
Abstract
Background and Objective
Proprotein convertase subtilisin/kexin type 9 inhibition with monoclonal antibodies such as alirocumab significantly reduces low-density lipoprotein-cholesterol levels ± other lipid-lowering therapies. We aimed to develop and qualify a population pharmacokinetics (PopPK) model for alirocumab in healthy subjects and patients, taking into account the mechanistic target-mediated drug disposition (TMDD) process.
Methods
This TMDD model was developed using a subset of the alirocumab clinical trial database, including nine phase I/II/III studies (n = 527); the model was subsequently expanded to a larger data set of 13 studies (n = 2870). Potential model parameters and covariate relationships were explored, and predictive ability was qualified using a visual predictive check.
Results
The TMDD model was built using the quasi-steady-state approximation. The final TMDD–quasi-steady-state model included a significant relationship between distribution volume of the central compartment and disease state: distribution volume of the central compartment was 1.56-fold higher in patients vs. healthy subjects. Separately, application of the model to the expanded data set revealed a significant relationship between linear clearance and statin co-administration: linear clearance was 1.27-fold higher with statins. The good predictive performance of the TMDD model was assessed based on graphical and numerical quality criteria, together with the visual predictive check and comparison of the predictions to those from a PopPK model with parallel linear and Michaelis–Menten clearances (i.e., simplification of the TMDD PopPK model).
Conclusions
This mechanistic TMDD PopPK model integrates the interaction of alirocumab with its target and accurately predicts both alirocumab and total proprotein convertase subtilisin/kexin type 9 concentrations in healthy subjects and patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The two-compartment, target-mediated drug disposition–quasi-steady-state (TMDD–QSS) population pharmacokinetics model of alirocumab accurately predicted both drug and target concentrations in 527 healthy volunteers or patients from nine phase I/II/III clinical studies. |
The final TMDD–QSS population pharmacokinetics model included only one covariate: the disease state (healthy subjects or patients) on the distribution volume of the central compartment. |
Successful application of the TMDD–QSS model on an expanded data set of 2870 subjects/patients from 13 clinical studies revealed a significant relationship between statin co-administration and linear clearance; this is the first published TMDD model developed on such a large population. |
1 Introduction
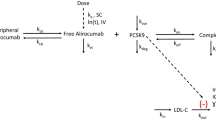
Proprotein convertase subtilisin/kexin type 9 (PCSK9), a major regulator of plasma low-density lipoprotein-cholesterol (LDL-C) metabolism, has received considerable attention in the last decade as a promising therapeutic target in the management of lipid disorders and the prevention of coronary heart disease [1–3]. In humans, PCSK9 is highly expressed in the liver, and to a lesser extent in the small intestine and kidneys [4, 5]. As shown in Fig. 1, secreted PCSK9 binds the LDL receptor (LDLR) on the surface of the hepatocyte, leading to its internalization, intracellular retention, and degradation in the lysosomes. PCSK9 secretion therefore prevents the receptor from being recycled to the plasma membrane for further clearing of LDL-C [2, 6], resulting in a decrease of LDLR on the cell surface of hepatocytes, which decreases the ability of the liver to remove LDL-C from circulation and causes higher levels of circulating LDL-C [2]. Human genetic studies have shown that gain-of-function PCSK9 mutations are associated with higher serum levels of LDL-C and premature incidences of coronary heart disease, whereas loss-of-function mutations are associated with low serum levels of LDL-C and reduced risk of coronary heart disease [7–10]. The complete loss of PCSK9 in humans has been shown to result in the low serum LDL-C concentration of about 15 mg/dL in healthy subjects [11, 12]. Inhibition of PCSK9 therefore offers a novel therapeutic mechanism for the lowering of LDL-C levels [13].

Mechanism of action of alirocumab, a PCSK9 inhibitor (adapted from Lambert et al. [2]). ASO antisense oligonucleotides, LDL low-density lipoprotein, LDLR LDL receptor, PCSK9 proprotein convertase subtilisin/kexin type 9, siRNA small interfering RNA, SREBP2 sterol-responsive element-binding protein 2
Monoclonal antibodies that exclusively target circulating PCSK9 prevent the association of PCSK9 with LDLR, thereby promoting LDL-C clearance and LDLR recycling back to the plasma membrane [2]. Alirocumab, one such monoclonal antibody recently approved by the US Food and Drug Administration and the European Medicines Agency, has been shown to significantly reduce LDL-C levels both alone and in combination with other lipid-lowering therapies in heterozygous familial hypercholesterolemia (FH) and non-FH dyslipidemic individuals [14–27].
