
Abstract
Cladribine Tablets (MAVENCLAD®) are used to treat relapsing multiple sclerosis (MS). The recommended dose is 3.5 mg/kg, consisting of 2 annual courses, each comprising 2 treatment weeks 1 month apart. We reviewed the clinical pharmacology of Cladribine Tablets in patients with MS, including pharmacokinetic and pharmacometric data. Cladribine Tablets are rapidly absorbed, with a median time to reach maximum concentration (Tmax) of 0.5 h (range 0.5–1.5 h) in fasted patients. When administered with food, absorption is delayed (median Tmax 1.5 h, range 1–3 h), and maximum concentration (Cmax) is reduced by 29% (based on geometric mean). Area under the concentration–time curve (AUC) is essentially unchanged. Oral bioavailability of cladribine is approximately 40%, pharmacokinetics are linear and time-independent, and volume of distribution is 480–490 L. Plasma protein binding is 20%, independent of cladribine plasma concentration. Cladribine is rapidly distributed to lymphocytes and retained (either as parent drug or its phosphorylated metabolites), resulting in approximately 30- to 40-fold intracellular accumulation versus extracellular concentrations as early as 1 h after cladribine exposure. Cytochrome P450-mediated biotransformation of cladribine is of minor importance. Cladribine elimination is equally dependent on renal and non-renal routes. In vitro studies indicate that cladribine efflux is minimally P-glycoprotein (P-gp)-related, and clinically relevant interactions with P-gp inhibitors are not expected. Cladribine distribution across membranes is primarily facilitated by equilibrative nucleoside transporter (ENT) 1, concentrative nucleoside transporter (CNT) 3 and breast cancer resistance protein (BCRP), and there is no evidence of any cladribine-related effect on heart rate, atrioventricular conduction or cardiac repolarisation (QTc interval prolongation). Cladribine Tablets are associated with targeted lymphocyte reduction and durable efficacy, with the exposure–effect relationship showing the recommended dose is appropriate in reducing relapse risk.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This review discusses the clinical pharmacology of Cladribine Tablets in patients with relapsing multiple sclerosis, presenting pharmacokinetic, pharmacodynamic and pharmacometric data. |
Cladribine Tablets are associated with a selective reduction in lymphocyte counts and durable efficacy relative to the fast disposition in plasma, and short-term treatment posology in each of the 2 treatment years. |
The recommended cumulative dose of Cladribine Tablets 3.5 mg/kg over 2 years is shown to be appropriate in reducing relapse risk. |
1 Introduction
Multiple sclerosis (MS) is a neurodegenerative disease, where a patient’s immune system attacks their central nervous system, resulting in demyelination, axonal damage and progressive disability [1, 2]. Cladribine Tablets (MAVENCLAD®; Merck Serono Europe Ltd), an oral formulation of cladribine, were shown to have significant efficacy for the treatment of relapsing MS in placebo-controlled, phase III trials [3,4,5]. A cumulative dose of 3.5 mg/kg body weight (consisting of 2 annual courses that are each comprised of 2 treatment weeks; 1 at the start of the first month and 1 at the start of second month of each year) has been approved for the treatment of adults with certain types of relapsing MS [6,7,8]. The short-term treatment posology of Cladribine Tablets has the potential to facilitate patient adherence [4], which is an ongoing challenge for the long-term treatment of MS [9].
Cladribine is a nucleoside analogue of deoxyadenosine. The cladribine prodrug is phosphorylated intracellularly to its active product, 2-chlorodeoxyadenosine triphosphate (Cd-ATP), by deoxycytidine kinase. This deoxynucleotide product is degraded in most cells, by 5ʹ-nucleotidase. Cells such as lymphocytes that contain a high deoxycytidine kinase activity but low 5ʹ-nucleotidase activity, i.e. a high deoxycytidine kinase to 5ʹ-nucleotidase activity ratio, accumulate deoxynucleotides to toxic concentrations, resulting in lymphocyte cell death. By this mechanism, Cladribine Tablets exert a selective mode of action on B and T lymphocytes [10, 11]. Variations in the expression levels of deoxycytidine kinase and 5ʹ-nucleotidase between immune cell subtypes explain differences in immune cell sensitivity to cladribine. Because of these expression levels, cells of the innate immune system are less affected than cells of the adaptive immune system [12, 13]. Cladribine Tablets have been described as a selective immune reconstitution therapy due to their selective effect on the adaptive versus innate immune system, together with the short and intermittent nature of the treatment courses [14, 15]. The current theory is that by inducing lymphopenia, Cladribine Tablets reset the immune system [15].
Cladribine was first established as a parenteral formulation for the treatment of B- and T cell lymphoid malignancies, including hairy cell leukaemia and chronic lymphocytic leukaemia [16, 17]. The clinical pharmacokinetics (PK) of parenteral cladribine in patients with malignancies were previously reported by Liliemark in 1997 [18], reflecting the state of knowledge at that time. Due to the recent approval of Cladribine Tablets, a decision that was based on a substantial amount of new study data, an up-to-date review of the clinical pharmacology of cladribine is warranted [3, 19, 20]. Here, we review the clinical PK and outcomes of pharmacometric analyses (PK and primary pharmacodynamics [PD]) of the oral tablet formulation of cladribine in patients with MS in order to provide a comprehensive and timely summary of the available data. This report represents a narrative review of data from publications, congress materials, label information, and unpublished data on file.
2 Pharmacokinetics (PK)
2.1 Overview of Studies
The PK of Cladribine Tablets has been investigated in 5 phase I studies in patients with MS, plus a subpopulation of the phase III CLARITY clinical trial. A summary of the phase I studies is presented in Table 1. In addition, data from 3 of the PK studies (studies 25803, 26127 and 26486) plus the PK subpopulation from the CLARITY clinical trial (see footnote in Table 2) were recently combined in a population PK analysis [19]. The population PK analysis was performed to characterise the concentration–time course of cladribine, to estimate interindividual variability in PK, and to identify covariates that explain such variability.
