Emax relationship) was not superior to a model using dose as the predictor of diarrhoea risk. Second, visual predictive checks based on exposure–diarrhoea models and further exploratory analyses indicated that the diarrhoea risk for patients with the same exposure but receiving different doses was not comparable. Finally, results from covariate analyses suggested that patient populations with different exposure levels seemed to have a comparable diarrhoea risk. Therefore, no change in diarrhoea risk might be expected for patients with altered nintedanib exposure due to co-medications or individual patient characteristics. However, nintedanib exposure tended to be associated with a higher risk of developing liver enzyme elevations (defined as alanine transaminase and/or aspartate transaminase elevation ≥ 3 × ULN). The observed relationship was weak, and the analysis was limited by the low number of observed events (i.e. 38 safety events out of 895 nintedanib-treated patients). The effect of covariates on the relationship between exposure and liver enzyme elevations has not been investigated at present. Overall, the exposure–response analyses provide a modelling framework for a quantitative benefit–risk assessment in patients with IPF with altered nintedanib exposure due to co-medication or patient characteristics. The analyses support the 150 mg twice daily starting dose of nintedanib resulting in efficacious exposure levels in the majority of IPF patients (with the median exposure level just approaching the plateau of the established Emax relationship). Due to a potentially higher frequency of adverse events (e.g. in terms of liver enzyme elevations), close monitoring for tolerability is warranted for patients with elevated nintedanib exposure (e.g. due to Asian race, low body weight, older age or combinations of these risk factors).
8 Summary/Conclusions
In patients with advanced solid tumours or IPF, Cmax values of nintedanib occur approximately 2–4 h after oral dosing and decline afterwards in an at least bi-exponential manner. Nintedanib is metabolised in the liver and intestine primarily by esterases and predominantly excreted in the faeces and about 1% in urine. The terminal half-life is about 10–15 h, justifying a twice-daily dosing regimen. Over the investigated dose range of 50–450 mg once daily and 150–300 mg twice daily the pharmacokinetics of nintedanib are dose proportional. Nintedanib is a high clearance drug showing a high volume of distribution. The main covariate for nintedanib exposure is hepatic function. Other covariates include ethnicity, body weight, age and smoking, while factors such as sex, patient population (NSCLC vs. IPF) and renal function have no significant influence.
Predicted exposure increases for Asian race subgroups, patients with low body weight or increased age are within the variability range of nintedanib exposure, suggesting that there is no need for a priori dose adjustment, but close monitoring for tolerability is warranted for these patients.
Based on the detected increase in nintedanib exposure in hepatically impaired patients, a dose reduction from 150 mg to 100 mg twice daily is recommended for patients with IPF associated with mild hepatic impairment (Child-Pugh A) [19, 20], while patients with NSCLC-associated mild hepatic impairment should be closely monitored and the dose adapted based on tolerability [22]. Administration of nintedanib in patients with moderate or severe hepatic impairment is not recommended. At the approved nintedanib doses of 150 and 200 mg twice daily, there is very low drug–drug interaction potential, especially with drugs metabolised via CYP. Only concomitant treatment with strong inhibitors or inducers of P-gp influences the pharmacokinetics of nintedanib, and caution is therefore advised with this combination. At an investigated dose of 200 mg, nintedanib does not have proarrhythmic potential. It can be combined with various IPF (e.g. bosentan and pirfenidone) and chemotherapy drugs.
In conclusion, the studies discussed in this review support the use of nintedanib as an efficacious treatment for patients in its approved indications with an acceptable tolerability profile.
References
Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824.
Martinez FJ, Collard HR, Pardo A, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. 2017;3:17074.
Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–52.
Selman M, King TE, Pardo A, for the American Thoracic Society Study Group, European Respiratory Society Study Group, and American College of Chest Physicians Study Group. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–51.
Chaudhary NI, Roth GJ, Hilberg F, et al. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. 2007;29:976–85.
Wollin L, Wex E, Pautsch A, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1434–45.
King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–92.
Richeldi L, du Bois RM, Raghu G, INPULSIS Trial Investigators, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82.
Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17:1359–70.
Wulkersdorfer B, Zeitlinger M, Schmid M. Pharmacokinetic aspects of vascular endothelial growth factor tyrosine kinase inhibitors. Clin Pharmacokinet. 2016;55:47–77.
Ferrara N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010;21:21–6.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307.
Roth GJ, Binder R, Colbatzky F, et al. Nintedanib: from discovery to the clinic. J Med Chem. 2015;58:1053–63.
Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68:4774–82.
Roth GJ, Heckel A, Colbatzky F, et al. Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120). J Med Chem. 2009;52:4466–80.
Wollin L, Maillet I, Quesniaux V, et al. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther. 2014;349:209–20.
FDA Center for Drug Evaluation and Research. Nintedanib pharmacology NDA review, Feb 2014. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205832Orig1s000PharmR.pdf. Accessed 15 Mar 2019.
Beyer C, Distler JH. Tyrosine kinase signaling in fibrotic disorders: translation of basic research to human disease. Biochim Biophys Acta. 2013;1832:897–904.
Boehringer Ingelheim. OFEV® (nintedanib) capsules, for oral use [package insert]. Ridgefield: Boehringer Ingelheim Pharmaceuticals; 2018. http://docs.boehringer-ingelheim.com/Prescribing.Information/PIs/Ofev/ofev.pdf. Accessed 15 Mar 2019.
European Medicines Agency. Ofev. Summary of product characteristics. 26 Nov 2018. https://www.ema.europa.eu/documents/product-information/ofev-epar-product-information_en.pdf. Accessed 19 Feb 2019.
Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–87.
European Medicines Agency. Vargatef. Summary of product characteristics. 3 Aug 2018. https://www.ema.europa.eu/documents/product-information/vargatef-epar-product-information_en.pdf. Accessed 19 Feb 2019.
Reck M, Kaiser R, Mellemgaard A, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014;15:143–55.
FDA Center for Drug Evaluation and Research. Nintedanib clinical pharmacology NDA review, Feb 2014. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205832Orig1s000ClinPharmR.pdf. Accessed 15 Mar 2019.
Mross K, Stefanic M, Gmehling D, et al. Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors. Clin Cancer Res. 2010;16:311–9.
Redente EF, Aguilar MA, Black BP, et al. Nintedanib reduces pulmonary fibrosis in a model of rheumatoid arthritis-associated interstitial lung disease. Am J Physiol Lung Cell Mol Physiol. 2018;314:L998–1009.
Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390:1685–99.
Huang J, Beyer C, Palumbo-Zerr K, et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis. 2016;75:883–90.
Huang J, Maier C, Zhang Y, et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis. 2017;76:1941–8.
Kutluk Cenik B, Ostapoff KT, Gerber DE, et al. BIBF 1120 (nintedanib), a triple angiokinase inhibitor, induces hypoxia but not EMT and blocks progression of preclinical models of lung and pancreatic cancer. Mol Cancer Ther. 2013;12:992–1001.
European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) assessment report for Vargatef (nintedanib), 25 September 2014. https://www.ema.europa.eu/en/documents/assessment-report/vargatef-epar-public-assessment-report_en.pdf. Accessed 15 Mar 2019.
European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) assessment report for Ofev (nintedanib), 20 November 2014. https://www.ema.europa.eu/en/documents/assessment-report/ofev-epar-public-assessment-report_en.pdf. Accessed 15 Mar 2019.
Therapeutic Goods Administration (Australia). Extract from the clinical evaluation report for nintedanib esilate, April 2015. https://www.tga.gov.au/sites/default/files/auspar-nintedanib-esilate-160208-cer.pdf. Accessed 15 Mar 2019.
Stopfer P, Rathgen K, Bischoff D, et al. Pharmacokinetics and metabolism of BIBF 1120 after oral dosing to healthy male volunteers. Xenobiotica. 2011;41:297–311.
Boehringer Ingelheim. Safety and pharmacokinetics/bioavailability of a single dose of 150 mg BIBF 1120 administered as soft gelatine capsules with and without food to healthy male volunteers in an open, randomised, intra-individual crossover comparison design, phase I trial. Trial no. 1199.17. https://trials.boehringer-ingelheim.com/public/trial_results_documents/1199/1199.17_U06-1411-02.pdf. Accessed 15 Mar 2019.
Boehringer Ingelheim. Safety and relative bioavailability of a single dose of 150 mg BIBF 1120 administered as soft gelatine capsules charge 1 compared to BIBF 1120 soft gelatine capsules charge 2 compared to BIBF 1120 administered as drinking solution following oral administration to healthy male volunteers in an open, randomised, intra-individual, crossover comparison design. Trial no. 1199.21. https://trials.boehringer-ingelheim.com/public/trial_results_documents/1199/1199.21_119921U717362COpdf.pdf#page=1. Accessed 15 Mar 2019.