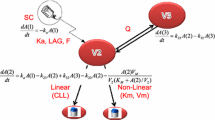
The pharmacokinetics (PK) and pharmacodynamics of alirocumab are well characterized [28–30]. The high specificity of monoclonal antibodies to bind to a specific target and subsequent turnover of the drug–target complex can significantly impact the disposition of these drugs. This target-mediated drug disposition (TMDD) model was first described by Mager and Jusko in 2001 [31] and is shown in Fig. 2. Monoclonal antibodies such as alirocumab that exhibit TMDD are predominantly characterized by nonlinear PK at low concentrations owing to the saturability of the target (as there are finite numbers of the target on the cell surface), resulting in faster than linear elimination of the drug. At high concentrations when the nonlinear target-mediated elimination pathway is saturated, linear clearance of alirocumab becomes more important, mostly because of catabolism.

General pharmacokinetic model of target-mediated drug disposition (adapted from Mager and Krzyzanski [48]). First described by Mager and Jusko in 2001 [31], this schematic shows the drug in the central compartment (C) binds to free receptors (R) at the second-order rate (K on) to form a drug–receptor complex (RC). This complex may then either dissociate at the first-order rate (K off), or be internalized via endocytosis and degraded at the first-order rate (K int). Free drug can also be eliminated from the system at the first-order rate (K el), or distributed to non-specific tissue-binding sites (AT) at first-order rates K tp and K pt. The zero-order rate of synthesis (K syn) and the first-order rate of degradation (K deg) of the free receptor, as well as the input rate [In(t)] to the free drug compartment, are also reflected. Free drug C, free receptor R, and the drug–receptor complex RC are expressed in molar concentrations, and AT denotes moles of nonspecifically tissue bound or distributed drug
In this analysis (performed as part of the Biologics License Application to the US Food and Drug Administration for alirocumab), we aimed to develop and qualify a population PK (PopPK) model for alirocumab, taking into account the mechanistic TMDD process, by integrating all of the information regarding the target (PCSK9) and the alirocumab–PCSK9 complex. Potential relationships between model parameters and demographic covariates, relevant co-administration, antibody drug status, disease state, administration site and device, and relevant biologic constants were explored.
2 Methods
2.1 Clinical Studies and Sample Analysis
This TMDD model was developed on a subset of the alirocumab clinical trial program database, including 527 healthy volunteers or patients from nine phase I/II/III studies [32–39]. Subsequently, the model was expanded to a larger data set (2870 subjects/patients from 13 studies) [15, 16, 18, 19, 24]. The doses, dosing regimens, and main characteristics of these clinical studies are presented in Table 1. The protocols were approved by the relevant ethics committees, and written informed consent was obtained from all participants.
Total alirocumab, total PCSK9, and free PCSK9 concentrations in serum samples were determined using an enzyme-linked immunosorbent assay; the upper and lower limit of quantification (ULOQ and LLOQ, respectively) for each of these parameters in the human serum matrix, and their corresponding values in undiluted human serum, are summarized in Table 2. Total PCSK9 concentrations were determined following an acid treatment of the serum samples that resulted in the dissociation of any complexed PCSK9 (including any PCSK9–alirocumab complexes), thereby permitting the quantification of all PCSK9 in the sample. To assess immunogenicity, the presence of anti-drug antibodies (ADAs, i.e., anti-alirocumab antibodies) was evaluated using a validated, non-quantitative, titer-based, bridging immunoassay at three different points: during the initial screen, the confirmation assay for drug specificity, and the assay in the serum samples. Parameters used for assay sensitivity and drug tolerance limits are summarized in Table 2.
2.2 Pharmacokinetic Exclusion Criteria
From the randomized and treated individuals, patients/subjects receiving placebo treatment were excluded. From the remaining patients/subjects receiving alirocumab, data were excluded from the database for any of the following reasons: missing observations, missing alirocumab concentrations, missing total PCSK9 concentrations, or values below the LLOQ. Missing covariate values were replaced by the previous values from the same individual or interpolated if time dependent. Completely missing covariate values (if not resolved) led to the exclusion of the patient/subject. Missing ADA status was not replaced with previous values but was always considered as missing; all baseline values for ADAs were set to a negative ADA status. For baseline values (e.g., baseline free and total PCSK9 concentrations), the mean was calculated based on the available data before the first dose administration.
