
Abstract
Purpose
The aims of this study were to characterize the concentration–time course of cladribine (CdA) and its main metabolite 2-chloroadenine (CAde), estimate interindividual variability in pharmacokinetics (PK), and identify covariates explaining variability in the PK of CdA.
Methods
This population PK analysis was based on the combined dataset from four clinical studies in patients with multiple sclerosis (MS): three phase I studies, including one food and one drug–drug interaction study, and one phase III clinical study. Plasma and urine concentration data of CdA and CAde were modeled simultaneously.
Results
The analysis comprised a total of 2619 CdA and CAde plasma and urine concentration observations from 173 patients with MS who received an intravenous infusion or oral tablet doses of CdA as a single agent or in combination with interferon (IFN) β-1a. CdA PK data were best described by a three-compartment model, while a one-compartment model best described the PK of CAde. CdA renal clearance (CLR) was correlated with creatinine clearance (CLCR), predicting a decrease in the total clearance of 19%, 30% and 40% for patients with mild (CLCR = 65 ml/min), moderate (CLCR = 40 ml/min) and severe (CLCR = 20 ml/min) renal impairment, respectively. Food decreased the extent of CdA absorption by 11.2% and caused an absorption delay. Coadministration with IFNβ-1a was found to increase non-CLR (CLNR) by 21%, resulting in an increase of 11% in total clearance.
Conclusions
Both CdA and CAde displayed linear PK after intravenous and oral administration of CdA, with CdA renal function depending on CLCR.
Trial registration number for study 25643: NCT00213135.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The pharmacokinetics of cladribine (CdA) and its main metabolite 2-chloroadenine (CAde) in patients with multiple sclerosis were described after intravenous and oral administration of CdA. |
Both renal and non-renal clearances were estimated for CdA and CAde. |
Covariate effects of food on CdA absorption and of coadministration of interferon β-1a on non-renal clearance were characterized. |
1 Introduction
Cladribine (2-chloro-2′-deoxyadenosine; CdA), is a synthetic deoxyadenosine analog that is activated by intracellular phosphorylation in specific cell types, resulting in a targeted reduction of circulating lymphocytes [1]. Injectable formulations of CdA have been approved in many countries for the treatment of hematological malignancies [2]. It was hypothesized that CdA may have beneficial effects in the treatment of autoimmune disorders such as multiple sclerosis (MS) due to its selective lymphocyte-reducing properties [2]. This led to the development of an oral formulation of CdA, cladribine tablets, for investigation in relapsing–remitting MS. Cladribine tablets demonstrated significant efficacy compared with placebo on clinical and neuroimaging measures in the large, phase III, 96-week CLARITY (CLAdRIbine Tablets Treating MS OrallY) study [3]. The development program for cladribine tablets, as well as long-term follow-up of patients exposed to cladribine tablets, is ongoing.
The population pharmacokinetics (PK) of CdA have been previously studied in patients with malignancies after administration of parenteral, intravenous and oral solutions [4]. We studied the population PK in patients with MS because physiological differences related to disease pathology in patients with malignancies and those with MS could lead to different kinetic and metabolic behavior for CdA. In addition, the dosing level and schedule and drug formulation that were proposed for use in MS are different from those routinely used in the oncology setting. Data from the main metabolite of CdA, 2-chloroadenine (CAde), were also available for this analysis; it has been documented that metabolite data can provide additional information about the kinetics of the parent drug itself [5].
The overall objective of our analysis was to assess the population PK of CdA and its main metabolite, CAde, after oral tablet administration in patients with relapsing–remitting MS. The specific aims of the analysis included quantifying different routes of CdA elimination (renal and non-renal); assessing the effect of food on CdA absorption and bioavailability; and assessing potential drug–drug interactions of CdA with interferon (IFN) β-1a.
2 Methods
2.1 Clinical Trials
This population PK analysis evaluated data obtained from four clinical studies of CdA (studies 25803, 26127, 26486 and 25643 [CLARITY; trial registration number: NCT00213135]).
Study 25803 was a phase I, open-label, randomized, two-period, two-sequence crossover study investigating the PK of CdA and its metabolites following the administration of a single oral tablet and a single intravenous dose in patients with MS. Sixteen patients were enrolled this study. Patients received CdA 3 mg intravenously as a single 1-h infusion or as a single 10 mg tablet, with a minimum 10-day washout between each treatment. In each treatment period, blood and urine samples were collected for the evaluation of CdA and CAde, pre-dose (within 5–30 min before dosing) and at 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 72, 96 and 120 h post-dose. Urine was collected pre-dose (within 2 h before dosing) and at 0–4, 4–8, 8–12, 12–24, 24–48 and 48–72 h post-dose.
