
Abstract
Background and Objectives
Upadacitinib is a selective Janus kinase (JAK) 1 inhibitor being developed as an orally administered treatment for patients with moderate to severe rheumatoid arthritis (RA) and other autoimmune disorders. These analyses characterized the population pharmacokinetics of upadacitinib across phase I–III clinical trials using data for immediate-release (IR) and extended-release (ER) formulations.
Methods
Pharmacokinetic data from 4170 subjects taking IR doses of 1–48 mg and ER doses of 7.5–30 mg across 12 studies spanning phase I–III clinical trials, with a total of 29,372 upadacitinib plasma concentrations, were analyzed using non-linear mixed-effects modeling. The model was evaluated using bootstrap analyses and visual predictive checks.
Results
A two-compartment model with first-order absorption with lag time for the IR formulation, mixed zero- and first-order absorption with lag time for the ER formulation, and linear elimination, adequately described upadacitinib plasma concentration–time profiles. Population estimates of upadacitinib apparent oral clearance and steady-state volume of distribution in healthy volunteers for the ER formulation were 53.7 L/h and 294 L, respectively. The relative bioavailability of the ER formulation compared with the IR formulation was estimated to be 76.2%. Statistically significant covariates were patient population (RA subjects vs. healthy subjects), creatinine clearance, and baseline bodyweight on apparent clearance (CL/F) and bodyweight on volume of distribution of the central compartment (Vc/F). The intersubject variability for upadacitinib CL/F and Vc/F were estimated to be 21% and 24%, respectively, in the phase I studies, and 37% and 53%, respectively, in the phase II/III studies. Upadacitinib area under the concentration–time curve (AUC) was estimated to be only 5% higher or lower for RA patients who were < 60 or > 100 kg, respectively, relative to subjects with a bodyweight of 60–100 kg. RA subjects with mild or moderate renal impairment had 13% and 26% higher AUC, respectively, compared with RA subjects with normal renal function. Sex, race, concomitant use of pH-modifying drugs, moderate cytochrome P450 3A inhibitors, or methotrexate use had no effect on upadacitinib exposure.
Conclusions
A robust population pharmacokinetic model was developed for upadacitinib using a large dataset from phase I–III clinical trials in healthy volunteers and subjects with RA. None of the identified covariates had a clinically meaningful effect on upadacitinib exposures. The model is appropriate to use for simulations and to evaluate the exposure–response relationship of upadacitinib.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Upadacitinib is a novel Janus kinase 1 inhibitor being developed for the treatment of patients with moderate to severe rheumatoid arthritis (RA). A population pharmacokinetic model was developed for upadacitinib using data from phase I–III clinical trials in healthy volunteers and subjects with RA. |
A two-compartment model with first-order absorption with lag time for the immediate-release formulation, mixed zero- and first-order absorption with lag time for the extended-release formulation, and linear elimination well-described upadacitinib plasma concentration versus time data. |
Bodyweight and mild or moderate renal impairment had statistically significant but non-clinically relevant effects on upadacitinib exposures. |
Sex, race, concomitant use of pH-modifying agents, moderate cytochrome P450 3A inhibitors, or methotrexate use had no effect on upadacitinib pharmacokinetics. |
1 Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disorder affecting approximately 0.5–1% of adults in industrialized countries [1], and is most common in women and elderly people. The Janus kinase/signal transducers and activators of transcription (JAK-STAT) pathway plays a major role in the pathogenesis and progression of RA, making it an attractive target for the development of RA treatments [2]. The JAK family includes four tyrosine kinases [JAK1, JAK2, JAK3, and the non-receptor tyrosine-protein kinase 2 (Tyk2)], which play key roles in cytokine signaling. These enzymes play an important role not only in proinflammatory cytokine signaling but also in regulation of important physiological functions of the immune system, as well of erythropoiesis [3, 4]. Activation of the JAK-STAT pathway leads to increased gene expression of proinflammatory cytokines, such as interleukin (IL)-6, which contribute to the chronic inflammation of RA.
Upadacitinib is a novel selective JAK 1 inhibitor being developed as an oral treatment of RA and other autoimmune diseases. Based on in vitro cellular assays, the selectivity of upadacitinib for JAK1 is approximately 60- and more than 100-fold greater over JAK2 and JAK3, respectively [5]. Selective inhibition of JAK1 leads to selective modulation of the subset of proinflammatory cytokines in the JAK-STAT pathway, which can potentially improve the benefit–risk profile compared with less-selective JAK inhibitors [6,7,8,9].