2.3 TMDD Modeling
The TMDD PopPK analysis was performed with nonlinear mixed-effects modeling (NONMEM) (ICON, Dublin, Ireland) software version 7.2 running on a LINUX cluster of multi-processor computers. The first-order conditional estimation with interaction option method was used for each step of the model development, including the detection of outliers in the preliminary model.
2.3.1 Preliminary and Pharmacostatistical Model Development
An outlier screening was performed using a preliminary model. The best preliminary model was chosen based on the examination of the objective function value (OFV), parameter precision, error estimates, shrinkage values, and the visual inspection of the goodness-of-fit plots. Following outlier screening and deletion, the pharmacostatistical model was developed based on the same criteria as the preliminary model selection. The population parameters (fixed effects and random effects) together with the individual estimates were computed assuming no dependency between model parameters and covariates.
2.3.2 Covariate Model Development
Demographic characteristics such as age, sex, race, body weight, body mass index, disease state (DISST, healthy subjects vs. patients), and albumin levels were tested as potential model covariates. Other covariates included the type of disease (FH vs. non-FH), baseline free and total PCSK9 levels, ADA levels, and co-administration of other anti-hypercholesterolemia drugs (fibrates, ezetimibe, rosuvastatin, atorvastatin, or simvastatin). Unless otherwise stated, time-varying values of covariates during the course of the trial, in addition to baseline values, were used.
Selected covariates were added individually to the model (forward selection method) and tested for statistical significance using the likelihood ratio test. The impact of the covariate inclusion in the model on the quality of individual fit was evaluated by visual inspection of the goodness-of-fit plots [conditional weighted residuals (CWRES) or individual weighted residuals (IWRES), population predicted (PRED) or individual predicted (IPRED) in comparison with observed concentrations (OBS)] before and after covariate inclusion. The covariates that produced significant changes (p < 0.05) in the OFV (ΔOFV of ≥3.84 units) were retained in the model. To verify that the covariates selected during the model building process were still relevant for the final population model, each term of the proposed final model was alternatively deleted (backward deletion), and its impact on the OFV was determined; covariates that decreased OFV by less than 10.8 (p = 0.001) would be excluded. Before qualification, model verification was performed by examination of the goodness-of-fit plots and by estimation of several quality criteria (mean prediction error, precision, and average fold error).
2.3.3 Qualification of the Model
The qualification of the predictive performance of the TMDD PopPK model was performed by examination of the goodness-of-fit plots, visual predictive check (VPC), and comparison of the predictions to those from a PopPK model with parallel linear and Michaelis–Menten clearances (a simplification of the current TMDD PopPK model).
To evaluate the impact of the removal of concentration time points after outliers’ detection, the final PopPK model was applied to the whole data set.
2.3.4 Application and Refinement of the TMDD Model to the Expanded Data Set
The expanded application of the developed model to the data set of 2870 healthy subjects and patients from 13 clinical studies allowed for the qualification of the previously mentioned assumptions on a much larger data set. Covariate screening was performed using the same approach, and the same criteria described above were used to select the pharmacostatistical model, the parameter–covariate relationships, and the final TMDD PopPK model with the expanded data set. Similarly, the model qualification was based on the goodness-of-fit plots, the computation of numerical quality criteria for both total alirocumab and total PCSK9, and the VPC method.
3 Results
3.1 Summary of Patient/Subject Characteristics
Descriptive statistics (baseline values) of potential covariates of the patients/subjects tested in the TMDD analysis are summarized in Table 3.
3.2 Pharmacostatistical Model Development
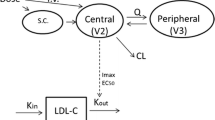
Based on the mechanism of action of alirocumab, the available clinical data, and the known quasi-instantaneous alirocumab–PCSK9 complex formation, a TMDD PopPK model with the quasi-steady-state (QSS) approximation was deemed to be the most appropriate model to describe the PK of alirocumab. The pharmacostatistical model was a TMDD–QSS PopPK model, occurring in the central compartment of a two-compartment model: the central compartment (V c, L) and the peripheral compartment (V p, L); the two compartments were linked by an inter-compartmental clearance (Q, L/day). The QSS approximation was parameterized by the association rate constant K on (nM−1·day−1), the PCSK9 degradation rate constant (K deg, day−1), and the complex internalization rate constant (K int, day−1). In addition, a linear clearance (CLL, L/day) from the central compartment, representing the catabolic clearance, was also parameterized. To describe the absorption process, a first-order absorption rate constant from the depot to the central compartment (K a, day−1) with a lagtime (LAG, day) and the bioavailability (F1) were estimated for subcutaneous administration. These variables composed the fixed-effects model parameters.