Study 26127 was a phase I, randomized, two-way, crossover study investigating the effects of food on the PK of CdA administered as oral tablets to patients with MS (16 patients).
Patients were randomized (1:1) to one of two treatment sequences. One group received the treatments in the ‘fed then fasted’ sequence, and the other group received the treatment in the ‘fasted then fed’ sequence. Each treatment consisted of a single 10 mg cladribine tablet. In both treatment periods, blood samples for the assessment of CdA concentration were collected pre-dose (within 5–30 min prior to dosing), and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48 and 72 h post-dose.
Study 26486 was an open-label, multiple-dose study assessing the effects of oral cladribine tablets on the PK of subcutaneous IFNβ-1a (Rebif®, Merck KGaA, Darmstadt, Germany) and vice versa, as well as the safety of combination treatment in patients with MS. Sixteen patients received one or two 10 mg cladribine tablets administered once-daily for 5 consecutive days (days 1–5). After at least 2 days, patients received IFNβ-1a as follows: 8.8 µg three times weekly (days 7–18; six doses), then 22 µg three times weekly (days 21–32; six doses), followed by 44 µg three times weekly (days 35–60; 12 doses). During the last week of IFNβ-1a treatment, therapy with cladribine tablets (50–100 mg in total) was coadministered for 5 consecutive days (days 56–60). Blood samples for PK analysis were taken pre-dose and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36 and 48 h post-dose at the following times: at the end of the initial treatment course of cladribine tablets (days 4–7); after 2 weeks of IFNβ-1a 44 µg (days 45–48); and after the last concomitant administration of cladribine tablets and IFNβ-1a (days 59–62).
Study 25643 (CLARITY) was a phase III, double-blind, placebo-controlled, 96-week study evaluating the safety and efficacy of cladribine tablets in patients with relapsing–remitting MS [3]. Patients were randomly assigned (1:1:1 ratio) to receive one of two cumulative doses of cladribine tablets or placebo. Depending on their actual body weight, patients took one or two cladribine 10 mg tablets (or matching placebo) per day over 4–5 days in either (1) weeks 1 and 5 of years 1 and 2, or (2) weeks 1, 5, 9, and 13 of year 1, followed by weeks 1 and 5 of year 2, for a cumulative dose of 3.5 and 5.25 mg/kg, respectively [3]. Detailed information on the dosing of cladribine tablets in the CLARITY study is provided in electronic supplementary Tables 1 and 2. Samples from 125 patients were available for population analysis.
2.2 Bioanalytical Analysis
Bioanalytical methods were developed and validated to support the clinical program for cladribine, as described briefly below. CdA and CAde were quantified by liquid chromatography-tandem mass spectrometry (LC-MSMS) methods, validated for human plasma and urine. The lower limit of quantification (LLOQ) for both CdA and CAde was 100 pg/mL, and precision and accuracy of quality controls were assessed for the range of 100–45,000 pg/mL of CdA and CAde. Intra- and interbatch precision was below 7.5%. Furthermore, intrabatch accuracy of quality controls was between −8.7 and 5.8%, and −1.3 and 4.8% for CdA and CAde, respectively, and interbatch accuracy of quality controls was included in the range −1.7–1.6%, and −0.1–3.3% for CdA and CAde, respectively. No interfering chromatographic peaks were observed in blank plasma samples for CdA, CAde and the internal standards.
2.3 Population Pharmacokinetic (PK) Analysis
2.3.1 Structural Model Development
The model-building procedure included the development of five key models/steps described below and summarized in Table 1.
-
1.
Development of a model for CdA and CAde, both in plasma and urine, after intravenous administration of CdA. Data used for this step were from study 25803 (after intravenous administration only)—Model I.
-
2.
An extension of Model I with the absorption model in the fasted state. Data were extended with the oral administration data from study 25803 (CdA and CAde, both in plasma and urine)—Model II.
-
3.
An extension of Model II with the absorption model in the fed state. Data were extended with the data from study 26127—Model III.
-
4.
An extension of Model III with the model for multiple dosing of CdA and its interaction with IFNβ-1a. Data were extended with the data from study 26486—Model IV.
-
5.
Extension of Model IV with the data from study 25643—Model V.