Upadacitinib was evaluated in early phase I studies and in phase II studies in RA in the form of an immediate-release (IR) formulation. Single doses of 1–48 mg and multiple doses of 3–24 mg twice-daily using the IR formulation were evaluated across these studies [10, 11]. Using the IR formulation, upadacitinib had a functional half-life of 3–4 h, which supported twice-daily administration of the IR formulation. To simplify the upadacitinib dosing regimen in patients in phase III studies, an extended-release (ER) tablet formulation was developed that enables once-daily dosing [11, 12]. Upadacitinib ER doses of 15 and 30 mg once daily were evaluated in global phase III studies in subjects with RA [13].
The pharmacokinetics of upadacitinib were previously characterized using non-compartmental analyses of individual phase I studies for the IR and ER formulations [10, 12], as well as using a population pharmacokinetic analysis across phase I and RA phase II studies after administration of the IR formulation [14]. Overall, upadacitinib showed linear pharmacokinetics over the range of doses evaluated in phase I and II studies. Upadacitinib peak plasma concentrations (Cmax) were reached within 1–2 h after IR dosing, and 2–4 h after ER dosing, followed by a bi-exponential decline of plasma concentrations. The terminal half-life of upadacitinib ranged from 6 to 16 h following administration of the IR or ER formulations. Upadacitinib doses of 15 and 30 mg once daily using the ER formulation provided equivalent daily area under the concentration–time curve (AUC) and comparable Cmax and minimum plasma concentration (Cmin) to 6 mg twice daily and 12 mg twice daily, respectively, using the IR formulation [12].
Upadacitinib is a non-sensitive substrate for cytochrome P450 (CYP) 3A, and in vitro data indicate a potential minor contribution to upadacitinib metabolism by CYP2D6. Approximately 20% of the upadacitinib dose is eliminated unchanged in urine. In a population analysis of phase I and RA phase II studies [14], CYP2D6 metabolic phenotype had no effect on upadacitinib apparent oral clearance. Additionally, sex, renal impairment (mild or moderate), and bodyweight had statistically significant but non-clinically relevant effects on upadacitinib pharmacokinetics. Upadacitinib typical oral clearance in RA patients was 24% lower compared with healthy subjects.
The efficacy and safety of upadacitinib have been evaluated in two phase IIb studies using the IR formulation, five global phase III studies using the ER formulation, and one regional phase IIb/III study in Japan using the ER formulation [11, 15]. Results from the five phase III studies demonstrated that upadacitinib, administered as monotherapy or in combination with conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), showed rapid and significant improvements in clinical signs and symptoms in subjects with moderate to severe RA compared with placebo or active comparators [13, 16,17,18,19,20].
Given the additional large data that recently became available from the global phase III and Japanese phase IIb/III studies using the upadacitinib ER formulation in subjects with RA, an update of the characterization of upadacitinib pharmacokinetics was warranted. Such a large analysis dataset provides greater power for characterizing the effect of covariates on upadacitinib pharmacokinetics. Additionally, this is the first characterization of upadacitinib pharmacokinetics from the ER formulation in RA patients. In the present analyses, available upadacitinib pharmacokinetic data from 12 studies spanning phase I–III clinical trials utilizing IR and ER formulations were combined and analyzed simultaneously using non-linear mixed-effects modeling. Various covariates of interest were evaluated to determine their potential effect on upadacitinib exposures.
2 Methods
2.1 Study Design and Population
Data from four phase I studies that enrolled healthy volunteers and RA patients, two phase II studies in RA patients, five phase III studies in RA patients, and one regional phase IIb/III study in Japanese RA patients were utilized in the population pharmacokinetic analyses. Healthy volunteers received upadacitinib as monotherapy; subjects with active RA received upadacitinib as monotherapy or in combination with methotrexate or other csDMARDs. An overview of study design, treatment, population, and pharmacokinetic sampling for the 12 studies included in these analyses is shown in Table 1. In these studies, upadacitinib was administered at doses of 1–48 mg using the IR formulation and 7.5–30 mg using the ER formulation. The studies were designed according to good clinical practice guidelines, the Declaration of Helsinki, and applicable regulations, and were approved by Institutional Review Boards. All participants provided written informed consent.
Blood samples for pharmacokinetic analyses were collected at a variety of time points across the 12 studies. Plasma upadacitinib concentrations were quantified using validated liquid chromatography methods with tandem mass spectrometric detection, as previously described [10]. Upadacitinib concentration values below the lower limit of quantification (LLOQ) observed during the studies were set to one-half of the LLOQ [21]; the second and all subsequent concentrations below the LLOQ recorded after the last dose were censored in the analyses. In addition, an outlier identification and exclusion rule was applied to avoid bias in the population and individual pharmacokinetic parameter estimates. Details of the outlier rules are provided in the electronic supplementary Methods. Only 0.9% of all dataset concentrations were flagged as clear outliers that likely resulted from inaccurate dosing records and were hence excluded from the analysis. To ensure that the exclusion rules applied did not bias the population pharmacokinetic model parameter estimates, the final model developed with the dataset applying the exclusion rules outlined above was re-run on the full dataset including the outliers to evaluate the impact of outliers on parameter estimates.