Other approximations of the TMDD model are explained in detail in Fig. 3. The quasi-equilibrium approximation was ruled out owing to the long elimination half-life of the alirocumab–PCSK9 complex (i.e., non-negligible K int). The irreversible binding approximation was also ruled out, given that the K D was measured in vitro to be about 0.58 nM. The QSSconst simplification was not made, given that the total PCSK9 was not constant, and the Michaelis–Menten approximation and Michaelis–Menten equation simplifications were not made, given that the alirocumab–PCSK9 complex concentration is not much smaller compared with free alirocumab.

Hierarchy of approximations of the target-mediated drug disposition (TMDD) model (adapted from Gibiansky et al. [49]). The first approximation of the general TMDD model, the quasi-equilibrium (QE), was suggested by Mager and Krzyzanski in 2005 [48]. The quasi-equilibrium approximation assumes the internalization and elimination rate constant (K int) of the drug (C)–receptor (R) complex (RC) is much smaller and negligible compared with the dissociation rate constant (K off); K D, the equilibrium dissociation constant, is the ratio of K off and the association rate constant of the complex (K on). Conversely, the irreversible binding (IB) approximation assumes that K off is much smaller than K int. The quasi-steady-state (QSS) approximation assumes that the binding process is nearly instantaneous, and the compound, target, and complex are in quasi-steady-state (K SS). The Michaelis–Menten (MM) approximation assumes that time derivatives of the free and total drug concentrations are similar, which allows this approximation to describe the system when RC is small relative to C. Further simplifications can be made to the quasi-equilibrium (QEconst), QSS (QSSconst), and Michaelis–Menten approximations/equations by assuming R tot (R + RC) is constant. K deg rate of degradation, K syn rate of synthesis
The inter-individual error term was evaluated for all fixed-effect parameters (including CLL, K int, Kdeg, Q, V c, K a, and F1) except K on and V p, which were fixed to a constant value following sensitivity analysis.
A preliminary TMDD PopPK model was developed for outliers’ detection and deletion. However, because of the good quality of both the model and the data set, the outliers’ deletion step did not need to be performed. No combination (among 56 tested) of estimated covariance between the seven estimated inter-individual variabilities (within an ω-block) improved the quality of the model.
The PopPK parameters obtained before covariate inclusion are presented in Table 4. As shown in Table 4, the relative standard errors of the estimates for each parameter were small enough so that the 95% confidence interval did not include zero. The maximum values of 7.08% for the fixed-effect parameters and 27.1% for the random-effect parameters confirmed the quality of the model.
Supplementary Table 1 summarizes the main quality criteria computed for the comparison of PRED and IPRED vs. OBS (the dependent variable in NONMEM) for the pharmacostatistical model for the pool of total alirocumab and total PCSK9, as well as for each of these dependent variables individually. The mean prediction errors for PRED vs. OBS and IPRED vs. OBS were slightly but significantly different from zero. The average fold error of the pool, total alirocumab, and total PCSK9 was 1.61, 1.94, and 1.37, respectively, for PRED vs. OBS, and 1.24, 1.28, and 1.21, respectively, for IPRED vs. OBS. In addition, among 9379 samples, only 11 |IWRES| and 0 |CWRES| values were higher than 8, and only 15 IWRES and 17 CWRES were out of the range [−4; +4]. Based on these numerical quality criteria, the model quality was found to be good for the pool of total alirocumab and total PCSK9. For each of these dependent variables separately, the quality of model estimation obtained for total PCSK9 was found to be slightly better than that of total alirocumab.
3.3 Screening and Selection of Covariates
Each significant covariate (listed in Table 3) was added sequentially (univariate model) and the developed model was evaluated for its effect on OFV, 95% CI, and inter-individual variability of the corresponding parameter estimates. Only one significant parameter–covariate relationship was retained: DISST on V c. The ΔOFV was found to be −88.4 according to the following equation:
where TVVC is the typical value of V c, COV1 is the DISST effect on V c, and DISST = 0 for healthy subjects and 1 for patients.