2.2 Absorption
Cladribine is rapidly absorbed after tablet administration (Fig. 1); oral administration of a single 10 mg tablet in a fasted state is associated with a median time to reach maximum concentration (Tmax) of approximately 0.5 h (range 0.5–1.5 h) [21]. The mean maximum concentration (Cmax) is in the range of 22–29 ng/mL, with the corresponding mean area under the concentration–time curve (AUC) in the range of 80–101 ng·h/mL (arithmetic means from various studies) [22]. Cladribine can be considered a Biopharmaceutics Classification System (BCS) class III compound (low permeability, high solubility) [20]. The oral bioavailability of Cladribine Tablets when administered in the fasted state is approximately 40% [22], possibly limited by breast cancer resistance protein (BCRP)-mediated intestinal efflux. In the phase III studies, patients were instructed to take their tablets after an overnight fast on an empty stomach, and, once administered, to wait at least 1 h before eating. If a dose was to be administered in the afternoon, or if a subject had mistakenly eaten before dosing, subjects were to wait at least 4 h after eating their last meal before dosing. In a phase I study, it was shown that taking a single 10 mg Cladribine Tablet after a high-fat breakfast results in a delay of the median Tmax from 0.5 h in the fasted state to 1.5 h (range 1–3 h) in the fed state [22]. This is associated with a 29% reduction in the maximum exposure of cladribine (geometric mean Cmax) compared with administration after an overnight fast, while the total exposure (estimated by noncompartment analysis) is minimally affected (geometric mean AUC from time zero to infinity (AUC∞) was 72.8 ng·h/mL for the fed state versus 75.7 ng·h/mL for the fasted state). Cladribine Tablets can therefore be administered without regard to food [22]. In the population PK analysis, absorption in the fasted state was described by a first-order process, and a transit-compartment model described the absorption delay in the data from the fed state. The fed/fasted status of phase III subjects was classified as ‘unknown’ for modelling purposes. For phase III subjects, the rate of absorption and bioavailability estimates were the same or very similar to the values for fed subjects, and absorption delay was more similar to the estimated value for fasted subjects. Oral bioavailability of Cladribine Tablets was estimated to be 45.6%, and coadministration with a high-fat meal resulted in a modest change in bioavailability to 40.5%, and a modest delay in absorption (Fig. 2). This was not expected to have a clinically meaningful impact [19]. Taken together, the noncompartmental and population PK analyses of cladribine bioavailability, rate of absorption, and food effects yielded consistent results.

Adapted from Munafo et al. [21]
Mean (standard deviation) plasma cladribine concentration by treatment. IV intravenous.

Adapted from Savic et al. [19]. © The Authors 2017
Population pharmacokinetic visual predictive checks for plasma cladribine concentrations in fasted and fed conditions. Light blue shaded area indicates simulated median with uncertainty; pink shaded area indicates simulated 5th and 95th percentiles with uncertainty; solid blue line indicates observed median; dashed blue line indicates observed 5th and 95th percentiles.
2.3 Distribution
The volume of distribution of cladribine is large, in the range of 480–490 L, which indicates extensive tissue distribution and intracellular uptake [19, 22]. Various transporter proteins facilitate the distribution of cladribine across biological membranes, including equilibrative nucleoside transporter (ENT) 1, concentrative nucleoside transporter (CNT) 3 and BCRP. Cladribine is most likely transported into lymphocytes by ENT1 and CNT3 [23, 24]. The ENT1 transporter protein is also thought to be an important contributor to the active efflux of cladribine from white blood cells [25]. The oral bioavailability of cladribine may be limited by the efflux transporter BCRP, which has an affinity with cladribine (BCRP overexpression has been shown to strongly reduce the rate of 2-CdA accumulation in human osteosarcoma cells) [26] and is expressed at high levels in the small intestine [27]. The contribution of P-glycoprotein (P-gp; ABCB1) to cladribine efflux is probably not important for the overall bioavailability of Cladribine Tablets, based on results of studies in MDCKII-MDR1 cells that show P-gp is not an efficient transporter of cladribine [22, 28]. Clinically relevant interactions with inhibitors of P-gp are therefore not expected.
Results of an uptake study of 14C-labelled cladribine into cryopreserved human hepatocytes suggest that transporter-mediated uptake of cladribine into human hepatocytes is negligible (data on file). Cladribine and/or its phosphorylated metabolites are substantially accumulated and retained in human lymphocytes. As shown in vitro, cladribine is rapidly distributed to, and retained in (either as parent drug or its phosphorylated metabolites), human lymphocytes, resulting in approximately 30- to 40-fold intracellular accumulation compared with extracellular concentrations, as early as 1 h after cladribine exposure (data on file). Cladribine has the potential to penetrate the blood–brain barrier [18]. A small study in another indication has shown a cerebrospinal fluid/plasma concentration ratio of approximately 0.25 [29]. In spiked human plasma, the plasma protein binding of cladribine was 20%, and independent of plasma cladribine concentration (data on file).
2.4 Metabolism
Cladribine metabolism was investigated in patients with MS following the administration of a single 10 mg tablet and a single 3 mg intravenous dose (study 25803) (Table 1). Following both oral and intravenous administration, the parent compound cladribine was the main component present in plasma and urine. The metabolite 2-chloroadenine was a minor metabolite both in plasma and in urine, i.e. amounting to ≤ 3% of the AUC of parent compound in plasma after oral administration. Only traces of other metabolites could be found in plasma and urine [30]. In hepatic in vitro systems, cladribine was only metabolised to a very low extent, with at least 90% of radiolabelled cladribine remaining unchanged (data on file).
Cladribine is not a relevant substrate to cytochrome P450 (CYP) enzymes, based on reaction phenotyping studies where cladribine was incubated with microsomes prepared from human recombinant lymphoblastoid cells that were genetically engineered to express specific human CYP enzymes that may be responsible for the metabolism of cladribine in vitro: CYP1A2, 2A6, 2C9, 2C19, 2D6, 2E1, and 3A4 (data on file).
After entering the target cells, cladribine is phosphorylated to cladribine monophosphate (Cd-AMP) by deoxycytidine kinase (and also by deoxyguanosine kinase in the mitochondria). Cd-AMP is further phosphorylated to chlorodeoxyadenosine diphosphate (Cd-ADP), and Cd-ADP is in turn phosphorylated by 5ʹ-nucleotidase to Cd-ATP [10, 11]. In a study of the intracellular PK of Cd-AMP and Cd-ATP in another indication, the levels of Cd-ATP were approximately half that of the Cd-AMP levels. The intracellular half-life of Cd-AMP was 15 h and the intracellular half-life of Cd-ATP was 10 h [31].