2.2 Pharmacokinetic Analysis
2.2.1 Model Development
A non-linear mixed-effects modeling approach was utilized to analyze the upadacitinib data. NONMEM® software (version 7.4.2) compiled with a GNU Fortran compiler was used for the analyses. The pharmacokinetic models were fit to the data using the first-order conditional estimation with interaction (FOCEI) method. The population pharmacokinetic model was built in a stepwise manner. An appropriate structural model was developed first and terms were added for intersubject variability (ISV) and residual error, and the significance of potential covariates was then tested. Model development was guided by goodness-of-fit plots, likelihood ratio tests [22], plausibility of parameter estimates, and previous learnings of upadacitinib pharmacokinetics [14].
Intersubject variability in pharmacokinetic parameters was modeled using an exponential error model (Eq. 1):
where Pi is the parameter estimate for ith individual, and ηi is the deviation of Pi from the population estimate θ. ηi is assumed to be normally distributed, with a mean of 0 and variance of ω2.
Residual unexplained variability was modeled using a combination of additive and proportional error terms (Eq. 2):
where Cij is the measured plasma concentration in subject i at time j, Ĉij is the corresponding model-predicted plasma concentration, and ε1ij and ε2ij are the proportional and additive components of the residual random error. The ε1ij and ε2ij values were assumed to be normally distributed, with a mean of 0 and variances of σ12 and σ22, respectively: εn ~ N(0, σn2).
Covariates assessed for their influence on pharmacokinetic parameters included:
-
For apparent clearance (CL/F): baseline serum bilirubin concentration, baseline creatinine clearance (CrCl), baseline total bodyweight, age, baseline aspartate aminotransaminase (AST), baseline alanine aminotransferase (ALT), baseline Disease Activity Score (DAS) 28–C-reactive protein (CRP), baseline high-sensitivity CRP (hsCRP), sex, race (White, Black, Hispanic, Asian), country (Taiwan, Japan, China, Korea), concomitant use of methotrexate, concomitant use of any pH-modifying medications, concomitant use of antacids, concomitant use of H2 blockers, concomitant use of proton-pump inhibitors, concomitant use of moderate or strong CYP3A inhibitors, and concomitant use of strong CYP3A inducers.
-
For apparent volume of distribution of the central compartment (Vc/F): sex, race (White, Black, Hispanic, Asian), baseline total bodyweight, and country (Taiwan, Japan, China, Korea).
-
For relative bioavailability of the ER formulation: upadacitinib dose, concomitant use of antacids, concomitant use of H2 blockers, concomitant use of proton pump inhibitors, concomitant use of any pH-modifying agents, concomitant use of moderate or strong CYP3A inhibitors, and concomitant use of strong CYP3A inducers.
For healthy subjects with missing DAS28–CRP or hsCRP at baseline, values of 0.96 and 0.15 mg/L, respectively, were imputed. For all other missing continuous covariates, the study median value was assigned. Use of concomitant medication was evaluated as a time-varying covariate. Missing co-medication information was imputed with no use. Continuous covariates were included in the model with a power function centered on the median covariate value. The fractional differences between groups for categorical covariates were determined using a multiplicative model. Relationships between individual pharmacokinetic parameter estimates and possible covariates were explored using graphical techniques. Stepwise inclusion of covariates in the model involved testing the effect of each covariate on the relevant model parameters in separate model runs. In the first step, all covariates were tested to improve the model fit. The most significant covariate among the significant covariates (p < 0.01) was included in the model, and all covariates that had been significant in the first step were tested again in a second step. This procedure was repeated until no covariates could be included anymore. The model resulting from this forward inclusion process is referred to as the full model. Once the full model was defined, it was used as the starting model for the backward elimination process. All included covariate relations were evaluated by comparing the starting model to a model without the particular covariate–parameter relationship. If insignificant covariates (p > 0.001) were found, the covariate relationship that resulted in the least significant increase in objective function value (OFV) was discarded, and the reduced model was used as the starting model for the next iteration of the backward elimination process. This process was repeated until no insignificant covariates were left in the model. The resulting model was referred to as the final model.
2.2.2 Final Model Evaluation
2.2.2.1 Bootstrap
To estimate confidence intervals (CI) of the model parameters, 500 bootstrap replicates were constructed by randomly sampling (with replacement) N subjects from the original dataset, where N is the number of subjects in the original dataset. Model parameters were estimated for each bootstrap replicate and the resulting values were used to estimate medians and CIs. Bootstrap statistics were based on replicates that converged successfully. The medians and 95% CIs for bootstrap model parameters were derived as the 50th percentile and the range from the 2.5th to the 97.5th percentiles of the results from individual replicates. Model parameters based on the original dataset were compared against the bootstrap results.