In the last step in the covariate model development, the relevance of the covariates selected during the model building process in the final population model was verified by performing backward deletion; as previously mentioned, covariates that decreased OFV by less than 10.8 (p = 0.001) would be excluded. However, because the DISST-V c relationship led to a decrease of 88.4 of the OFV, it was retained in the final model.
3.4 Final Population Model
The final TMDD PopPK parameters are presented in Table 4 alongside those for the pharmacostatistical model. As previously mentioned, the final model included only one significant parameter–covariate relationship: DISST–V c. The V c was estimated to be 3.16 L for healthy subjects and 4.93 L for patients. Therefore, V c was estimated to be 1.56-fold higher in patients (n = 377) compared with healthy subjects (n = 150), with a 17.4% decrease (from 37.3 to 30.8%) in the inter-subject variability. The highest absolute value of the correlation between parameter estimates (0.715) was lower than 0.95, and the %RSEs were small enough so that 95% CIs did not include zero: the highest %RSE value was 7.79% for the fixed-effect parameters and 42.5% for the inter-individual error terms.
Supplementary Table 1 summarizes the main quality criteria computed for the comparison of PRED and IPRED vs. OBS for the final model alongside the pharmacostatistical model. As shown in Supplementary Table 1, there was a clear improvement in the model quality from the pharmacostatistical model to the final model for all of the dependent variables (pool, total alirocumab, total PCSK9). The mean prediction error for all of the dependent variables was low but significantly different from zero for both PRED vs. OBS and IPRED vs. OBS. The mean prediction errors of the mean observed value was 7.79 and 4.94% for the pool, 7.50 and 2.96% for the total alirocumab concentrations, and 8.51 and 9.81% for the total PCSK9 concentrations for PRED vs. OBS and IPRED vs. OBS, respectively. The average fold error was 1.56 and 1.24 for the pool, 1.85 and 1.28 for the total alirocumab concentrations, and 1.34 and 1.21 for the total PCSK9 concentrations for PRED vs. OBS and IPRED vs. OBS, respectively. The correlation characteristics for the pool were excellent for both PRED and IPRED vs. OBS, as shown by the correlation coefficients of greater than 0.96 (0.967 for PRED and 0.995 for IPRED). Based on these numerical quality criteria, the model quality was found to be good for the pool of total alirocumab and total PCSK9. For each of these dependent variables separately, as above, the quality of model estimation obtained for total PCSK9 was found to be slightly better than that of total alirocumab. The CWRES vs. PRED and IWRES vs. IPRED for the final model are plotted in Fig. 4. The PRED and IPRED vs. OBS for the pool, for total alirocumab and for total PCSK9 are plotted in Fig. 5 to evaluate the global quality of the model fitting.

Relationship between a conditional (population) weighted residuals and population predicted concentrations and b individual weighted residuals and individual predicted concentrations, for the pool after covariate inclusion. Linear scale. The tendency line is indicated in red

Relationship between population predicted and observed concentrations (left panels), and between individual predicted and observed concentrations (right panels) in the total data set after covariate inclusion (linear scale) for the pool (a), total alirocumab (b), and total proprotein convertase subtilisin/kexin type 9 (c). The tendency line is indicated in red
3.5 Model Qualification
VPCs were used to evaluate the performance of the final model. The VPC of each study is presented for total alirocumab in Supplementary Fig. 1 and for total PCSK9 in Supplementary Fig. 2. The results of the VPC for both total alirocumab and total PCSK9 showed that the overwhelming majority of the observed concentrations were included in the 5th–95th percentile range, illustrating the good qualification of the developed TMDD–QSS PopPK model. As only one covariate was included in the final model (DISST on V c) and each study included either only healthy subjects or patients, it was not necessary to compute and plot the prediction-corrected VPC.