2.5 Elimination
The population PK analysis showed that renal and non-renal routes of cladribine elimination are of approximately equal importance; the median values were estimated to be 22.2 L/h for renal clearance and 23.4 L/h for non-renal clearance. Renal clearance correlated with creatinine clearance (CLCR), and CLCR was therefore used to predict renal cladribine clearance in patients with renal impairment using the population PK model, with the assumption that changes in CLCR only affect the renal component of total cladribine clearance. Intersubject variability for non-renal clearance was estimated to be 7.6%. The nonrenal proportion of cladribine clearance (approximately 50%) comprises negligible hepatic metabolism with extensive intracellular distribution and trapping of Cd-ATP within lymphocytes, and the subsequent elimination of intracellular Cd-ATP according to lymphocyte elimination pathways and lifecycle [22].
Renal clearance appears to exceed glomerular filtration rate, indicating net tubular excretion in addition to glomerular filtration [19]. As cladribine was shown not to be a substrate of the kidney-specific basolateral transporters OCT2, OAT1, and OAT3, nor of the kidney-specific apical transporter OAT4 (data on file), only BCRP [26, 28], ENT1/2 and CNT2/3 [32] remain as conceivable candidate transporters for active tubular secretion of cladribine. Basolateral-located ENT1 is the most likely candidate to facilitate basolateral uptake of cladribine as none of the other basolateral candidates tested actually facilitated cladribine uptake. In the apical membrane of renal tubular cells, of the 3 transporters implicated in renal transport of cladribine, only BCRP has been unambiguously shown to transport cladribine efficiently in multiple expression systems. P-gp-mediated transport does not seem to be efficient, and, for MRP4, transport was not shown in either of the test systems studied (HEK293-MRP4, MDCKII-MRP4; data on file). Based on these findings, the apical efflux is likely driven by BCRP, with some contribution of ENT1.
A decline in total cladribine clearance by 21% is predicted for patients with a CLCR of 60 mL/min (lower bound of mild renal impairment range), by 30% in patients with a CLCR of 40 mL/min (moderate renal impairment), and by 40% in patients with a CLCR of 20 mL/min (severe renal impairment), when compared with patients with normal renal function. As a result, an increase in AUC of 25, 45 and 65% is to be expected for an otherwise typical patient with mild, moderate, and severe renal impairment, respectively. There is good confidence in the predicted clearance value for patients with mild renal impairment since the clinical studies included a substantial proportion of patients (25%) with mild impairment (CLCR of ≥ 60 to < 90 mL/min). In contrast, calculations for moderate and severe renal impairment suffer from more uncertainty, given the low number of patients with moderate or severe renal impairment in the population PK analysis (Table 3) [19, 20, 22].
2.6 Dose and Time Dependency of PK
Cmax and AUC increased in a dose-proportional fashion after oral administration of Cladribine Tablets across a range of doses from 3 to 20 mg, suggesting that absorption is not affected by rate- or capacity-limited processes up to a 20 mg oral dose [21, 22]. No significant accumulation of cladribine concentration in plasma after once-daily repeated dosing has been observed [22, 33]. There is no indication that cladribine PK might be time-dependent after repeated administration of Cladribine Tablets. PK were observed in both the mid-term (measurements at weeks 5, 9 and 13) and the long-term (measurements at weeks 48 and 52) in the CLARITY study [3]. There were very similar distributions of the observations between visits, and no apparent trend in terms of either monotonic increases or decreases of cladribine concentrations with time (Fig. 3).

Measured cladribine plasma concentration by visit: a 3.5 mg/kg dose group; b 5.25 mg/kg dose group (pharmacokinetic population). D day, W week
2.7 Influence of Intrinsic Factors on PK
Due to the long-lasting effects on lymphocytes, PK studies have only been conducted in patients with MS, therefore studies in special populations (including children or elderly patients, or patients with hepatic impairment) have not been performed. However, the influence of demographic covariates on PK parameters was studied as part of the population PK analysis. This did not show any effect of age (studies in patients aged 19–65 years) or sex on the PK of Cladribine Tablets beyond what is accounted for by the effect of CLCR on renal clearance [19]. As safety and efficacy in patients with moderate or severe renal impairment have not been established, Cladribine Tablets should not be used in such patients.
No studies have been conducted in patients with hepatic impairment, therefore, the use of Cladribine Tablets is not recommended in patients with moderate or severe hepatic impairment (Child–Pugh score > 6) due to a lack of data [22].
2.8 Drug Interactions
Only a few clinically relevant drug interactions with Cladribine Tablets are expected. The absence of drug–drug interactions with Cladribine Tablets is of particular benefit due to the high rates of comorbid conditions associated with MS [34], which are likely to require concomitant pharmaceutical treatment.
2.8.1 Pharmacodynamic (PD) Interactions
There is a risk of mutual additive/synergistic effects on the immune system if Cladribine Tablets are used concomitantly with immunosuppressive or myelosuppressive therapies. Similarly, an additive effect on haematological adverse reactions is likely if Cladribine Tablets are used concomitantly with therapies that affect the haematological profile, such as carbamazepine [22]. Results from modelling analyses that examined the dose–exposure–response relationship with respect to absolute lymphocyte count (ALC) following administration of oral cladribine (see Sect. 4.1) demonstrated no statistically significant effect of concomitant glucocorticoids on the ALC response to cladribine [20].
2.8.2 PK Interactions
Cladribine as a Perpetrator Drug of PK Interactions
Cladribine Tablets contain hydroxypropyl betadex, and there is the potential for complex formation between free cyclodextrin, released from the Cladribine Tablets formulation, and other medicinal products [22]. Complex formation potentially leads to an increase in bioavailability of the concomitantly administered product (especially medicinal products with poor solubility and low bioavailability). The risk of complex formation with cyclodextrin can be minimised by separating the administration of any other oral pharmaceutical from the administration of Cladribine Tablets by at least 3 h, during the limited number of days in a treatment year in which Cladribine Tablets are administered [22].