2.2.2.2 Visual Predictive Checks
For visual predictive checks (VPCs), 400 simulated replicates of the pharmacokinetic dataset were generated using NONMEM and PsN 4.7. Subsequently, the simulated data were compared with the observed data by superimposing the median, 2.5%, and 97.5% of the observed data with 95% prediction bands of each of these percentiles from the simulations. In addition, prediction-corrected VPCs were generated for the ER formulation [23].
2.2.3 Evaluation of the Impact of Statistically Significant Covariates on Upadacitinib Exposures
After identifying the statistically significant covariates for upadacitinib pharmacokinetic parameters, simulations were performed to explore their clinical relevance or impact on upadacitinib AUC and Cmax at steady state. Simulations with 200 replicates of the dataset using the demographics of the RA patient population from the phase II and III studies were performed. Simulations for each of the statistically significant covariates of interest were carried out separately while fixing the other covariates to the reference value. The mean of the ratios across the replicates and the 90% CI for the mean (5th and the 95th percentiles of the ratios) were calculated and summarized graphically using a forest plot.
3 Results
3.1 Dataset and Demographic Summary
Evaluable pharmacokinetic data were available from 4170 subjects, with a total of 29,372 upadacitinib plasma concentrations collected following 1–48 mg doses using the IR formulation and 7.5–30 mg doses using the ER formulation. Approximately 3.6% of data points (1087 records) were below the LLOQ (see electronic supplementary material). Given the small fraction of concentrations below the LLOQ, the M5 imputation method was used by imputing below the limit of quantification (BLQ) concentrations with the LLOQ/2 [21]. Given the nature of data collection in phase II and III trials, an outlier identification and exclusion rules were applied as described in the electronic supplementary Methods. Using these rules, only 0.9% of all dataset concentrations were flagged as clear outliers that likely resulted from inaccurate dosing records and were hence excluded from the analysis.
A summary of the key baseline demographics of the subjects who participated in the studies is shown in Table 2. The subject population was predominantly White (80%) and female (76%), with a mean age of 54 years and mean bodyweight of 76 kg. The majority of subjects had moderate to severe RA (96%), while approximately 65% of subjects had background methotrexate use. Approximately 3% and 40% of subjects included in the analysis dataset were reported to have used a moderate CYP3A or pH-modifying agents concomitantly, respectively, while receiving upadacitinib. The calculated CrCl for subjects included in the dataset ranged from 30.2 to 391 mL/min.
3.2 Population Pharmacokinetic Model
Based on the prior analysis of upadacitinib pharmacokinetic data from phase I and II trials [14], model development started with a two-compartment pharmacokinetic model with linear absorption and lag time for the IR formulation. The starting model included ISV on apparent clearance (CL/F) and volume of distribution of the central compartment (Vc/F). For the ER formulation, a mixed zero- and first-order absorption model with lag time was assumed. Alternative absorption models for the ER formulation (mixed zero- and first-order absorption without lag time, linear absorption with and without lag time) were explored but provided a worse OFV compared with the previous model. Subsequently, ISV on the different absorption parameters was tested to improve the fit, and adding ISV on the absorption rate constant (Ka) for the ER formulation was selected for further development. Additional model refinements were explored to improve the model fit. A model with different ISV on CL/F and Vc/F between phase I and phase II/III studies was found to reduce the OFV by 152 points. A split of the proportional residual error between phase I and phase II/III studies was tested and was found to further improve the fit by a 1108-point reduction in the OFV. The estimation of ISV variability on the Ka of the IR formulation was not numerically feasible with the current dataset. A covariate effect of the subject population (RA, yes/no) on CL/F was included in the model prior to testing other covariates, based on the findings from the previous population analyses of upadacitinib phase I and II clinical trials [14]. This led to further improvement of the model by a 233-point reduction in the OFV and was thus included in the base model.
The effect of covariates on upadacitinib pharmacokinetics was evaluated using a forward inclusion and backward elimination approach, as described in the Methods section. After completion of stepwise forward inclusion, the full model included RA, CrCl, bodyweight on CL/F, and bodyweight and sex on Vc/F. When backward elimination was performed on the full model, sex on Vc/F did not maintain statistical significance (p > 0.001) and was hence removed from the model. Other evaluated covariates (sex, race, concomitant use of pH-modifying agents, moderate CYP3A inhibitors, strong CYP3A inducers, or background methotrexate) did not correlate significantly with upadacitinib pharmacokinetic parameters.