PK modeling began with a three-compartment model with first-order (linear) elimination and first-order absorption for oral administration. More complex PK models with respect to absorption and disposition were evaluated for both CdA and CAde.
2.3.2 Statistical Model Development
Additive, log-additive, proportional and combined-error models were explored for residual variability. Between-patient variability in residual error was evaluated, and additive and/or exponential error models were explored for between-patient variability in the model parameters. A diagonal Ω-structure was initially employed and the inclusion of off-diagonal elements was investigated.
2.3.3 Covariate Model Development
The identification of covariates was undertaken using ‘Stepwise Covariate Model-Building’ using Perl-speaks NONMEM (PsN; version 2.3.2 and higher) [6]. This method involved stepwise testing of linear and non-linear relationships in a forwards inclusion (change in objective function value [ΔOFV] of 6.63; p < 0.01 for 1 degree of freedom [DF]) and backwards exclusion (ΔOFV of 10.83; p < 0.001 for 1 DF) procedure [7]. The resultant final model contained covariates that met the predefined statistical criteria. In addition, covariates would only be retained on the basis of their relevance, in view of the purpose of the model. The covariates tested were concomitant IFNβ-1a administration (16 yes/157 no), food state (16 fed/16 fasted/141 unknown), and markers of renal function, i.e. creatinine clearance [CLCR], based on the Cockcroft–Gault equation, sex, age and body weight.
2.3.4 Modeling Methodology and Software
The population PK analysis was performed using the non-linear mixed effects modeling approach using NONMEM (version VI and higher; Icon Development Solutions, Hanover, MD, USA) [8]. The R-based version of Xpose (version 4.0 and higher) was used to produce standard goodness-of-fit plots [6]. Perl (version 5.8.8; http://www.perl.org) and PsN were used for model evaluation and automatic covariate model-building [6].
The NONMEM estimation methods used were first-order conditional (FOCE) and FOCE with interaction (FOCEI). The stability of NONMEM models was assessed on the basis of acceptable basic goodness-of-fit plots, number of significant digits ≥3 for all estimated parameters, successful covariance step, estimates of typical patient parameters (Ө’s) not close to a boundary, and stability check performed for a selected basic model (the model finds the global minimum when the initial values are altered in each direction [i.e. each parameter, one at a time] by a large factor [10 in this analysis]). Model selection was based on the comparison of full versus reduced models using the log-likelihood criterion (the difference in the minimum OFV between hierarchical models was assumed to be Chi-square distributed with degrees of freedom equal to the difference in the number of parameters between models), goodness-of-fit plots (e.g. relevant residuals against time randomly distributed around zero), and scientific plausibility of the model.
2.3.5 Model Evaluation
To evaluate whether the estimated fixed- and random-effect parameters adequately describe data, 1000 Monte Carlo simulation replicates of the original dataset were generated using each of the five key population PK models (Model I through Model V). The data were plotted versus time along with the summary statistics computed from the simulated data with 5th, 50th, and 95th percentiles including uncertainty. The coincidence between the original data and simulated data demonstrated the predictive ability of fixed effects parameters in the final model. The visual predictive check was stratified on variables of interest, such as route of administration, food state and coadministration with IFNβ-1a.
A stratified non-parametric bootstrap procedure was performed to assess the uncertainty and stability of all key models. Confidence intervals were calculated from the successfully converged runs, and a large, non-parametric bootstrap (n = 1000) was performed for Models I and II only due to long run times. A shorter non-parametric bootstrap including 30 and 100 samples was performed to assess standard errors for Models IV and V, respectively.
3 Results
3.1 Data
A summary of the studies included in this analysis is provided in Table 2. PK profiles from a total of 173 patients were used in this analysis and a summary of the study demographics is shown in Table 3. The original dataset contained 4790 concentration–time records; 45% of records were excluded for various reasons, such as being below the limit of quantification (BLQ) and missing sampling times, leaving 2619 concentration records for population analysis. Most of the excluded data (96.5%) were BLQ records, coming mostly from two studies: approximately one-third of the BQL samples from study 25803 with an extended follow-up period (up to 120 h), and approximately two-thirds of the BQL samples from study 25643, where approximately half the samples were collected before the administration of the CdA dose.