The potential of cladribine to alter the PK of coadministered object drugs is considered low. With regard to metabolism-based drug interactions, cladribine does not show potential to act as an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4, based on studies in pooled human liver microsomes (data on file). Therefore, cladribine is unlikely to contribute to any inhibitory drug–drug interactions in vivo that are mediated by CYP450 enzymes. Cladribine appears to have no clinically meaningful inductive effect on CYP1A2, CYP2B6 and CYP3A4 enzymes, although the results from 2 in vitro studies of pooled human liver microsomes were not entirely consistent and conclusive (data on file). There is sufficient in vitro information available to support the view that cladribine does not inhibit the most important ABC transporters in vivo, i.e. P-gp (ABCB1) [in Caco-2 cells], BCRP (ABCG2) [in MDCKII-BCRP cells], and MRP2 (ABCC2), MRP4 (ABCC4) and MRP5 (ABCC5) [in membrane vesicle preparations], nor any of the examined organic anion transporters, i.e. OATP1B1 and OATP1B3 (in transfected HEK cells), OAT1 and OAT3 (in membrane vesicle preparations) and OAT4 (in vector-transfected S2 cells), and organic cation transporters, i.e. OCT1 (in transfected HEK cells) and OCT2 (in membrane vesicle preparations) [data on file] [20]. Data on inhibition of ENTs (ENT1 and ENT2), CNTs (CNT1, CNT2, CNT3), and MATE transporters by cladribine are currently incomplete or lacking.
Cladribine as an Object Drug of PK Interactions
A drug interaction study with pantoprazole has been conducted to investigate whether elevated gastric pH may alter the oral bioavailability of, and thus exposure to, Cladribine Tablets. Repeated single doses of oral pantoprazole 40 mg were administered 15 h and 3 h prior to administration of cladribine in patients with MS. The estimated geometric mean ratios of cladribine with pantoprazole versus cladribine alone were near unity for all parameters (1.006 and 0.980 for the primary endpoints AUC∞ and Cmax, respectively). The 90% confidence intervals (CIs) of the geometric mean ratios were 0.907–1.116 for AUC∞ and 0.804–1.194 for Cmax, both within the standard bioequivalence acceptance limits of 0.8–1.25. It was therefore concluded that coadministration of pantoprazole with Cladribine Tablets had no effect on the rate and extent of the absorption of cladribine [19].
As cladribine is only marginally metabolised by CYP-based metabolism, inhibition of any of the major CYP enzymes or genetic polymorphisms thereof (e.g. CYP2D6, CYP2C9 or CYP2C19) are not expected to result in clinically significant effects on cladribine PK or exposure.
Cladribine is a BCRP substrate (see Sect. 2.3), therefore BCRP inhibitors may have an effect on cladribine exposure. However, marketed BCRP inhibitors (including the tyrosine kinase inhibitors imatinib, sunitinib and gefitinib) are altogether contraindicated in cladribine-treated patients, together with the older-generation antihypertensive drug reserpine, which has been discontinued from most markets, as well as the estrogen receptor antagonist tamoxifen.
Based on the findings of a recent database search (University of Washington Drug–Drug Interaction Database [35]), the susceptibility of cladribine to become a victim/object of transporter-based drug interactions is limited in clinical practice to a few marketed products with ENT1 or BCRP inhibitory capacity, which are not contraindicated for cladribine-treated patients. Coadministration of potent ENT1 or BCRP inhibitors may theoretically alter the oral absorption/bioavailability of Cladribine Tablets. To apply a conservative approach, coadministration of these products should be avoided during each 4- to 5-day treatment period of Cladribine Tablets [22].
Cladribine may theoretically become the object/victim of potent CNT2 and CNT3 inhibitors. However, the database search was unable to identify any relevant marketed CNT2 inhibitors, only the cytotoxic nucleoside analogues fludarabine and clofarabine as potent CNT3 inhibitors, which are contraindicated in cladribine-treated patients. Identified ENT1 inhibitors include the P2Y12 antagonist ticagrelor, the adenosine reuptake inhibitors dipyridamole and dilazep, the dihydropyridine calcium channel blockers nimodipine, nifedipine and nitrendipine, and the PDE3 inhibitor cilostazol.
It has been reported that lamivudine can inhibit the phosphorylation of cladribine intracellularly, and thereby the therapeutic efficacy of cladribine [36]. Therefore, compounds that require intracellular phosphorylation to become active, such as lamivudine, zalcitabine, ribavirin, stavudine, and zidovudine should not be administered concomitantly with cladribine. Since cladribine therapy must not be initiated in patients with acute or chronic infections, this group of antiviral and antiretroviral drugs will not be used in patients treated with Cladribine Tablets.
The population PK analysis included 1 study on drug interactions with interferon-β1a, and no clinically relevant PK interaction was found [19, 22]. However, it should be noted that Cladribine Tablets are not intended for use with other disease-modifying drugs for MS as their safety and efficacy with concomitant treatment has not been established [22].
2.8.3 Summary of Drug Interactions
Due to the posology of Cladribine Tablets (and its relatively fast disposition), cladribine is only present in the body for a few days of every treatment year. This means that treatments that have the potential to influence the absorption of cladribine, e.g. inhibitors of transporters such as ENT1, CNT3 and BCRP (e.g. eltrombopag, dilazep, nifedipine, nimodipine, cilostazol, sulindac or reserpine) may be taken safely/effectively outside of the treatment window for Cladribine Tablets. The absence of drug–drug interactions with Cladribine Tablets is of particular benefit due to the high rates of comorbid conditions associated with MS [34], which are likely to require concomitant pharmaceutical treatment.
3 Cardiac Safety
3.1 Preclinical Data
Available nonclinical data do not suggest any clinically significant effects of cladribine on cardiac repolarisation. In vitro, only a very high concentration of cladribine (10−4 M) resulted in a marginal inhibition (i.e. 13%) of the human ether-a-go-go-related gene (hERG) tail current. Based on this, the safety margin for IKr block of the highest free cladribine concentration observed with Cladribine Tablets in humans is estimated to be 275-fold. In addition, no effect on action potential duration, in particular on action potential duration at 90% repolarisation (APD90), was observed in isolated canine Purkinje fibres at cladribine concentrations up to 10−4 M [20].
Cladribine did not exert any effect on heart rate, mean, systolic or diastolic systemic arterial blood pressure or any electrocardiogram (ECG) outcomes in dogs. In particular, the duration of the heart rate-corrected QT intervals were not affected during an observation period of 4 h postdose. The cladribine exposure of animals exceeded the exposure in patients with relapsing MS at the highest daily dose of 20 mg by 23-fold (data on file). In addition, a 3-month toxicology study in monkeys did not indicate any effect on the QTc interval following administration of 1.5, 3 or 6 mg/kg/day oral cladribine or 0.3 mg/kg/day subcutaneous cladribine (data on file).