Simulations were performed to evaluate the impact of DISST on V c on total alirocumab, total PCSK9, and free PCSK9 levels after treatment for up to 12 weeks with alirocumab 75 mg every 2 weeks (Q2W; Fig. 6a) or alirocumab 150 mg Q2W (Supplementary Fig. 3a). A summary of the predicted exposures is also shown in Supplementary Table 3. These simulations predicted a lower Area Under the Curve between Week 10 and Week 12 (i.e. following the 6th Q2W administration) (AUCW10–W12) and maximum plasma concentration (C max) for total alirocumab and total PCSK9 after 75- and 150-mg Q2W administration in patients compared with healthy volunteers. For free PCSK9 concentrations, the simulations predicted a 1.42-fold higher AUCW10–W12 and a 1.09-fold higher C max after the 75-mg Q2W dose in patients compared with healthy volunteers, while a 2.89-fold higher AUCW10–W12 and a 3.62-fold higher C max was predicted after the 150-mg Q2W dose; this illustrates a more pronounced saturation of the TMDD elimination in healthy volunteers compared with patients at the higher alirocumab dose.

Predicted total alirocumab, total PCSK9, and free PCSK9 levels using the final model on alirocumab 75 mg Q2W for up to weeks 10–12. a shows a typical healthy volunteer (red) vs. a typical patient (blue) with the patient disease state (DISST) effect on V c. b shows the statin effect on CLL (with statin co-administration in blue vs. without statin co-administration in red). CLL linear clearance, PCSK9 proprotein convertase subtilisin/kexin type 9, Q2W every 2 weeks, V c volume of distribution of central compartment
3.6 Post-Submission Analysis: TMDD–QSS Model Expanded to 2870 Healthy Subjects/Patients from 13 Studies
Descriptive statistics (baseline values) of potential covariates of the patients/subjects tested in the TMDD analysis in the expanded data set are summarized in Table 3, and the obtained PopPK parameters are presented in Table 4. After many attempts, the TMDD–QSS model converged with very acceptable quality criteria (Supplementary Table 1). The pharmacostatistical model with the expanded data set included theory-based allometric scaling on CLL, Q, and V c (the V p being fixed similarly to the previous TMDD–QSS model), as previously described [40]. The body weight (WT) was included on those parameters as follows:
where P i is the individual parameter, TVP is the typical value of the parameter, WTmed is the median value of body weight in the expanded data set, and the exponent EXP equals 1 for V c and 0.75 for CLL and Q. The inter-patient/subject variability was estimated for seven model parameters (CLL, K int, K deg, Q, V c, K a, and F1) and ranged between 22.7 and 85.0% in the expanded model. The residual variability was moderate with a coefficient of variation of 15.2% in the expanded model for the proportional error term.
The final model of the expanded data set included one significant relationship between statin co-administration (STATIN) and CLL according to the following equation:
where TVCLL is the typical value of CLL, COV1 is the STATIN effect on CLL, and STATIN = 1 with statin co-administration and 0 without statin co-administration. The CLL was estimated to be 0.176 L/day without statins and 0.224 L/day with statins (i.e., 1.27-fold higher CLL with statin co-administration). In addition, the comparison of the main quality criteria reflecting the predictive performance of the TMDD–QSS and TMDD–Michaelis–Menten models are summarized in Supplementary Table 2. This comparison highlights the very similar predictive performance of the two models toward the prediction of total alirocumab concentrations. The model was qualified using the VPC (see Fig. 7 for alirocumab and Fig. 8 for total PCSK9, stratified by study).

Visual predictive check results for total alirocumab (n = 14097) per study (STUD) for the expanded data set. Linear scale (observations, median, 5th and 95th percentiles, confidence intervals of median and centiles). Dark blue dots indicate observations; the solid red line is the median of observations; solid dashed lines are the 5th and 95th percentiles of observations; pink and blue areas are the confidence intervals of the median and 5th and 95th percentiles of predictions

Visual predictive check results for total PCSK9 (n = 14877) per study (STUD) for the expanded data set. Linear scale (observations, median, 5th and 95th percentiles, confidence intervals of median and centiles). Dark blue dots indicate observations; the solid red line is the median; solid dashed lines are the 5th and 95th percentiles; pink and blue areas indicate confidence intervals of the median and 5th and 95th percentiles. PCSK9 proprotein convertase subtilisin/kexin type 9
As above, simulations were performed to evaluate the impact of statin co-administration on CLL on total alirocumab, total PCSK9, and free PCSK9 levels after treatment for up to 12 weeks with alirocumab 75 mg Q2W (Fig. 6b) or alirocumab 150 mg Q2W (Supplementary Fig. 3b), and a summary of the predicted exposures are also shown in Supplementary Table 3. These simulations predicted a lower AUCW10–W12 and C max for total alirocumab after 75- and 150-mg Q2W administration in individuals with statin co-administration compared with those without statin co-administration. For free PCSK9 concentrations, the simulations predicted a 2.08-fold higher AUCW10–W12 and a 1.67-fold higher C max after the 75-mg Q2W dose in individuals taking statins compared with those not taking statins, while AUCW10–W12 and C max were predicted to be 3.57-fold and 5.91-fold higher, respectively, after the 150-mg Q2W dose, suggesting an amplified saturation of the TMDD elimination at the higher dose in individuals without statin co-administration compared with those taking statins.