3.2 Population PK Model
3.2.1 Structural Model Development
Model I A three-compartment model best described the PK of CdA after intravenous administration, and a one-compartment model best described the PK of the main CdA metabolite, CAde. Available urine data enabled estimation of renal clearance (CLR) for both CdA and CAde. For CdA, CLR was implemented as a linear function of CLCR, while renal elimination of the metabolite was best described with a saturable model. Separate non-CLR (CLNR) and CLR for both the parent drug and metabolite could be estimated. The fraction of metabolized drug could be assessed as CLNR/total clearance = 0.536. Renal elimination of CdA accounted for approximately 50% of total elimination, while renal elimination of the metabolite was minor. Metabolite profiles were well described, as indicated by the low residual variability (13.4%), even though no separate interindividual variability was estimated in the metabolite disposition parameters.
Model II A first-order absorption model was sufficient to describe the absorption of CdA in the fasted state. The introduction of absorption delay models, such as a lag-time model and a transit-compartment model, offered some improvement in the fit. However, the observed delay could be assigned to only a few individuals and therefore it was not kept in the model. Bioavailability was estimated to be 45.5%.
Model III Inclusion of the data from the fed state, with the aim of assessing the effect of food on absorption parameters, required elaboration of the absorption model to describe the effect of food on the absorption delay. The transit-compartment model best described the absorption delay in the data from the fed state. This model offered an improvement compared with first-order absorption with a lag time. Mean transit time was estimated to be 1.04 h, and the number of transit compartments was estimated to be 2.44. Bioavailability decreased with food by approximately 14%.
Model IV Data from the multiple-dosing schedule (study 26486) did not contain information on food state and therefore absorption parameters were modeled separately. A potential effect of IFNβ-1a coadministration on CdA bioavailability and CLNR was tested. The bioavailability of CdA in study 26486 was generally lower compared with other studies; however, no significant differences in bioavailability were found between CdA administered alone and CdA administered with IFNβ-1a. IFNβ-1a coadministration led to an increase in CLNR of CdA of approximately 22%, which was statistically significant (p < 0.05) and associated with a slight improvement in predictive checks.
Model V The addition of phase III data only required the addition of a lag time, which improved the fit during the absorption phase. Overall, with the addition of phase III data, parameters changed very little compared with the parameters from the final phase I model. Due to sparse sampling at late time points, the phase III data were lacking information relating to CLR; however, due to the presence of metabolite data, they supported good estimation of CLNR and bioavailability. Good correspondence between phases I and III was observed for almost all parameters, although some changes in the rate of absorption were observed. The population parameter point estimates and their uncertainty, assessed via non-parametric bootstrap (n = 100), are shown in Table 4, and the structure of the final model is shown in Fig. 1.

Schematic representation of the final model. CdA cladribine, Ka absorption rate constant, Ktr transfer rate constant, F bioavailability, TC2 second transit compartment, TCn: nth transit compartment, per 1 first peripheral compartment, per 2 second peripheral compartment, CL clearance, CRCL creatinine clearance, CAde 2-chloroadenine
The final model was used to generate cladribine profiles following 5 days of cladribine treatment (dose = 10 mg) for a typical patient with normal renal function. CdA concentrations were then predicted using three different values of CLCR, mimicking 3 degrees of severity of renal impairment, by incorporating those values into the developed model. The predicted decrease in total clearance was 19%, 30% and 40% for patients with mild (CLCR = 65 ml/min), moderate (CLCR = 40 ml/min) and severe (CLCR = 20 ml/min) renal impairment, respectively.
3.2.2 Covariate Model Development
Covariate analysis revealed no significant influence of demographic covariates (body weight, age and sex) on PK parameters, in addition to their impact on renal function mediated though CLCR. Additional investigation of individual predicted CLNR against aspartate aminotransferase (AST), alanine aminotransferase (ALT) and bilirubin did not show any association between liver function markers and CLNR.
3.2.3 Model Evaluation
The final model was evaluated by a visual predictive check stratified on variables of interest, such as route of administration, food state and coadministration with IFNβ-1a (Figs. 2, 3, 4). The stratified non-parametric bootstrap (n = 30–1000) was performed for all key models in order to compute relative standard errors (RSE) for parameters.