3.2 Clinical Data
The cardiac safety of Cladribine Tablets was assessed in an ECG substudy of the CLARITY study in a subpopulation of 143 patients (data on file). To allow for a concentration–effect analysis of QT/QTc interval data (QTc/PK modelling), subjects who underwent ECG recording also took part in the population PK sampling (see Sect. 3.2.2 for results).
ECG data (analyses of central tendencies and outlier analysis according to ICH E14) did not show evidence of any cladribine-related effect on heart rate, atrioventricular (AV) conduction or cardiac depolarisation, as measured by the PR and QRS interval durations. Acute effects on QT/QTc (i.e. comparison of visit baseline [mean of the 3 predose ECGs] versus postdose ECGs) at all visits and across all treatment groups, including the placebo group, were consistent and showed a temporal, small, postdose increase versus visit baselines in QTcF ranging between 1 and 4 ms. The reason for this uniform finding for all treatments is not entirely clear but would be consistent with circadian effects on the QTc interval. However, the point estimates and upper limits of 90% CIs for placebo-corrected (i.e. differences to placebo) postdose QTcF changes from visit baseline were, on all occasions and for both cladribine treatment groups, < 7 ms (point estimates) and < 8 ms (upper limit one-sided 90% CI).
3.2.1 Correlation of Cladribine Plasma Concentration and QTcF Interval Data
The lack of a drug effect on QTc is further supported by the PK/PD analysis. The relationship between the placebo-adjusted QTcF differences to baseline (∆∆QTcF) and the corresponding cladribine concentrations was analysed according to the approach of Garnett et al. [37]. This linear regression analysis resulted in slopes very close to zero, and non-significant p values, indicating that there is no correlation between cladribine plasma concentrations and the corresponding ∆∆QTcF values (Fig. 4).

Cladribine plasma concentration versus ΔΔQTcF using visit baseline (ECG population of the CLARITY study). Placebo-adjusted difference of QTcF to visit baseline and cladribine concentrations, including regression line: number of subjects = 93; number of non-missing observations = 434; intercept = − 0.09, slope + 6.30E−06; p value = 0.761. ECG electrocardiogram
3.2.2 Population PK/PD Modelling and Simulation Analysis
Overall, the results of the PK/QTcF modelling and simulation analysis were highly consistent with the results of the traditional descriptive and ANOVA data analyses by treatment groups, as detailed above. The predose QTcF was estimated to be 405 ms for women and 385 ms for men. The interindividual and interoccasion variability in the baseline QTcF were estimated to be 3.5% (coefficient of variation [CV]) and 1.7% (CV), respectively, and the additive residual error variability was 8.7 ms. There was a significant postdose increase of 2.0 ms in QTcF 0.5–4 h after treatment administration for both placebo- and cladribine-treated patients, a finding that is consistent with circadian variations of the QTc interval. There were no other significant increases with time, or a relationship between QTcF and concentration of cladribine, concentration of the 2-chloroadenine metabolite, or cladribine dose. PK sampling provided important additional robust data for the overall interpretation of longitudinal ECG data in a heterogeneous relapsing MS patient population. The use of modelling is a contemporary approach and is in line with the type of data that many health authorities have begun to request for benefit–risk assessments.
4 PD Modelling
PD modelling has been performed using data from the Cladribine Tablets clinical development program in order to characterise the dose–exposure–response relationship and to develop integrated longitudinal models to better understand disease progression and assess the predictive value of ALC with respect to clinical response. Clinical trial simulations were further performed to assess the impact of various treatment algorithms, driven by cladribine-induced lymphopenia, on the efficacy of Cladribine Tablets in patients with MS.
4.1 Absolute Lymphocyte Count (ALC) Modelling
The most common adverse event reported with Cladribine Tablets is lymphopenia, consistent with the mechanism of action of cladribine. In the CLARITY study, a minority of patients treated with a 3.5 mg/kg cumulative dose of Cladribine Tablets developed transient grade 3 (24.9%) or grade 4 (0.7%) lymphopenia at any time during the 2-year study period [3]. Patients who completed the CLARITY study could enter the CLARITY Extension study if they had a normal ALC. The CLARITY Extension study was not a preplanned study and, as a consequence, there was a variable bridging interval between studies for each patient (median 40.3 weeks) [4]. A pooled analysis of patients treated with Cladribine Tablets in the CLARITY or CLARITY Extension studies found that ALC rapidly decreases after treatment with Cladribine Tablets, up to approximately 2 months after dosing, followed by gradual returns towards baseline counts. In patients treated with a cumulative dose of 3.5 mg/kg over 2 years, median ALC had returned to the normal range in 75% of patients by 144 weeks after the first dose of Cladribine Tablets [38].
A model was developed to assess the dose–exposure–response relationship with respect to ALC using data from subjects treated with Cladribine Tablets in the CLARITY, CLARITY Extension and ORACLE-MS trials (see Table 2 for trial characteristics) [39]. From the developed model, inferences about the time for return to a given lymphopenia grade from a certain ALC nadir can be made and visualised, which is of use for dosing decisions. This ALC model was further developed to incorporate measures of efficacy, as described below.
4.2 Efficacy Endpoint Modelling: Expanded Disability Status Scale and Relapse Rate
The Kurtzke Expanded Disability Status Scale (EDSS) is the most commonly reported measure of disease progression as an indicator of treatment efficacy in MS. Seven functional systems, plus walking ability and reliance on aid, are assessed by a neurologist to give an overall score of between 1 and 10, with 10 representing death due to MS [40]. In patients in the CLARITY study receiving Cladribine Tablets 3.5 mg/kg over 2 years, followed by 2 years of placebo in the CLARITY Extension study, the risk of 3-month confirmed EDSS progression was lower than in the placebo group during active treatment. The proportion of patients free from sustained disability progression was maintained for more than 2 years after the last dose of active treatment, confirming the durability of the efficacy of Cladribine Tablets [4].
