
Abstract
Background and Objective
Semaglutide is a glucagon-like peptide-1 analogue in development for the once-weekly treatment of type 2 diabetes mellitus. Its effect on the rate and extent of absorption of concomitant oral medications (metformin, warfarin, atorvastatin and digoxin) was evaluated in healthy subjects.
Methods
Subjects received metformin (500 mg twice daily for 3.5 days), warfarin (25 mg, single dose), atorvastatin (40 mg, single dose) or digoxin (0.5 mg, single dose) before and with subcutaneous semaglutide treatment at steady state (1.0 mg). Lack of drug–drug interaction was concluded if the 90% confidence intervals for the area under the plasma concentration–time curve ratio before and with semaglutide were within a pre-specified interval (0.80–1.25).
Results
Overall, metformin, warfarin, atorvastatin and digoxin pharmacokinetics were not affected to a clinically relevant degree with semaglutide co-administration. Estimated area under the plasma concentration–time curve ratios for all concomitant medications before and with semaglutide treatment were within the pre-specified interval. In addition, semaglutide did not affect maximum plasma concentration of concomitant medications to a relevant degree. Furthermore, no clinically relevant change in international normalised ratio response to warfarin was observed with semaglutide co-administration. Most adverse events with semaglutide treatment were mild or moderate. Adverse events with semaglutide and co-administered medication were comparable to those reported during treatment with semaglutide alone, and were mostly gastrointestinal related.
Conclusions
No clinically significant pharmacokinetic or pharmacodynamic interactions were identified and no new safety issues observed with combined treatment with semaglutide. This suggests that no dose adjustments should be required when semaglutide is administered concomitantly with these medications.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Semaglutide did not cause any clinically significant changes in the area under the plasma concentration–time curve of metformin, warfarin, atorvastatin or digoxin. Furthermore, semaglutide did not produce a clinically significant change in the anticoagulant effect of warfarin (as measured by changes in international normalised ratio response) in healthy subjects. |
Metformin, warfarin, atorvastatin and digoxin were well tolerated in combination with semaglutide 1.0 mg, and no new safety issues were identified. |
On the basis of these findings, these medications can be used without dose adjustment when taken concomitantly with semaglutide. |
1 Introduction
A range of pharmacotherapies, in addition to lifestyle changes, are available for the treatment of type 2 diabetes mellitus (T2D). These include both oral and injectable treatment options [1]. Owing to the progressive nature of the disease, most patients ultimately require a combination of pharmacological therapies to achieve glycaemic control [1,2,3]. Furthermore, although glucose control remains a central objective, it should be managed in the context of a comprehensive cardiovascular (CV) risk factor reduction programme, including smoking cessation, blood pressure control, lipid management and antiplatelet therapy [1]. Warfarin, digoxin and atorvastatin are indicated to prevent and treat CV disease and are commonly prescribed in patients with T2D who have a CV condition. Warfarin is a widely used anticoagulant to reduce the risk of thromboembolism [4]. Digoxin is used to treat atrial fibrillation, and atorvastatin is a lipid-lowering drug used to treat patients with high cholesterol and to lower the risk of CV disease in patients who are at an increased risk of heart disease [5, 6]. Hence, many individuals with T2D and co-morbidities, such as CV disease, may be taking multiple medications, which may have an increased risk of drug interactions [7].
One class of agents that is available for managing T2D is the glucagon-like peptide-1 receptor agonists (GLP-1RAs). In addition to their anti-hyperglycaemic effects, these agents have been associated with CV protection in two large CV outcome trials [8, 9]. Semaglutide is a GLP-1 analogue suitable for once-weekly administration, and is in development for the treatment of T2D. Structurally, it has 94% homology to native GLP-1, but with important structural modifications that make semaglutide suitable for once-weekly subcutaneous administration, with fully retained potency [10].