Boehringer Ingelheim. Safety and tolerability of single rising doses of 1 mg, 3 mg, 10 mg and 20 mg of BIBF 1120 as intravenous infusion (single-blind, placebo controlled at each dose group) and absolute bioavailability of 100 mg BIBF 1120 as soft gelatine capsule (intra-individual comparison). Trial no. 1199.75. http://www.trials.boehringer-ingelheim.com/public/trial_results_documents/1199/1199.75_U10-1400-02.pdf. Accessed 15 Mar 2019.
Dallinger C, Trommeshauser D, Marzin K, et al. Pharmacokinetic properties of nintedanib in healthy volunteers and patients with advanced cancer. J Clin Pharmacol. 2016;56:1387–94.
Marzin K, Kretschmar G, Luedtke D, et al. Pharmacokinetics of nintedanib in subjects with hepatic impairment. J Clin Pharmacol. 2018;58:357–63.
Palmer DH, Ma YT, Peck-Radosavljevic M, et al. A multicentre, open-label, phase-I/randomised phase-II study to evaluate safety, pharmacokinetics, and efficacy of nintedanib vs. sorafenib in European patients with advanced hepatocellular carcinoma. Br J Cancer. 2018;118:1162–8.
Yen CJ, Kim TY, Feng YH, et al. A phase I/randomized phase II study to evaluate the safety, pharmacokinetics, and efficacy of nintedanib versus sorafenib in Asian patients with advanced hepatocellular carcinoma. Liver Cancer. 2018;7:165–78.
Ogura T, Taniguchi H, Azuma A, et al. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1382–92.
Stopfer P, Roth W, Mross K, et al. Pharmacokinetic characterization of BIBF 1120, an orally active triple angiokinase inhibitor (VEGFR, PDGFR, FGFR) in advanced cancer patients [abstract no. 73]. Eur J Cancer. 2006;4:26.
Kropff M, Kienast J, Bisping G, et al. An open-label dose-escalation study of BIBF 1120 in patients with relapsed or refractory multiple myeloma. Anticancer Res. 2009;29:4233–8.
Okamoto I, Kaneda H, Satoh T, et al. Phase I safety, pharmacokinetic, and biomarker study of BIBF 1120, an oral triple tyrosine kinase inhibitor in patients with advanced solid tumors. Mol Cancer Ther. 2010;9:2825–33.
Reck M, Kaiser R, Eschbach C, et al. A phase II double-blind study to investigate efficacy and safety of two doses of the triple angiokinase inhibitor BIBF 1120 in patients with relapsed advanced non-small-cell lung cancer. Ann Oncol. 2011;22:1374–81.
Eisen T, Shparyk Y, Macleod N, et al. Effect of small angiokinase inhibitor nintedanib (BIBF 1120) on QT interval in patients with previously untreated, advanced renal cell cancer in an open-label, phase II study. Investig New Drugs. 2013;31:1283–93.
Lee CP, Taylor NJ, Attard G, et al. Phase I study of nintedanib incorporating dynamic contrast-enhanced magnetic resonance imaging in patients with advanced solid tumors. Oncologist. 2015;20:368–9.
Schmid U, Liesenfeld KH, Fleury A, et al. Population pharmacokinetics of nintedanib, an inhibitor of tyrosine kinases, in patients with non-small cell lung cancer or idiopathic pulmonary fibrosis. Cancer Chemother Pharmacol. 2018;81:89–101.
Schmid U, Doege C, Dallinger C, et al. Population pharmacokinetics of nintedanib in patients with idiopathic pulmonary fibrosis. Pulm Pharmacol Ther. 2018;48:136–43.
Pugh RN, Murray-Lyon IM, Dawson JL, et al. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–9.
Luedtke D, Marzin K, Jungnik A, et al. Effects of ketoconazole and rifampicin on the pharmacokinetics of nintedanib in healthy subjects. Eur J Drug Metab Pharmacokinet. 2018;43:533–41.
Ellis PM, Kaiser R, Zhao Y, et al. Phase I open-label study of continuous treatment with BIBF 1120, a triple angiokinase inhibitor, and pemetrexed in pretreated non-small cell lung cancer patients. Clin Cancer Res. 2010;16:2881–9.
Daga H, Takeda K, Okada H, et al. Phase I study of nintedanib in combination with pemetrexed as second-line treatment of Japanese patients with advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2015;76:1225–33.