An item response theory (IRT) model was developed to characterise disease progression in MS using data from the CLARITY study, and based on the individual functional measures of the EDSS. Simulations representing the CLARITY study showed that the typical patient receiving placebo treatment will progress 0.16 EDSS points over 2 years. The final model assessed both exposure-dependent and -independent drug effects, with the exposure-independent effect suggesting that treatment with Cladribine Tablets significantly slows disease progression rate by 20% compared with placebo over 2 years across a wide range of cumulative doses. In addition, an exposure-dependent reduction of 45% in the rate of disease progression is suggested in a typical patient receiving a dose of 240 mg [41]. IRT allows for quantification of the information content of the individual components of the EDSS. Four of 8 EDSS components contained 80% of the total information in the studied population. Simulations have demonstrated that a proposed reduction in the number of components will not hamper determination of the proportion of patients experiencing a 3-month sustained disease progression. Furthermore, IRT has demonstrated the advantage of increasing the detection power of potential drug effects in MS clinical trials (data on file).
In addition, a repeated time-to-event model of relapse rate (RR) data from the CLARITY, CLARITY Extension and ORACLE-MS studies was developed to characterise the dose–exposure–response relationship of Cladribine Tablets. The RR model predicted an exposure-dependent decrease in relapse hazard versus placebo. The hazard of experiencing a relapse during the study was positively related to baseline disease activity (i.e. the annualised number of relapses prior to the study), which supports the findings of previous studies showing that the number of relapses prior to entry into clinical trials is a predictor of on-study RR in individual patients [39]. The dose response is illustrated in Fig. 5, depicting the model-derived relationship between drug exposure (in an effect compartment) and the effect on the hazard of developing a relapse. It shows that for high exposure, the likelihood of having a relapse may be reduced by up to 72% at the end of year 2. Superimposed on top of the relationship is the range of effect–compartment exposure associated with either a 3.5 or 5.25 mg/kg cumulative dose of Cladribine Tablets (coloured horizontal bars). This highlights that the recommended dose of 3.5 mg/kg over 2 years is truly appropriate in reducing the risk of relapses; increasing the dose would not substantially increase the effect on the underlying hazard, while decreasing the dose would move the exposure to the steeper part of the dose–response curve, which is likely to be associated with a marked drop in efficacy (data on file).

Model-derived exposure–effect relationship, together with the range of cladribine effect compartment exposure at the end of year 2 of the CLARITY study. The black line represents the model-derived relationship between cladribine exposure in the effect compartment (x-axis) and the effect (y-axis), which is the drug-dependent factor multiplying the underlying hazard of having a relapse that exists in the absence of treatment. The grey line represents the Emax, estimated at 72%. The 5th–95th range of effect compartment exposure at the end of year 2 (based on subjects in the CLARITY trial) following the 3.5 mg/kg (blue line) and 5.25 mg/kg (green line) regimens is also shown, with the black dots representing the respective median exposure. Emax maximum obtainable effect
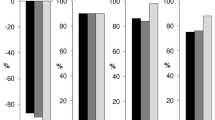
4.3 Relationship between ALC and Efficacy of Cladribine Tablets
The relationship of ALC with the 2 efficacy endpoints described in the previous section (EDSS and RR) were analysed using data from the CLARITY and CLARITY Extension studies. Predictions from the model of 4-year hazard of relapse, and survival for experiencing a first relapse, for a patient with 1 relapse in the previous 12 months for placebo and each of the 5 cladribine treatment sequences across the CLARITY and CLARITY Extension studies are shown in Fig. 6a, b. Similarly, predictions from the model of 4-year progression of EDSS are shown in Fig. 6c. These figures demonstrate that a clear drug effect of Cladribine Tablets is accounted for via individual changes from baseline of ALC. Decreasing ALC relative to baseline is linked to a clinical benefit in terms of the hazard of occurrence of a relapse (77% decrease in ALC will produce 25% maximal effect) and disease progression, as measured by EDSS (33% decrease in ALC is needed to reach 50% of maximal effect).

Adapted from Novakovic et al. [39]. © The American College of Clinical Pharmacology 2018
Predictions of 4-year hazard and survival for experiencing a first relapse in patients with 1 relapse in the previous 12 months, together with predictions of 4-year EDSS time-course for the typical subject. The lines represent predictions from the model for a the hazard, b survival (fraction relapse-free patients), and c EDSS progression over 4 years following treatment according to five treatment sequences: (1) 4 years of placebo (red); (2) 4 years of Cladribine Tablets, 7 mg/kg cumulative dose (brown dashes); (3) 2 years of Cladribine Tablets followed by 2 years of placebo, 3.5 mg/kg cumulative dose—represents the recommended treatment sequence (green); (4) 2 years of placebo followed by 2 years of Cladribine Tablets, 3.5 mg/kg cumulative dose (blue dashes); and (5) 2 years of Cladribine Tablets followed by 2 years of placebo, 5.25 mg/kg cumulative dose (purple). EDSS Expanded Disability Status Scale.
The relative reduction in ALC from baseline (i.e. at the individual level) was found to be a significant predictor of both time to relapse and EDSS time-course. The effect of Cladribine Tablets on the reduction of RR, and slowing disease progression as measured by EDSS, was therefore demonstrated to be mediated, at least partially, through ALC dynamics. These findings suggest that ALC could play a role as a biomarker related to the probability of relapses [39].
5 Measures to Reduce the Risk of Severe Lymphopenia
A clinical trial simulation of data from the CLARITY study was conducted using a generated virtual population of patients treated with a 3.5 mg/kg cumulative dose of Cladribine Tablets [42]. The objective of this analysis was to investigate the impact of cladribine treatment guidelines on the occurrence of qualifying relapses in subjects with relapsing–remitting MS who would require a longer time to recover from grade 2–4 lymphopenia before receiving the second year of cladribine treatment. Simulation of RR dynamics in virtual patients under such dosing scenarios, i.e. taking into account ALC values, showed that the postponement of treatment with Cladribine Tablets due to lymphopenia up to 6 months would not impact the effect of Cladribine Tablets on the probability of relapses during the second year of treatment (Fig. 7). In addition, in those who qualified for postponements, the proportion reaching grade 3/4 lymphopenia at any time in the study was decreased when the mitigation rule was applied. This supports treatment guidelines for Cladribine Tablets for managing the risk of severe lymphopenia, while preserving the efficacy of Cladribine Tablets.

Adapted from Terranova et al. [42]. © The Authors 2018
Repeated time-to-event model predictions of relapse risk for the typical patient (weight = 69.3 kg; creatinine clearance = 104.5 mL/min; number of relapses in the 12 months preceding study entry = 1) treated without postponement and with postponements of different month blocks (from 1 month, up to 9 months) during year 2.