A previous study has shown that semaglutide is metabolised prior to excretion, with semaglutide-related material excreted in both urine and faeces (only 3% elimination of intact semaglutide in urine) [11]. In addition, cytochrome P450 (CYP) enzymes and transporters are not expected to be inhibited or induced by semaglutide [12]. However, similarly to native GLP-1, semaglutide delays gastric emptying during the first hour after a meal [13]. A delay in gastric emptying could potentially alter the gastrointestinal (GI) absorption kinetics of concomitantly administered therapies, thereby requiring dose adjustment.
Two studies were carried out in healthy subjects to investigate the effects of semaglutide on the pharmacokinetics of four therapies that are commonly co-administered with GLP-1RAs [14,15,16] and are characterised by differing properties of solubility and intestinal permeability [17]. Study 1 evaluated the effect of semaglutide 1.0 mg at steady state on the pharmacokinetics of concomitant metformin and warfarin. The pharmacodynamic (PD) profile of concomitant warfarin was also investigated. Metformin has high solubility and low permeability, and is a widely used dimethylbiguanide for the treatment of T2D. It is absorbed in the small intestine and has an oral bioavailability of 50–60% [18]. The absorption of metformin decreases with food, and may be altered when co-administered with compounds affecting gastric emptying [19]. Peak plasma concentrations of single-dose metformin occur 2–3 h after dosing [20]. Warfarin has high permeability and high/low solubility properties and is completely absorbed after oral administration, with a peak concentration occurring after 2–6 h [21, 22]. Warfarin comprises two enantiomers, one of which (R-warfarin) is metabolised by CYP1A2 and CYP3A4, while the other (S-warfarin; five times more potent than R-warfarin) is primarily metabolised by CYP2C9 [23].
Study 2 evaluated the effect of semaglutide on the pharmacokinetics of, respectively, concomitant atorvastatin and digoxin. Atorvastatin has low solubility and high permeability, and shows complete intestinal absorption when orally administered, but its systemic bioavailability is only 14% as it is subject to first-pass metabolism in the gut and liver. Atorvastatin is metabolised by CYP3A4, and drug–drug interactions with inhibitors of this system have been demonstrated [24]. Digoxin has low solubility and low permeability; it is incompletely (70%) absorbed from the GI tract and attains its maximum effect 6 h after oral dosing [5, 16]. Both warfarin and digoxin have a narrow therapeutic window, thus there is a relatively high possibility of drug–drug interactions. The purpose of these studies was to assess whether semaglutide altered the exposure of co-administered drugs commonly prescribed for the treatment of various co-morbidities associated with T2D.
2 Methods
2.1 Study Population
Male and female adults were eligible for inclusion if they met the following criteria: healthy; 18–55 years of age at the time of signing informed consent; body mass index 23.0–30.0 kg/m2 (Study 1) or 20.0–30.0 kg/m2 (Study 2); and glycosylated haemoglobin <6.5%. Key exclusion criteria included: any clinically significant disease history or systemic or organ disease; use of prescription or non-prescription systemic or topical medicinal products.
2.2 Study Design and Treatment
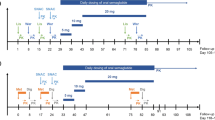
Two open-label, one-sequence, crossover clinical pharmacology studies investigated the effect of once-weekly subcutaneous semaglutide 1.0 mg steady state on the pharmacokinetics of metformin, warfarin, atorvastatin and digoxin, and the pharmacodynamics of warfarin (NCT02022254, NCT02243098). Semaglutide was administered subcutaneously once weekly in escalating doses of 0.25 mg (4 weeks), 0.5 mg (4 weeks) and 1.0 mg (4 weeks, to steady state). Dosing was continued for an additional 2 weeks to ensure drug–drug interactions were assessed at semaglutide steady state (Fig. 1). Interaction assessments [pharmacokinetic (PK) profiles] were carried out during in-house visits both before and with semaglutide treatment (1.0 mg).