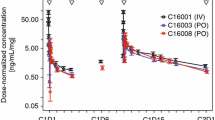
Visual predictive checks for CdA and CAde in plasma of intravenously and orally administered CdA. Light blue shaded area indicates simulated median with uncertainty; pink shaded area indicates simulated 5th and 95th percentiles with uncertainty; solid blue line indicates observed median; dashed blue line indicates observed 5th and 95th percentiles. a Based on 193 samples from 16 subjects; b based on 211 samples from 16 subjects; c based on 470 samples from 125 subjects; d based on 81 samples from 16 subjects; e based on 108 samples from 16 subjects; and f based on 466 samples from 125 subjects. CdA cladribine, md multiple dosing, CAde 2-chloroadenine, IV intravenous

Visual predictive checks for CdA and CAde in urine of intravenously and orally administered CdA. Light blue shaded area indicates simulated median with uncertainty; pink shaded area indicates simulated 5th and 95th percentiles with uncertainty; solid blue line indicates observed median; dashed blue line indicates observed 5th and 95th percentiles. a Based on 83 samples from 16 subjects; b based on 83 samples from 16 subjects; c based on 40 samples from 16 subjects; d based on 49 samples from 16 subjects. CdA cladribine, CAde 2-chloroadenine, IV intravenous

Visual predictive checks for CdA in plasma after oral administration of CdA in a fasted and fed conditions, and b with and without IFNβ-1a coadministration. Light blue shaded area indicates simulated median with uncertainty; pink shaded area indicates simulated 5th and 95th percentiles with uncertainty; solid blue line indicates observed median; dashed blue line indicates observed 5th and 95th percentiles. (1) Based on 417 samples from 16 subjects; (2) based on 210 samples from 16 subjects; (3) based on 210 samples from 16 subjects; (4) based on 209 samples from 16 subjects. CdA cladribine, IFN interferon
4 Discussion
This analysis characterized the population PK of both CdA and CAde following oral and intravenous administration of CdA in patients with relapsing–remitting MS. The final population PK model provided a good description of the PK data of CdA in patients with MS. Goodness-of-fit criteria revealed that the final model was consistent with the observed data and no systematic bias remained. The model evaluation results provided evidence that both the fixed and random effect components of the final model were reflective of the observed data.
The PK data were described reasonably well using a three-compartment PK model, which is consistent with previous findings [4]. Availability of the urine data enabled separation of the CLR and CLNR, and the typical population parameter estimates from the final model for these two elimination routes were CLR = 22.2 L/h and CLNR = 23.4 L/h, indicating that both elimination pathways are equally represented. The previous population PK analysis of CdA reported a value for total clearance of 39.3 L/h [4]; however, that analysis was carried out in terminally ill cancer patients, who are likely to have decreased function of vital organs and therefore lower clearance values. Once the differences in the mean CLCR between two populations are taken into account (4.86 L/h in oncology patients compared with 6.31 L/h in the MS population), the estimates of clearance become comparable (39.3 vs. 40.5 L/h). Moreover, the previous analysis was a retrospective analysis where the data were collected over long time periods and using different bioanalytical methods, which may not provide fully comparable results. CLR appeared to be higher than the glomerular filtration rate, indicating that other mechanisms, such as active secretion, are involved in CdA renal elimination, which is also consistent with previous reports [9].
Bioavailability of CdA after oral administration was 45.6%, which is consistent with previous findings [10, 11]. Coadministration of CdA with food appeared to decrease bioavailability to 40.5%, which is not expected to have a clinically meaningful impact. In addition, food also appeared to delay absorption.
Coadministration of CdA with IFNβ-1a appeared to have an effect on CdA clearance, which increased CLNR by 21%, resulting in an increase of total clearance by approximately 11.1%. However, although the modeling pointed to this effect on CLNR, it could also have been modeled on its bioavailability. Given the data available, it was not really possible to discriminate between the two. Furthermore, the observed effect could also be due to a period effect or interoccasion variability; however, these effects were confounded (owing to the study design) and further elaboration could not be supported with the available data. PK profiles of patients receiving CdA alone or coadministered with pantoprazole were also evaluated and no clinically relevant drug–drug interaction was found.
Covariates included in the model were fasted/fed state on absorption parameters, IFNβ-1a effect on CLNR, and CLCR on CLR of CdA. Sex, age and body weight have also been investigated and were not associated with the PK of CdA (except for their impact on renal function mediated though CLCR). A graphical investigation of individual predicted CLNR against AST, ALT and bilirubin did not show any association between liver function markers and CLNR, and the previous population analysis of CdA in cancer patients also revealed no significant covariate effect [4].