6 Conclusions
This review of the clinical pharmacology of Cladribine Tablets in patients with MS is complementary to the previous review of parenteral cladribine in oncology patients by Liliemark [18]. The PK of Cladribine Tablets are well-described in the indicated patient population, and the application of PD modelling represents an innovative approach to data generation that is becoming increasingly relevant for health authorities and practitioners. Cladribine Tablets have a low drug–drug interaction liability, which is important for patients with comorbidities receiving polypharmacotherapy; the cardiac safety characteristics of Cladribine Tablets are favourable.
Cladribine Tablets lead to a selective reduction in lymphocyte counts and are associated with durable efficacy relative to the fast disposition in plasma, as well as short-term treatment posology in each of the 2 treatment years. The short-term treatment posology of Cladribine Tablets represents a potential convenience to patients requiring chronic MS therapy and will likely improve treatment adherence, which is a common problem with long-term treatment of MS; however, prospective adherence studies would be needed to confirm this assumption.
Treatment guidelines for Cladribine Tablets based on ALC can be used to manage the risk of severe lymphopenia, while preserving efficacy.
Change history
01 August 2018
The cladribine prodrug is phosphorylated intracellularly to its active product, 2-chlorodeoxyadenosine triphosphate (Cd-ATP), by deoxycytidine kinase.
References
Giovannoni G, Butzkueven H, Dhib-Jalbut S, Hobart J, Kobelt G, Pepper G, et al. Brain health: time matters in multiple sclerosis. Mult Scler Relat Disord. 2016;9(Suppl 1):S5–48.
Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–17.
Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Soelberg Sorensen P, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):416–26.
Giovannoni G, Soelberg-Sorensen P, Cook S, Rammohan K, Rieckmann P, Comi G, et al. Safety and efficacy of Cladribine Tablets in patients with relapsing–remitting multiple sclerosis: results from the randomized extension trial of the CLARITY study. Mult Scler. https://doi.org/10.1177/1352458517727603(Epub 1 Aug 2017).
Leist TP, Comi G, Cree BA, Coyle PK, Freedman MS, Hartung HP, et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol. 2014;13(3):257–67.
Merck Serono Australia Pty Ltd. MAVENCLAD® Tablets: Product Information. 2017 [11 Jan 18]. https://www.ebs.tga.gov.au/ebs/picmi/picmirepository.nsf/pdf?OpenAgent&id=CP-2010-PI-07339-3.
Merck Serono Europe Limited. MAVENCLAD 10 mg tablets: summary of product characteristics. 2017. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004230/WC500234561.pdf.
EMD Serono. MAVENCLAD Cladribine 10 mg Tablet: Product Monograph. 2017 [12/12/17]. https://pdf.hres.ca/dpd_pm/00042413.PDF.
Menzin J, Caon C, Nichols C, White LA, Friedman M, Pill MW. Narrative review of the literature on adherence to disease-modifying therapies among patients with multiple sclerosis. J Manag Care Pharm. 2013;19(1 Suppl A):S24–40.
Beutler E. Cladribine (2-chlorodeoxyadenosine). Lancet. 1992;340(8825):952–6.
Comi G, Hartung HP, Kurukulasuriya NC, Greenberg SJ, Scaramozza M. Cladribine Tablets for the treatment of relapsing-remitting multiple sclerosis. Expert Opin Pharmacother. 2013;14(1):123–36.
Salvat C, Curchod M, Guedj E, Peixoto H, Guerrier M, Wojcik J, et al. Cellular expression profiling of genes involved in the cladribine metabolic pathway: insights into mechanism of action in multiple sclerosis. Mult Scler. 2009;15:S74–5.
Soelberg-Sorensen P, Dangond F, Hicking C, Giovannoni G. P1141: innate immune cell counts in patients with relapsing-remitting multiple sclerosis (RRMS) treated with Cladribine Tablets 3.5 mg/kg in CLARITY and CLARITY extension. Mult Scler. 2017;23(S3):598.
Giovannoni G. Disease-modifying treatments for early and advanced multiple sclerosis: a new treatment paradigm. Curr Opin Neurol. 2018;31(3):233–43.
Wiendl H. Cladribine: an old newcomer for pulsed immune reconstitution in MS. Nat Rev Neurol. 2017;13:573–4.
Lindemalm S, Savic RM, Karlsson MO, Juliusson G, Liliemark J, Albertioni F. Application of population pharmacokinetics to cladribine. BMC Pharmacol. 2005;5:4.
Janssen-Cilag Ltd. Leustat injection: summary of product characteristics. 2014.
Liliemark J. The clinical pharmacokinetics of cladribine. Clin Pharmacokinet. 1997;32(2):120–31.
Savic RM, Novakovic AM, Ekblom M, Munafo A, Karlsson MO. Population pharmacokinetics of cladribine in patients with multiple sclerosis. Clin Pharmacokinet. 2017;56(10):1245–53.
European Medicines Agency. European Public Assessment Report: Mavenclad. European Medicines Agency; 2017.
Munafo A, Tran D, Marcus S, Ammoury N. An open-label randomized three-way crossover study on the absolute oral bioavailability of Cladribine Tablets administered to subjects with multiple sclerosis. Poster presented at the 21st Congress of the European Committee for Treatment and Research in Multiple Sclerosis and the 10th Annual Meeting of the Americas Committee for Treatment and Research in Multiple Sclerosis; 2005.
Merck Serono Europe Limited. MAVENCLAD 10 mg tablets: summary of product characteristics. 2017 [13/09/17]. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004230/WC500234561.pdf.
Fernandez-Calotti PX, Colomer D, Pastor-Anglada M. Translocation of nucleoside analogs across the plasma membrane in hematologic malignancies. Nucleos Nucleot Nucl. 2011;30(12):1324–40.
Pastor-Anglada M, Molina-Arcas M, Casado FJ, Bellosillo B, Colomer D, Gil J. Nucleoside transporters in chronic lymphocytic leukaemia. Leukemia. 2004;18(3):385–93.
Wright AMP, Gati WP, Paterson ARP. Enhancement of retention and cytotoxicity of 2-chlorodeoxyadenosine in cultured human leukemic lymphoblasts by nitrobenzylthioinosine, an inhibitor of equilibrative nucleoside transport. Leukemia. 2000;14(1):52–60.