Study design. Atorv atorvastatin, Dig digoxin, Met metformin, Ortho-OH ortho-hydroxylated atorvastatin metabolite, Para-OH para-hydroxylated atorvastatin metabolite, SS steady state, War warfarin. Asterisk Metformin was administered 3.5 days prior to the metformin pharmacokinetic sampling visit. Dagger Administered 48 h after the fifth dose of semaglutide 1.0 mg. Double dagger Administered 48 h after the sixth dose of semaglutide 1.0 mg. Section sign In Study 2, follow-up visit was at visit 12. The subjects received metformin and warfarin in Study 1, and atorvastatin and digoxin in Study 2
Two drugs were evaluated in each of the trials, separated by a 7-day washout, the length of which was based on the elimination half-life (t 1/2) of metformin (5–6 h, in Study 1) or atorvastatin (14 h; 20–30 h for metabolites, in Study 2) and the semaglutide dosing interval. Pharmacokinetic evaluations for atorvastatin were carried out before digoxin, as the t 1/2 of digoxin is 30–50 h, corresponding to a 10-day washout. Medication was taken following an overnight fast of ≥8 h, and no food or liquid (excluding water) was allowed until 4 h post-dose, when a standardised meal was served. All other oral drugs (other than trial drugs) were disallowed ±4 h during dosing visits. The dosing conditions, schedule and washout period in both studies ensured there was no effect of food or drug–drug interaction between the two drugs evaluated in each study. Single doses of warfarin (Coumadin® 25 mg, dispersed as five tablets of 5 mg; anticipated ratio of 50% S-warfarin and 50% R-warfarin), atorvastatin (one tablet of Hennig® 40 mg) and digoxin (two tablets of Digacin® 0.25 mg) were given before and with semaglutide treatment. Metformin was dosed twice daily (one tablet of Glucophage® 500 mg) in two periods of 3.5 days before and with semaglutide, to obtain steady-state conditions and minimise the potential of within-subject variation in metformin bioavailability [20].
The selected dose range of the study drugs was as recommended for standard therapeutic maintenance treatment, and subjects were dose escalated to semaglutide 1.0 mg steady state to investigate potential interactions at the highest intended treatment dose. Drug–drug interactions were also evaluated near the expected time of maximum semaglutide concentration at steady state. Adherence to specific food restrictions was also required, prior to the PK dosing days, to avoid potential interactions with CYP-metabolising enzymes. Both studies were conducted in compliance with the International Conference on Harmonisation Good Clinical Practice guidelines [25] and the Declaration of Helsinki [26].
2.3 Endpoints and Assessments
The primary objective was to evaluate whether semaglutide 1.0 mg at steady state altered the steady-state exposure of metformin and the single-dose exposure of warfarin, atorvastatin and digoxin. Primary endpoints were assessed before and with semaglutide treatment and included: area under the plasma concentration–time curve (AUC) for metformin during a dosing interval 0–12 h after the last of seven repeated doses; AUC for S- and R-warfarin 0–168 h after a single dose of warfarin; AUC for atorvastatin 0–72 h after a single dose of atorvastatin; and AUC for digoxin 0–120 h after a single dose of digoxin.
Secondary endpoints included: maximum plasma concentration (C max) and time to C max (t max) for metformin, S- and R-warfarin, atorvastatin and digoxin; incremental area under the international normalised ratio (INR) curve for warfarin 0–168 h (iAUCINR,0–168 h); maximum observed INR response; time to maximum observed INR response; and the C max, t max and t 1/2 of semaglutide at steady state. Safety and tolerability were also assessed.
Samples for PK analysis were collected at specific timepoints for each drug, all before and with semaglutide. Pharmacokinetic sampling for metformin was performed from pre-dose to 30 h post-dose (after the last of seven doses administered over 3.5 days); for warfarin from pre-dose to 168 h post-dose; for atorvastatin from pre-dose to 72 h post-dose; and for digoxin from pre-dose to 120 h post-dose. Semaglutide sampling was at regular time intervals ranging between 0 and 840 h after the last dose of 1.0 mg of semaglutide.
Plasma concentrations of metformin, warfarin and semaglutide were measured by a validated liquid chromatography-tandem mass spectrometry method after precipitation of the plasma protein. Atorvastatin and digoxin were measured by high-performance liquid chromatography with mass-spectrum detection. All known active metabolites of atorvastatin were measured, while metabolites with unconfirmed activity were not. The INR was determined from prothrombin time using a validated assay method.