The PK of the main CdA metabolite, CAde, were well described by a one-compartment model. Apparent CLNR and volume of distribution for CAde were found to be 653 L/h and 365 L, respectively. The availability of urine data for CAde enabled separation of the renal and non-renal elimination pathways for CAde. Renal elimination appeared to be saturable and to represent a minor elimination pathway. The visual predictive check of Model II indicated a possible second compartment for CAde; however, this further extension of the metabolite model was not supported by the limited data. Furthermore, the indication of a second compartment could be spurious, driven by sporadic visible observations above the quantification limit (most of the metabolite observations were below the LLOQ at these late time points).
The estimated variability in most CdA PK parameters was modest. Moreover, the rather low variability in bioavailability demonstrated the consistent performance of the tablet formulation with regard to drug release and dissolution. Residual variability of urinary data was rather high (close to 90%). Urinary data are often more erratic than plasma, and, in these profiles, some inconsistent patterns were observed, which may have made it difficult to separate the variability sources. The estimated variability in CLR is low, and, in the final model, almost all variability in the urinary output is attributed to residual error.
5 Conclusions
The PK of CdA and its main metabolite, CAde, in patients with MS were described after both intravenous and oral administration of CdA, with estimation of CLR and CLNR for both compounds. Food appeared to delay CdA absorption and to slightly decrease the extent of absorption. Coadministration of IFNβ-1a slightly increased the CLNR of CdA, without a significant impact on CdA exposure, owing to the evenly split clearance between non-renal and renal components. The proposed model explained the PK of CdA in great detail thanks to the informative design of several clinical studies.
References
Leist TP, Weissert R. Cladribine: mode of action and implications for treatment of multiple sclerosis. Clin Neuropharmacol. 2011;34:28–35.
Leist TP, Vermersch P. The potential role for cladribine in the treatment of multiple sclerosis: clinical experience and development of an oral tablet formulation. Curr Med Res Opin. 2007;23:2667–76.
Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Soelberg Sorensen P, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362:416–26.
Lindemalm S, Savic RM, Karlsson MO, Juliusson G, Liliemark J, Albertioni F. Application of population pharmacokinetics to cladribine. BMC Pharmacol. 2005;5:4.
Svensson E, Acharya C, Clausson B, Dooley K, Karlsson MO. Pharmacokinetic interactions for drugs with a long half-life-evidence for the need of model-based analysis. AAPS J. 2016;18:171–9.
Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol. 2013;2:e50.
Jonsson EN, Karlsson MO. Automated covariate model building within NONMEM. Pharm Res. 1998;15:1463–8.
Beal SL, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM user’s guides (1989–2009). Ellicott City: Icon Development Solutions; 2009.
Liliemark J. The clinical pharmacokinetics of cladribine. Clin Pharmacokinet. 1997;32:120–31.
Lindemalm S, Liliemark J, Juliusson G, Larsson R, Albertioni F. Cytotoxicity and pharmacokinetics of cladribine metabolite, 2-chloroadenine in patients with leukemia. Cancer Lett. 2004;210:171–7.
Saven A, Cheung WK, Smith I, Moyer M, Johannsen T, Rose E, et al. Pharmacokinetic study of oral and bolus intravenous 2-chlorodeoxyadenosine in patients with malignancy. J Clin Oncol. 1996;14:978–83.
Acknowledgements
Editorial assistance in the preparation of this article was provided by Samantha Holmes at ACUMED® (Tytherington, UK) and was funded by Merck Serono S.A., Geneva, Switzerland.
At the time of preparation of this study, Radojka M. Savic was employed at the Department of Pharmaceutical Biosciences, Uppsala University, Uppsala, Sweden, and Marianne Ekblom was employed at Merck Serono S.A., Geneva, Switzerland.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by Merck Serono S.A., Geneva, Switzerland, an affiliation of Merck KGaA, Darmstadt, Germany.
Conflict of interest
Dr. Savic and Dr. Karlsson were/are, respectively, employees of the Uppsala University, which received financial support for performing this analysis. Mrs Novakovic has received financial support from Merck Serono for attending a symposia. Dr. Ekblom was an employee of Merck Serono S.A., Geneva, Switzerland, an affiliation of Merck KGaA, Darmstadt, Germany, at the time of the analysis. Dr. Munafo is an employee of Merck Institute for Pharmacometrics, Lausanne, Switzerland.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the studies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Savic, R.M., Novakovic, A.M., Ekblom, M. et al. Population Pharmacokinetics of Cladribine in Patients with Multiple Sclerosis. Clin Pharmacokinet 56, 1245–1253 (2017). https://doi.org/10.1007/s40262-017-0516-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-017-0516-6