The final model was re-run without excluding the outliers identified, as described in the electronic supplementary Methods section. Pharmacokinetic parameter estimates obtained from the models with or without inclusion of the outliers are compared in electronic Supplementary Table 1. The fixed-effects pharmacokinetic parameters, as well as residual variability, showed negligible differences between the models with and without outliers. As expected, the ISV estimates get inflated by up to 21% and show bias in their distribution after inclusion of outliers relative to the model without the outliers. The final model without outliers was used for further assessment.
The estimated pharmacokinetic parameter values, the effect of the covariates, and their associated variability for the final upadacitinib pharmacokinetic model are listed in Table 3. A bootstrap of the final model was performed with replicated datasets stratified by formulation [stratification categories: (1) phase I and II studies with the immediate-release formulation; (2) phase III studies with the extended-release formulation; and (3) a phase I study that evaluated both formulations] to guarantee a sufficient number of subjects to estimate the parameters for the IR formulation. A total of 456/500 runs (91.2%) converged successfully in the bootstrap analysis. The estimated pharmacokinetic parameter values based on the original dataset were in good agreement with the medians of the parameter values estimated from the bootstrap (Table 3). The bootstrap confirmed the robustness of the parameter estimates and the 95% CI did not include the value for no effect for any of the included covariate effects.
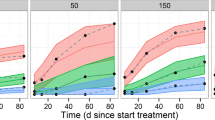
The goodness-of-fit plots for the final model are depicted in Electronic Supplementary Figs. 1 and 2. The plots of individual-predicted, population-predicted versus observed concentrations indicated that the model adequately describes most of the upadacitinib observations and no model misspecification. The conditional weighted residuals (CWRES) did not show a systematic trend when plotted against time since last dose or versus the population-predicted concentration, indicating a lack of systematic bias. A cluster of high CWRES values was observed close to 24 h since the last dose and at very low population-predicted concentrations. Both observations could be a result of a less than optimal recording of dosing times relative to sampling in the phase II and III setting. Overall, the model performance in describing the observed upadacitinib exposures based on these goodness-of-fit plots was adequate. VPCs with 400 replicates for the upadacitinib concentrations plotted versus time since the last dose, stratified by dose group and formulation used, show good agreement between simulated and observed data across the doses and formulations evaluated with respect to overall trend and variability (Figd. 1, 2). An additional prediction-corrected VPC (pcVPC) for upadacitinib concentrations from the ER dose groups confirmed that the model adequately described the central tendency and variability of the data (Fig. 3).

Visual predictive checks of upadacitinib plasma concentrations plotted versus time since last dose for the immediate-release formulation, stratified by dose. The shaded blue areas represent the 95% prediction interval of the 2.5th and 97.5th percentiles of observed concentrations, the red–blue areas represent the 95% of the 50th percentile of observed concentrations, the solid red line represents the median of the observed concentrations, and the dashed red lines represent the 2.5th and 97.5th percentiles of the observed concentrations

Visual predictive checks of upadacitinib plasma concentrations plotted versus time since last dose for the extended-release formulation, stratified by dose. The shaded blue areas represent the 95% prediction interval of the 2.5th and 97.5th percentiles of observed concentrations, the red–blue areas represent the 95% of the 50th percentile of observed concentrations, the solid red line represents the median of the observed concentrations, and the dashed red lines represent the 2.5th and 97.5th percentiles of the observed concentrations

Prediction-corrected visual predictive check plotted versus time since last dose using data for the extended-release formulation. The shaded blue areas represent the 95% prediction interval of the 2.5th and 97.5th percentiles of the prediction-corrected observed concentrations, the red–blue areas represent the 95% prediction interval of the 50th percentile of the prediction-corrected observed concentrations, the solid red line represents the median of the prediction-corrected observed concentrations, and the dashed red lines represent the 2.5th and 97.5th percentiles of the prediction-corrected observed concentrations
3.2.1 Impact of Significant Covariates on Upadacitinib Exposures
The impact of statistically significant covariates identified in the population pharmacokinetic analyses on upadacitinib steady-state AUC and Cmax are presented in Fig. 4. The forest plot was created using the methodology described in the Methods section. For bodyweight, the test group was 60–100 kg and the reference groups were < 60 kg and > 100 kg. For CrCl, the reference group was ≥ 90 mL/min and the test groups were 60 to < 90 mL/min and 30 to < 60 mL/min.