Safety and tolerability assessments included treatment-emergent adverse events (AEs) and hypoglycaemic episodes. Hypoglycaemia was evaluated according to the American Diabetes Association classification [27]. Probable symptomatic hypoglycaemia was defined as an episode during which symptoms of hypoglycaemia were not accompanied by a low plasma glucose determination but presumably caused by a low plasma glucose level [27].
2.4 Analytical and Statistical Methods
2.4.1 Determination of Sample Size
For 20 completing subjects, Study 1 was calculated to have a statistical power of 89%, assuming ratios of 0.97, and within-subject standard deviations on the log scale of 0.1 for warfarin and 0.2 for metformin. For 24 completing subjects, Study 2 was calculated to have a statistical power of 90%, assuming ratios of 0.95, and within-subject standard deviations on the log-scale of 0.17 for atorvastatin and 0.18 for digoxin. For both studies, the statistical power was calculated using two one-sided t tests with a 5% significance level and a pre-specified interval of (log [0.80]; log [1.25]).
2.4.2 Definition of Analysis Sets and Statistical Analysis of Endpoints
Pharmacokinetic, PD and safety assessments were based on all subjects who received one or more doses of trial product. For PK and PD analysis, subjects had to have evaluable profiles before and with semaglutide treatment.
The primary endpoints were calculated using standard non-compartmental methods, by means of the linear trapezoidal method on the observed concentrations, using actual sampling timepoints. The AUCs were analysed using linear normal models, based on the log-transformed values, including semaglutide exposure (before or with) and subject as fixed effects. Estimated differences in exposure are presented on the original scale as ratios with 90% confidence intervals (CIs). No clinically significant effect on AUC was concluded if the 90% CI was within the interval of 0.80–1.25.
Maximum plasma concentration for metformin, S- and R-warfarin, atorvastatin and digoxin was analysed using the same model as that for the primary endpoints. For metformin and warfarin, the 90% CIs for ratios were evaluated against the pre-specified interval in the same way as for the primary endpoints. The PD endpoint for INR (iAUC) was also analysed using the same model as that for the primary endpoints. Adverse events were coded using Medical Dictionary for Regulatory Activities version 17.0 and summarised descriptively [28].
3 Results
3.1 Baseline Characteristics
Baseline characteristics were similar between the two studies (Table 1); 24 subjects were exposed to metformin and warfarin in Study 1, and 31 subjects to atorvastatin and digoxin in Study 2. One subject withdrew from Study 1 after the first dose of 0.25 mg of semaglutide (consent withdrawal). One subject was excluded from the PK and PD analyses in Study 1 owing to non-compliance with semaglutide administration and self-induced vomiting shortly after warfarin dosing. Five subjects withdrew from Study 2 (four because of AEs; one because of consent withdrawal).
3.2 Pharmacokinetics
The mean PK profiles for all drugs tested are shown in Fig. 2a–d. The estimated exposure ratios (ERs) for AUC and C max for all concomitant medications are shown in Table 2 and Fig. 3. Overall, the 90% CIs of the ERs for AUC for all concomitant medications were within the pre-specified interval. The C max values for metformin, S-warfarin and R-warfarin were slightly reduced when co-administered with semaglutide 1.0 mg; however, these were well within the pre-specified interval (Table 2). The C max for atorvastatin was reduced by 38% (ER 0.62) when co-administered with semaglutide, but was similar for digoxin.

Mean pharmacokinetic profiles of metformin (a), S-warfarin* (b), atorvastatin (c) and digoxin (d) before and with semaglutide treatment. Reference line for the lower limit of quantification. Values below the lower limit of quantification are imputed. Asterisk R-warfarin curves are not included here but followed a similar pattern to the S-warfarin curves

Estimated exposure ratios for metformin, warfarin, atorvastatin and digoxin before and with semaglutide treatment. Estimated ratios, before vs. with semaglutide treatment and 90% confidence intervals are shown. AUC area under the plasma concentration–time curve, C max maximum plasma concentration (C max limits for atorvastatin and digoxin were not predefined)
The t max values for metformin and t 1/2 of metformin, S- and R-warfarin, and digoxin were comparable when administered before and with semaglutide (data not shown). The observed t max for warfarin was delayed with semaglutide treatment (median t max 3.0 vs. 1.0 h). Likewise, C max occurred later for atorvastatin with semaglutide treatment, vs. before semaglutide treatment (median t max 2.0 vs. 0.7 h).