Takenaka K, Morgan JA, Scheffer GL, Adachi M, Stewart CF, Sun D, et al. Substrate overlap between Mrp4 and Abcg2/Bcrp affects purine analogue drug cytotoxicity and tissue distribution. Cancer Res. 2007;67(14):6965–72.
Chan LMS, Lowes S, Hirst BH. The ABCs of drug transport in intestine and liver: efflux proteins limiting drug absorption and bioavailability. Eur J Pharm Sci. 2004;21(1):25–51.
de Wolf C, Jansen R, Yamaguchi H, de Haas M, de Wetering KV, Wijnholds J, et al. Contribution of the drug transporter ABCG2 (breast cancer resistance protein) to resistance against anticancer nucleosides. Mol Cancer Ther. 2008;7(9):3092–102.
Kearns CM, Blakley RL, Santana VM, Crom WR. Pharmacokinetics of cladribine (2-chlorodeoxyadenosine) in children with acute leukemia. Cancer Res. 1994;54(5):1235–9.
Scheible H, Laisney M, Wimmer E, Javornik A, Dolgos H. Comparison of the in vitro and in vivo metabolism of Cladribine (Leustatin, Movectro) in animals and human. Xenobiotica. 2013;43(12):1084–94.
Albertioni F, Lindemalm S, Reichelova V, Pettersson B, Eriksson S, Juliusson G, et al. Pharmacokinetics of cladribine in plasma and its 5ʹ-monophosphate and 5ʹ-triphosphate in leukemic cells of patients with chronic lymphocytic leukemia. Clin Cancer Res. 1998;4(3):653–8.
Elwi AN, Damaraju VL, Kuzma ML, Mowles DA, Baldwin SA, Young JD, et al. Transepithelial fluxes of adenosine and 2′-deoxyadenosine across human renal proximal tubule cells: roles of nucleoside transporters hENT1, hENT2, and hCNT3. Am J Physiol Renal Physiol. 2009;296(6):F1439–51.
Saven A, Cheung WK, Smith I, Moyer M, Johannsen T, Rose E, et al. Pharmacokinetic study of oral and bolus intravenous 2-chlorodeoxyadenosine in patients with malignancy. J Clin Oncol. 1996;14(3):978–83.
Marrie RA, Patten SB, Tremlett H, Wolfson C, Warren S, Svenson LW, et al. Sex differences in comorbidity at diagnosis of multiple sclerosis: a population-based study. Neurology. 2016. https://doi.org/10.1212/WNL.0000000000002481. (Epub 9 Mar 2016).
University of Washington. Drug interaction database program. 2017 [08/11/2017]. https://www.druginteractioninfo.org/.
Chtioui H, Millius C, Lammle B, Lauterburg BH. Concomitant treatment with lamivudine renders cladribine inactive by inhibition of its phosphorylation. Br J Haematol. 2009;144(1):136–7.
Garnett CE, Beasley N, Bhattaram VA, Jadhav PR, Madabushi R, Stockbridge N, et al. Concentration-QT relationships play a key role in the evaluation of proarrhythmic risk during regulatory review. J Clin Pharmacol. 2008;48(1):13–8.
Soelberg-Sorensen P, Dangond F, Hicking C, Giovannoni G. Absolute lymphocyte count recovery in patients with relapsing-remitting multiple sclerosis (RRMS) treated with Cladribine Tablets 3.5 mg/kg in CLARITY and CLARITY Extension (P5.379). Neurology. 2017;88(16):P5.379.
Novakovic AM, Thorsted A, Schindler E, Jönsson S, Munafo A, Karlsson MO. Pharmacometric Analysis of the relationship between absolute lymphocyte count and expanded Disability Status Scale and relapse rate, efficacy end points, in Multiple Sclerosis Trials. J Clin Pharmacol. https://doi.org/10.1002/jcph.1136(Epub 10 May 2018).
Kurtzke JF. Rating neurologic impairment in multiple-sclerosis: an Expanded Disability Status Scale (EDSS). Neurology. 1983;33(11):1444–52.
Novakovic AM, Krekels EHJ, Munafo A, Ueckert S, Karlsson MO. Application of item response theory to modeling of Expanded Disability Status Scale in multiple sclerosis. AAPS J. 2017;19(1):172–9.
Terranova N, Hicking C, Dangond F, Munafo A. Effects of postponing treatment in the second year of cladribine administration: clinical trial simulation analysis of absolute lymphocyte counts and relapse rate in patients with relapsing-remitting multiple sclerosis (RRMS). Clin Pharmacokinet. 2018. https://doi.org/10.1007/s40262-018-0693-y.
Acknowledgements
The authors would like to thank the patients, investigators, and co-investigators involved in the Cladribine Tablets clinical trial programme, as well as study teams at the participating centres at Merck KGaA, Darmstadt, Germany, and Merck Serono SA, Switzerland. The authors also thank Annick Seithel-Keuth for her contribution to the database search of transporter proteins in particular, and Marc Laisney for the initial analytical work in drug metabolism and PK. Additionally, we thank the personnel at Uppsala University, and Radojka Savic, for their contributions to the population PK analysis and PD modelling. The authors acknowledge Duncan Marriott of inScience Communications, Chester, UK, for medical writing support, funded by Merck KGaA, Darmstadt, Germany.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The studies summarised in this report were sponsored by EMD Serono Inc., a business of Merck KGaA, Darmstadt, Germany (in the US), and Merck Serono SA, Geneva, an affiliate of Merck KGaA, Darmstadt, Germany (rest of the world). Medical writing assistance was funded by Merck KGaA, Darmstadt, Germany.
Conflict of interest
Robert Hermann served as an external clinical pharmacology expert advisor for various aspects of several cladribine studies, and received financial support for research, consulting and training services from Merck KGaA, Darmstadt, Germany. Mats Karlsson is an employee of Uppsala University, which has performed contractual research for Merck KGaA, Darmstadt, Germany. Ana Novakovic and Markus Fluck are employees of Merck KGaA, Darmstadt, Germany, and Nadia Terranova and Alain Munafo are employees of Merck Serono SA, Switzerland, an affiliate of Merck KGaA, Darmstadt, Germany.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hermann, R., Karlsson, M.O., Novakovic, A.M. et al. The Clinical Pharmacology of Cladribine Tablets for the Treatment of Relapsing Multiple Sclerosis. Clin Pharmacokinet 58, 283–297 (2019). https://doi.org/10.1007/s40262-018-0695-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-018-0695-9