Model-predicted covariate effect on upadacitinib Cmax and AUC for different RA subpopulations relative to the reference RA population. Cmax maximum concentration, AUC area under the concentration–time curve, RA rheumatoid arthritis
Based on the simulations, subjects with RA with a bodyweight < 60 or > 100 kg were predicted to have 5% higher or lower AUC, on average, and 18% higher or lower Cmax, on average, respectively, compared with subjects with a bodyweight of 60–100 kg. Subjects with mild (CrCl 60 to < 90 mL/min) or moderate (30 to < 60 mL/min) renal impairment are predicted to have approximately 13 and 26% higher AUC, respectively, compared with subjects with normal renal function.
4 Discussion
Upadacitinib is a novel selective JAK 1 inhibitor being developed for the treatment of patients with moderate to severe RA. The recent results from phase III studies demonstrated that upadacitinib monotherapy or in combination with csDMARDs significantly improved the clinical signs and symptoms in subjects with moderate to severe RA [13, 16,17,18,19,20]. The current article describes a population pharmacokinetic model that integrates upadacitinib plasma concentration data from five global phase III studies, one regional phase IIb/III study in Japan, two phase II studies, and four phase I studies conducted in healthy subjects or subjects with moderate to severe RA. This comprehensive pharmacokinetic analysis used data from 4170 subjects, with a total of 29,372 plasma upadacitinib concentrations, with doses ranging from 1–48 mg using the IR formulation and 7.5–30 mg using the ER formulation. As part of the model-building process, demographics and clinical characteristics that may influence upadacitinib exposures were evaluated. This is the first report of upadacitinib pharmacokinetics from the ER formulation in RA patients.
Upadacitinib pharmacokinetics were adequately described by a two-compartment model with first-order absorption with lag time for the IR formulation, mixed zero- and first-order absorption with lag time for the ER formulation, and linear first-order elimination. The inclusion of data from several phase I, II, and III studies enabled the development of a robust population pharmacokinetic model, with all the structural parameters estimated with good precision (Table 3). The estimated apparent oral clearance and steady-state volume of distribution (represented as the sum of the central and peripheral volume of distribution) in healthy subjects were 53.7 L/h and 294 L, respectively, for the ER formulation. The ISV for upadacitinib apparent oral clearance and apparent central volume of distribution were estimated to be 21% and 24%, respectively, in the phase I population, and 37% and 53%, respectively, in the phase II/III population. The ISV in the phase II/III population was slightly higher compared with the phase I population, which was expected given the wider inclusion criteria of phase II and III studies compared with phase I studies, as well as the sparse and less-controlled pharmacokinetic assessment for phase II and III trials. The oral bioavailability of the ER formulation relative to the IR formulation was estimated to be 76%. Upadacitinib was not evaluated with intravenous administration in humans, therefore the absolute bioavailability of upadacitinib was not estimated in the present analysis. The administration of pH-modifying medications had no effect on upadacitinib absorption from the ER formulation. Upadacitinib bioavailability was dose-proportional over the evaluated ER dose range of 7.5–30 mg. The pharmacokinetic structural parameters for the upadacitinib IR formulation were consistent between the previously reported model utilizing data from phase I and II trials [14] and the current model with the larger dataset including phase III RA trials. Additionally, upadacitinib pharmacokinetics for the IR and ER formulations in the presented analyses are consistent with prior non-compartmental assessments in healthy subjects [10, 12].
The effect of various factors of interest on upadacitinib pharmacokinetics was evaluated within the framework of the population pharmacokinetic model. An influence of subject population (RA subjects vs. healthy subjects) on upadacitinib clearance was observed. RA subjects were estimated to have 25% lower upadacitinib clearance (leading to 33% higher estimated upadacitinib AUC) compared with healthy subjects. This estimate is consistent with previous upadacitinib population pharmacokinetics [14], as well as non-compartmental analyses [10], and is similar to prior observations with other JAK 1 inhibitors [24, 25]. IL-6 is commonly elevated in RA subjects and has been shown to suppress CYP3A metabolic activity [26]. The observed slightly higher upadacitinib clearance in RA subjects could be hypothesized due to the combination of the IL-6 effect and characteristics of the RA disease population. However, baseline hsCRP (a marker for inflammation) was not correlated with upadacitinib clearance. Additionally, there was no time-dependent change in upadacitinib clearance with continued upadacitinib treatment, as demonstrated by the lack of trend for change in CWRES versus time from the model fit (Electronic Supplementary Fig. 2). It is possible that the observed small difference in upadacitinib clearance between subjects with RA and healthy subjects is due to a combination of factors presented collectively in the RA population (e.g. older age, lower metabolic capacity, and possibly inflammation), as we previously noted [14]. Bodyweight and creatinine clearance were identified as significant covariates affecting upadacitinib oral clearance; however, the covariate effects of bodyweight and creatinine clearance only explained approximately 2.4% and 6.6% of the ISV on clearance in phase I and phase II/III studies, respectively. Subjects with RA who have mild and moderate renal impairment are estimated to have 13% and 26% higher AUC, respectively, compared with subjects with normal renal function. The prior analysis of data from a phase I study indicated that only a small percentage (approximately 20%) of administered upadacitinib doses is eliminated via the renal pathway [10]; hence, the small effect on upadacitinib exposure does not warrant any adjustment of upadacitinib dose. Estimates for the effect of renal impairment on upadacitinib exposures from a recent dedicated phase I study are consistent with the estimated effects based on population pharmacokinetic analyses [27].