CYP2C9 genotyping was performed to account for any variability in the pharmacokinetics and pharmacodynamics of CYP2C9-metabolised S-warfarin. Results of this analysis showed an even distribution between intermediate and extensive metabolisers (Table 1). The PK properties of semaglutide observed in these studies (C max 43.1–48.6 nmol/L; median t max 36 h; and t 1/2 156–160 h) are in line with those from previous studies [12], indicating a high level of compliance with the treatment regimen during the trial.
3.3 Effect of Semaglutide on International Normalised Ratio Response to Warfarin
For warfarin, the INR iAUC and maximum INR response were similar before and with semaglutide treatment [response ratio 1.05 (90% CI 0.87–1.28) and 1.04 (90% CI 0.99–1.10), respectively; Fig. 4]. The median time to maximum INR response was 48 h (range 36–60 h) before semaglutide and 36 h (range 36–60 h) with semaglutide (Fig. 4).

Mean international normalised ratio (INR) profile for warfarin
3.4 Safety
No deaths or serious AEs were reported. In Study 1, 121 treatment-emergent AEs were reported in 23 of 24 subjects: all were mild (n = 106) or moderate (n = 15). No withdrawals because of AEs were reported. In Study 2, 271 treatment-emergent AEs were reported in 28 of 31 subjects; most were mild (n = 209) or moderate (n = 59). Three severe AEs were reported (one each during atorvastatin-only treatment, semaglutide-only treatment, and digoxin co-administered with semaglutide treatment), and were attributed to atrioventricular block, nausea and diarrhoea, respectively. Adverse events leading to withdrawal were reported in two subjects during atorvastatin-only treatment (atrioventricular block and ankle fracture) and in two subjects during semaglutide-only treatment (both GI AEs).
The most common AEs across both studies were GI related (Table 3). Gastrointestinal-related AEs were observed during treatment with both semaglutide alone and with all four concomitant medications: nausea was the most frequent. Overall, AEs reported with semaglutide and co-administered medication were comparable to those reported during semaglutide-only treatment. No bleeding was observed with warfarin.
No severe hypoglycaemic episodes were reported. In Study 1, there was one probable symptomatic hypoglycaemic episode during semaglutide-only treatment. In Study 2, four hypoglycaemic episodes were reported: one asymptomatic episode during semaglutide-only treatment, and three probable symptomatic episodes (atorvastatin alone, n = 1; semaglutide alone, n = 2).
4 Discussion
Drug–drug interaction studies are needed to identify exposure, safety and tolerability issues related to the concomitant use of typically prescribed oral medications. In these studies, the absorption pharmacokinetics of four separately administered oral drugs was assessed before and with semaglutide treatment, to evaluate the potential for semaglutide to affect the absorption of co-administered oral drugs. For all of the concomitant medications evaluated in this study, the ratio for total exposure (AUC) was within the pre-specified limits (0.80–1.25), suggesting that there are no drug–drug interactions between semaglutide and the co-administered drugs.
Overall, AUCs were similar for digoxin and warfarin with ratios between AUCs close to 1.0. Peak plasma concentrations for digoxin were similar, and slightly reduced for warfarin, when co-administered with semaglutide; however, these were within pre-specified limits. Likewise, metformin and atorvastatin plasma profiles had AUC ratios close to 1.0 and shared the same characteristics of being slightly right-shifted and with lowered maximum concentrations (10 and 38%, respectively).