Bousquet G, Alexandre J, Le Tourneau C, et al. Phase I study of BIBF 1120 with docetaxel and prednisone in metastatic chemo-naive hormone-refractory prostate cancer patients. Br J Cancer. 2011;105:1640–5.
Okamoto I, Miyazaki M, Takeda M, et al. Tolerability of nintedanib (BIBF 1120) in combination with docetaxel: a phase 1 study in Japanese patients with previously treated non-small-cell lung cancer. J Thorac Oncol. 2015;10:346–52.
Gordon MS, Springett GM, Su YB, et al. A Phase I dose-escalation study of afatinib combined with nintedanib in patients with advanced solid tumors. Future Oncol. 2015;11:1479–91.
Bahleda R, Hollebecque A, Varga A, et al. Phase I study of afatinib combined with nintedanib in patients with advanced solid tumours. Br J Cancer. 2015;113:1413–20.
Doebele RC, Conkling P, Traynor AM, et al. A phase I, open-label dose-escalation study of continuous treatment with BIBF 1120 in combination with paclitaxel and carboplatin as first-line treatment in patients with advanced non-small-cell lung cancer. Ann Oncol. 2012;23:2094–102.
Forster M, Hackshaw A, De Pas T, et al. A phase I study of nintedanib combined with cisplatin/gemcitabine as first-line therapy for advanced squamous non-small cell lung cancer (LUME-Lung 3). Lung Cancer. 2018;120:27–33.
Van Cutsem E, Prenen H, D’Haens G, et al. A phase I/II, open-label, randomised study of nintedanib plus mFOLFOX6 versus bevacizumab plus mFOLFOX6 in first-line metastatic colorectal cancer patients. Ann Oncol. 2015;26:2085–91.
Richeldi L, Fletcher S, Adamali H, et al. No relevant pharmacokinetic drug–drug interaction between nintedanib and pirfenidone. Eur Respir J. 2019;53:1801060.
Vancheri C, Kreuter M, Richeldi L, et al. Nintedanib with add-on pirfenidone in idiopathic pulmonary fibrosis: results of the INJOURNEY trial. Am J Respir Crit Care Med. 2018;197:356–63.
Flaherty KR, Fell CD, Huggins JT, et al. Safety of nintedanib added to pirfenidone treatment for idiopathic pulmonary fibrosis. Eur Respir J. 2018;52:1800230.
Distler O, Brown KK, Distler JHW, SENSCIS trial investigators, et al. Design of a randomised, placebo-controlled clinical trial of nintedanib in patients with systemic sclerosis-associated interstitial lung disease (SENSCIS). Clin Exp Rheumatol. 2017;35(Suppl 106):75–81.
Wind S, Simons G, Bertulis J, et al. Coadministration of bosentan has no effect on the pharmacokinetics of nintedanib [abstract no. FRI0411]. Ann Rheum Dis. 2017;76(Suppl 2):642–3. https://doi.org/10.1136/annrheumdis-2017-eular.5849.
Corte T, Bonella F, Crestani B, et al. Safety, tolerability and appropriate use of nintedanib in idiopathic pulmonary fibrosis. Respir Res. 2015;16:116.
Hylton N. Dynamic contrast-enhanced magnetic resonance imaging as an imaging biomarker. J Clin Oncol. 2006;24:3293–8.
Schmid U, Weber B, Dallinger C, et al. Relationship between nintedanib exposure, patient characteristics and clinical efficacy in patients with idiopathic pulmonary fibrosis [abstract]. Am J Respir Crit Care Med. 2016;193:A5000.
Schmid U, Weber B, Dallinger C, et al. Relationship between nintedanib exposure and adverse events in patients with idiopathic pulmonary fibrosis [abstract]. Eur Respir J. 2016. https://doi.org/10.1183/13993003.congress-2016.OA49631.
Acknowledgements
The authors would like to thank their clinical oncology colleague Rolf Kaiser, MD, for his review and input to the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funds were used in the preparation of this manuscript.
Conflict of interest
Sven Wind, Ulrike Schmid, Matthias Freiwald, Kristell Marzin, Ralf Lotz, Thomas Ebner, Peter Stopfer and Claudia Dallinger are full-time employees of Boehringer Ingelheim.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the independent ethics committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual human participants included in the described studies.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wind, S., Schmid, U., Freiwald, M. et al. Clinical Pharmacokinetics and Pharmacodynamics of Nintedanib. Clin Pharmacokinet 58, 1131–1147 (2019). https://doi.org/10.1007/s40262-019-00766-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00766-0