Upadacitinib is a non-sensitive substrate for CYP3A, and a study with a strong CYP3A inhibitor, ketoconazole, showed a limited (approximately 75%) increase in upadacitinib exposures [28]. In the current population pharmacokinetic analyses, CYP3A modulators were not identified as significant covariates affecting upadacitinib apparent oral clearance; however, it should be noted that only a small percentage of the subjects used CYP3A modulators in the studies included in the population pharmacokinetic analysis dataset (3%: moderate CYP3A inhibitors; approximately 0.4%: strong CYP3A inhibitors; approximately 0.3%: strong CYP3A inducers). Methotrexate remains the mainstay therapy for RA [22,23,24] and upadacitinib may be used in combination with methotrexate, hence the effect of methotrexate was evaluated in this analysis and was confirmed to not affect upadacitinib pharmacokinetics. Approximately 40% of subjects have reported using pH-modifying agents concomitantly while receiving upadacitinib. The effect of these agents was evaluated in the population pharmacokinetic analysis and was found to not affect upadacitinib pharmacokinetics.
One of the challenges that occur when modeling the data using phase II and III clinical trials is that the less than optimal recording of dosing times relative to sampling can lead to concentrations outside the expected range for time since the last dose. Therefore, an outlier identification and exclusion rule was applied to avoid bias in the population and individual pharmacokinetic parameter estimates. A re-run of the final model on data, including previously excluded outliers, resulted in negligible changes in pharmacokinetic parameter estimates, indicating a lack of potential bias from outlier exclusions. Overall, these findings confirm the robustness of the fixed-effects pharmacokinetic parameter estimates. VPCs indicated that the final model described the central tendency and variability of the data reasonably well. The bootstrap analysis indicated robustness of the final model.
5 Conclusions
A robust population pharmacokinetic model was developed for upadacitinib using data from phase I–III clinical trials in healthy volunteers and subjects with RA who received upadacitinib IR or ER formulations. The model assessed the impact of intrinsic and extrinsic factors of interest on upadacitinib exposure. A slightly higher upadacitinib AUC was estimated in RA patients than in healthy subjects. Sex, race, concomitant use of pH-modifying agents, moderate CYP3A inhibitors, or methotrexate use had no effect on upadacitinib pharmacokinetics. Bodyweight and mild or moderate renal impairment had statistically significant but non-clinically relevant effects on upadacitinib exposures. The developed model was evaluated using diagnostic plots, bootstrap, and VPCs, which all demonstrated its adequacy and robustness for future use in pharmacokinetic simulations or exposure–response analyses in RA.
References
Scott DL, Wolfe F, Huizinga TWJ. Rheumatoid arthritis. Lancet. 2010;376:1094–108.
Malemud CJ. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther Adv Musculoskelet Dis. 2018;10:117–27.
Leonard WJ. Role of Jak kinases and STATs in cytokine signal transduction. Int J Hematol. 2001;73:271–7.
O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36:542–50.
Voss J, Graff C, Schwartz A, Hyland D, Argiriadi M, Camp H, et al. Pharmacodynamics of a novel Jak1 selective inhibitor in rat arthritis and anemia models and in healthy human subjects. Arthritis Rheum. 2013;65:1.
Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13:234–43.
Kontzias A, Kotlyar A, Laurence A, Changelian P, O’Shea JJ. Jakinibs: a new class of kinase inhibitors in cancer and autoimmune disease. Curr Opin Pharmacol. 2012;12:464–70.
Neubauer H, Cumano A, Müller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93:397–409.
Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin Investig Drugs. 2014;23:1067–77.
Mohamed M-EF, Camp HS, Jiang P, Padley RJ, Asatryan A, Othman AA. Pharmacokinetics, safety and tolerability of ABT-494, a novel selective JAK 1 inhibitor, in healthy volunteers and subjects with rheumatoid arthritis. Clin Pharmacokinet. 2016;55:1547–58.
Genovese MC, Smolen JS, Weinblatt ME, Burmester GR, Meerwein S, Camp HS, et al. Efficacy and safety of ABT-494, a selective JAK-1 inhibitor, in a phase IIb study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. 2016;68:2857–66.