The effect of semaglutide on gastric emptying has been assessed by the paracetamol absorption technique during a standardised meal [13, 29, 30]. The results indicate that there is a minor delay of gastric emptying during the early postprandial phase for subjects when treated with semaglutide, compared with placebo, but there is no overall effect on gastric emptying over a 5-h postprandial period [13]. A delayed gastric emptying may result in slower absorption, delayed t max and decreased C max of concomitant oral drugs, which would account for the trends observed in this and previous studies with GLP-1RAs [15, 31, 32]. Metformin is primarily absorbed in the small intestine (metformin concentration in the jejunum is 30–300 times higher than in plasma), thus a delayed gastric emptying might result in slower absorption [18]. In line with data reported for dulaglutide [33], the overall absorption of metformin was unchanged when co-administered with semaglutide, while the delayed gastric emptying resulted in a somewhat prolonged absorption, and thereby a slightly lowered C max.
The absorption pharmacokinetics of atorvastatin also appeared to be influenced by the GI transit time, showing a slightly prolonged absorption rate and a lowered C max; nevertheless, this did not influence overall exposure. The observed effect of semaglutide on peak concentration of atorvastatin was assessed to be of unlikely clinical relevance as the efficacy of atorvastatin has been shown to be poorly correlated with C max [34]. Similar absorption pharmacokinetics on C max have been observed for co-administration of atorvastatin with liraglutide [31], dulaglutide [33] and lixisenatide [35]. As with semaglutide, gastric emptying is decreased during the first hour after a meal with liraglutide [36]. On this basis, C max was expected to be lower with semaglutide. In contrast, delayed gastric emptying gives medications with low solubility or dissolution rate (such as digoxin) additional time to dissolve, which may increase exposure. This effect was observed for overall and peak exposure of co-dosing with albiglutide [15], while co-administration of liraglutide 1.8 mg lowered digoxin exposure [31]. However, for semaglutide and other GLP-1RAs, an unchanged exposure of concomitant digoxin has been observed [32, 33].
No clinically relevant change in the anticoagulant effect of warfarin was observed when it was co-administered with semaglutide, as measured by changes in INR. This finding, in addition to the observation that PK profiles of S- and R-warfarin are unaffected, supports the absence of a drug–drug interaction between semaglutide and warfarin. The INR of warfarin has also been shown to be unaffected by albiglutide administration [15]. A small change in the INR of warfarin was observed with exenatide administration, but was not clinically significant [32].
Overall, no new safety or tolerability issues related to semaglutide, either when administered alone or when co-administered with metformin, warfarin, atorvastatin or digoxin, were identified in these studies. On the basis of the results, no clinically relevant change in the tolerability and safety profiles of semaglutide, metformin, warfarin, atorvastatin or digoxin is anticipated when co-dosing at steady state.
In general, these findings confirm that semaglutide has a minor effect on the absorption of the drugs investigated, a group of drugs with various degrees of solubility, permeability and narrow therapeutic windows, and are consistent with results from previous studies with GLP-1RAs [31, 37,38,39,40]. The overall risk of clinically relevant interactions with semaglutide is therefore considered low, in the case of compounds that share the same properties as these studied drugs, including compounds with narrow therapeutic indexes; thus no dose adjustment in the clinical setting should be required.
5 Conclusion
Semaglutide treatment did not affect the overall exposure or C max of metformin, warfarin, atorvastatin or digoxin to a clinically relevant degree. In addition, semaglutide was well tolerated in combination with these drugs, and no new safety issues were identified. Therefore, these drugs can be used without dose adjustment when prescribed concomitantly with semaglutide.
References
Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38(1):140–9.
Fox CS, Golden SH, Anderson C, et al. Update on prevention of cardiovascular disease in adults with type 2 diabetes mellitus in light of recent evidence: a scientific statement from the American Heart Association and the American Diabetes Association. Diabetes Care. 2015;38(9):1777–803.
Bailey CJ, Blonde L, Del Prato S, et al. Global Partnership for Effective Diabetes Management. What are the practical implications for treating diabetes in light of recent evidence? Updated recommendations from the Global Partnership for Effective Diabetes Management. Diab Vasc Dis Res. 2009;6(4):283–7.