Mohamed M-EF, Zeng J, Marroum PJ, Song I-H, Othman AA. Pharmacokinetics of upadacitinib with the clinical regimens of the extended-release formulation utilized in rheumatoid arthritis phase 3 trials. Clin Pharmacol Drug Dev. 2019;8(2):208–16. https://doi.org/10.1002/cpdd.462.
Genovese MC, Fleischmann R, Combe B, Hall S, Rubbert-Roth A, Zhang Y, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet. 2018;391:2513–24.
Klünder B, Mohamed M-EF, Othman AA. Population pharmacokinetics of upadacitinib in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I and II clinical trials. Clin Pharmacokinet. 2018;57:977–88.
Kremer JM, Emery P, Camp HS, Friedman A, Wang L, Othman AA, et al. A phase IIb study of ABT-494, a selective JAK-1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti-tumor necrosis factor therapy. Arthritis Rheumatol. 2016;68:2867–77.
Burmester GR, Kremer JM, Van den Bosch F, Kivitz A, Bessette L, Li Y, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2018;391:2503–12.
Roy F. A phase 3, randomized, double-blind study comparing upadacitinib to placebo and to adalimumab, in patients with active rheumatoid arthritis with inadequate response to methotrexate [abstract no. 72780]. American College of Rheumatology Meeting Abstracts; 2018.
van Vollenhoven R. A phase 3, randomized, controlled trial comparing upadacitinib monotherapy to MTX monotherapy in MTX-naïve patients with active rheumatoid arthritis [abstract no. 75147]. American College of Rheumatology Meeting Abstracts; 2018.
Smolen JS. Upadacitinib As Monotherapy: A phase 3 randomized controlled double- blind study in patients with active rheumatoid arthritis and inadequate response to methotrexate. American College of Rheumatology Meeting Abstracts; 2018.
Tanaka Y. A Phase2b/3 randomized, placebo-controlled, double-blind study of upadacitinib, a selective JAK1 inhibitor, in Japanese patients with active rheumatoid arthritis and inadequate response to conventional synthetic dmards. EULAR18-4302. European League Against Rheumatism; 2018.
Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28:481–504.
Sheiner LB, Ludden TM. Population pharmacokinetics/dynamics. Annu Rev Pharmacol Toxicol. 1992;32:185–209.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13:143–51.
Namour F, Diderichsen PM, Cox E, Vayssière B, Van der Aa A, Tasset C, et al. Pharmacokinetics and pharmacokinetic/pharmacodynamic modeling of filgotinib (GLPG0634), a selective JAK1 inhibitor, in support of phase IIB dose selection. Clin Pharmacokinet. 2015;54:859–74.
Us FDA. Clinical pharmacology and biopharmaceutics review(s)—tofacitinib. Silver Spring: Center for Drug Evaluation and Research, US FDA; 2011.
Jover R, Bort R, Gómez-Lechón MJ, Castell JV. Down-regulation of human CYP3A4 by the inflammatory signal interleukin-6: molecular mechanism and transcription factors involved. FASEB J. 2002;16:1799–801.
Mohamed M-EF, Coppola S, Feng T, Anderson J, Othman AA. Characterization of the effect of renal impairment on upadacitinib pharmacokinetics. J Clin Pharmacol. https://doi.org/10.1002/jcph.1375.
Mohamed M-EF, Jungerwirth S, Asatryan A, Jiang P, Othman AA. Assessment of effect of CYP3A inhibition, CYP induction, OATP1B inhibition, and high-fat meal on pharmacokinetics of the JAK1 inhibitor upadacitinib. Br J Clin Pharmacol. 2017;83:2242–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical Approval and Informed Consent
The studies were designed according to good clinical practice guidelines, the Declaration of Helsinki, and applicable regulations, and were approved by Institutional Review Boards. All participants provided written informed consent.
Funding
This study was sponsored by AbbVie Inc., who contributed to the study design; research; data interpretation; and writing, review and approval of the abstract for publication.
Conflict of Interest
Ben Klünder, Rajendar K. Mittapalli, Mohamed-Eslam F. Mohamed, Anna Friedel, Peter Noertersheuser and Ahmed A. Othman are employees or former employees of AbbVie Inc., and may hold AbbVie stocks or options.
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis datasets), as well as other information (e.g. protocols and clinical study reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.The clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Klünder, B., Mittapalli, R.K., Mohamed, ME.F. et al. Population Pharmacokinetics of Upadacitinib Using the Immediate-Release and Extended-Release Formulations in Healthy Subjects and Subjects with Rheumatoid Arthritis: Analyses of Phase I–III Clinical Trials. Clin Pharmacokinet 58, 1045–1058 (2019). https://doi.org/10.1007/s40262-019-00739-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00739-3