Warfarin. Summary of product characteristics. https://www.medicines.org.uk (2017). Accessed 6 Feb 2017.
Digoxin. Summary of product characteristics. https://www.medicines.org.uk (2017). Accessed 6 Feb 2017.
Atorvastatin. Summary of product characteristics. https://www.medicines.org.uk (2017). Accessed 6 Feb 2017.
Ibrahim IA, Kang E, Dansky KH. Polypharmacy and possible drug-drug interactions among diabetic patients receiving home health care services. Home Health Care Serv Q. 2005;24(1–2):87–99.
Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375(4):311–22.
Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375(19):1834–44.
Lau J, Bloch P, Schaffer L, et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J Med Chem. 2015;58(18):7370–80.
Roffel AF, Jensen L, van Lier JJ, et al. Absorption, metabolism and excretion of 3H-semaglutide following a single, subcutaneous dose in healthy male subjects. Diabetologia. 2015;58(suppl 1):S1–607 (#787).
Kapitza C, Nosek L, Jensen L, et al. Semaglutide, a once-weekly human GLP-1 analog, does not reduce the bioavailability of the combined oral contraceptive, ethinylestradiol/levonorgestrel. J Clin Pharmacol. 2015;55(5):497–504.
Hjerpsted J, Axelsen MB, Brooks A, et al. Semaglutide improves postprandial glucose and lipid metabolism and delays first-hour gastric emptying in subjects with obesity. In: American Diabetes Association 76th Scientific Sessions. New Orleans, LA; 2016 (#763).
Thomsen RW, Baggesen LM, Sogaard M, et al. Early glycaemic control in metformin users receiving their first add-on therapy: a population-based study of 4,734 people with type 2 diabetes. Diabetologia. 2015;58(10):2247–53.
Bush M, Scott R, Watanalumlerd P, et al. Effects of multiple doses of albiglutide on the pharmacokinetics, pharmacodynamics, and safety of digoxin, warfarin, or a low-dose oral contraceptive. Postgrad Med. 2012;124(6):55–72.
Hurren KM, Pinelli NR. Drug-drug interactions with glucagon-like peptide-1 receptor agonists. Ann Pharmacother. 2012;46(5):710–7.
Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20.
McCreight LJ, Bailey CJ, Pearson ER. Metformin and the gastrointestinal tract. Diabetologia. 2016;59(3):426–35.
Marathe PH, Wen Y, Norton J, et al. Effect of altered gastric emptying and gastrointestinal motility on metformin absorption. Br J Clin Pharmacol. 2000;50(4):325–32.
Graham GG, Punt J, Arora M, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50(2):81–98.
Holford NH. Clinical pharmacokinetics and pharmacodynamics of warfarin: understanding the dose-effect relationship. Clin Pharmacokinet. 1986;11(6):483–504.
Lindenberg M, Kopp S, Dressman JB. Classification of orally administered drugs on the World Health Organization Model list of Essential Medicines according to the biopharmaceutics classification system. Eur J Pharm Biopharm. 2004;58(2):265–78.
Sansone RA, Sansone LA. Warfarin and antidepressants: happiness without hemorrhaging. Psychiatry (Edgmont). 2009;6(7):24–9.
Lennernas H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet. 2003;42(13):1141–60.
European Agency for the Evaluation of Medicinal Products. International Conference on Harmonisation-World Health Organization. Guideline for good clinical practice. ICH Harmonised Tripartite Guideline. Good clinical practice. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002874.pdf (2017). Accessed 6 Feb 2017.
World Medical Association. Declaration of Helsinki. Ethical principles for medical research involving human subjects. 52nd WMA General Assembly, Edinburgh, October 2000. Last amended with Note of Clarification on Paragraph 29 by the WMA General Assembly, Washington, 2002; and Note of Clarification on Paragraph 30 by the WMA General Assembly, Tokyo, 2004. http://www.wma.net/en/30publications/10policies/b3/index.html.pdf. Accessed 6 Feb 2017.
Seaquist ER, Anderson J, Childs B, et al. Hypoglycemia and diabetes: a report of a workgroup of the American Diabetes Association and the Endocrine Society. Diabetes Care. 2013;36(5):1384–95.
Medical Dictionary for Regulatory Activities. MedDRA version 17.0 complex changes. http://www.meddra.org/sites/default/files/guidance/file/whatsnew_17_0_english.pdf (2017). Accessed 6 Feb 2017.
Willems M, Quartero AO, Numans ME. How useful is paracetamol absorption as a marker of gastric emptying? A systematic literature study. Dig Dis Sci. 2001;46(10):2256–62.
Sanaka M, Kuyama Y, Yamanaka M. Guide for judicious use of the paracetamol absorption technique in a study of gastric emptying rate of liquids. J Gastroenterol. 1998;33(6):785–91.
Malm-Erjefalt M, Ekblom M, Vouis J, et al. Effect on the gastrointestinal absorption of drugs from different classes in the biopharmaceutics classification system, when treating with liraglutide. Mol Pharm. 2015;12(11):4166–73.
Kothare PA, Soon DK, Linnebjerg H, et al. Effect of exenatide on the steady-state pharmacokinetics of digoxin. J Clin Pharmacol. 2005;45(9):1032–7.
Dulaglutide. Summary of product characteristics. https://www.medicines.org.uk (2017). Accessed 6 Feb 2017.
Cilla DD Jr, Whitfield LR, Gibson DM, et al. Multiple-dose pharmacokinetics, pharmacodynamics, and safety of atorvastatin, an inhibitor of HMG-CoA reductase, in healthy subjects. Clin Pharmacol Ther. 1996;60(6):687–95.
Lixisenatide. Summary of product characteristics. https://www.medicines.org.uk (2017). Accessed 6 Feb 2017.
van Can J, Sloth B, Jensen CB, et al. Effects of the once-daily GLP-1 analog liraglutide on gastric emptying, glycemic parameters, appetite and energy metabolism in obese, non-diabetic adults. Int J Obes (Lond). 2014;38(6):784–93.
Buse JB, Rosenstock J, Sesti G, et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet. 2009;374(9683):39–47.
Buse JB, Nauck M, Forst T, et al. Exenatide once weekly versus liraglutide once daily in patients with type 2 diabetes (DURATION-6): a randomised, open-label study. Lancet. 2013;381(9861):117–24.
Garber A, Henry R, Ratner R, et al. Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet. 2009;373(9662):473–81.
Marre M, Shaw J, Brandle M, et al. Liraglutide, a once-daily human GLP-1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with type 2 diabetes (LEAD-1 SU). Diabet Med. 2009;26(3):268–78.
Author contributions statement
All authors participated in the trial design. FW and GG took part in the conduct of the trial and the data collection. HH, JDK, AGH, JBJ and TWA took part in the data analysis. All authors interpreted the data and participated in writing the manuscript together with medical writing services provided by the sponsor. All authors have read the report critically and approved the submitted version.
Acknowledgements
We thank all the participants, investigators and trial-site staff who were involved in the conduct of the trials. We also thank Bue F. Ross Agner and Mikkel Agersnap for their review and input to the manuscript, and Lisa Langley (AXON Communications) for medical writing and editorial assistance. All contributors received compensation from Novo Nordisk.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
FW and GG have no conflicts of interest to declare. HH, DK, AH, JJ and TA are full-time employees of Novo Nordisk.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Funding
This study was funded by Novo Nordisk. AXON Communications provided editorial and medical writing assistance, which was funded by Novo Nordisk.
Informed consent
Informed consent was obtained from all individual participants included in the study. Additional informed consent was obtained from all individual participants for whom identifying information is included in this article.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hausner, H., Derving Karsbøl, J., Holst, A.G. et al. Effect of Semaglutide on the Pharmacokinetics of Metformin, Warfarin, Atorvastatin and Digoxin in Healthy Subjects. Clin Pharmacokinet 56, 1391–1401 (2017). https://doi.org/10.1007/s40262-017-0532-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-017-0532-6