4 Discussion
This TMDD PopPK analysis was first conducted in a data set of 527 subjects and patients enrolled in nine phase I/II/III studies, and subsequently on a larger data set of 2870 individuals in 13 studies. To our knowledge, this expanded model is the first published TMDD model developed on more than 1564 individuals [41]. The great benefit of developing and qualifying a TMDD model is that it allows the prediction of the PK of alirocumab, by integrating all available information regarding the target (PCSK9) and taking into account the mechanistic behavior of the system. A TMDD PopPK model was developed with the QSS approximation, occurring in the central compartment of a two-compartment model (V c and V p) linked by an inter-compartmental clearance (Q, L/day). The QSS approximation was parameterized by K on, K deg, K int, and CLL from the central compartment (representing the catabolic clearance). The absorption process was described by a first-order K a from the depot to the central compartment with a LAG and F1 for subcutaneous administration.
The TMDD–QSS PopPK model developed in this analysis accurately predicted both alirocumab and total PCSK9 concentrations observed in patients and healthy subjects. This was demonstrated at each step of the model building process and, at the end, was assessed using the VPC approach. The inter-individual variability was modeled through an exponential error model and was estimated for all parameters except LAG, K on, and V p (which were fixed to a constant value). A combined (additive + proportional) error model was used to model the residual variability. The available potential sources of variability on the population parameters were extensively investigated. Among all the covariates tested, only one significant parameter–covariate relationship was retained in the TMDD–QSS PopPK model: the estimated typical value of V c was 3.16 L for healthy volunteers vs. 4.93 L for patients.
Of the demographic characteristics tested for effect on the TMDD PopPK parameters (age, weight, body mass index, sex, and race), none resulted in a significant improvement of the model fit in the initial population of 527 subjects. Additionally, there was no relationship between albuminemia, the presence of ADAs, the site of injection, the baseline levels of total and free PCSK9, the type of disease (FH vs. non-FH), co-medication (ezetimibe, high-dose statin, low-dose statin), and the parameters of the TMDD model in this population.
At first sight, the link between DISST and V c seems surprising. There is, however, a huge collinearity between DISST and statin co-medication. In the data set including 527 individuals, 321/377 patients were co-administered statins while only 56/377 patients and all of the 150 healthy volunteers received alirocumab alone. At the first covariate screening step, the links between DISST or statins and V c were very similar in terms of OFV decrease (ΔOFV <1 unit) with a similar impact on V c: 3.16 L for healthy volunteers vs. 4.93 L for patients and 4.13 L without statins vs. 6.73 L with statins. However, the model with inclusion of statins seemed over-parameterized (conditional number of 22,333, given that a value higher than 1000 suggests an over-parameterization of the model) and explained less inter-individual variability on V c compared with the model with inclusion of the DISST (10.2 vs. 17.2%, respectively). When looking at the empirical Bayesian estimates of the final model (taking into account the relationship between DISST and V c), the median estimated V c is 5.11 L for patients undergoing statin therapy (n = 321), 4.23 L for patients that were not taking statins (n = 56), and 3.17 L for the healthy volunteers (n = 150). These figures, in addition to the good quality criteria and predictive performance of the model using numerical and graphical qualification, strongly suggest that despite the huge collinearity between the two covariates, the DISST provided the most relevant information using the data set of the present analysis. Of note, numerous covariates are often not included in such mechanistic models, as the most important information is provided by the second dependent variable (which, in this case, is total PCSK9 concentrations).
Further to the Biologics License Application submission, this TMDD–QSS model was applied and refined on an expanded data set of 13 clinical studies including 2870 individuals. The pharmacostatistical model and the corresponding model parameters were very similar to those of the TMDD–QSS model, except for the inclusion of theory-based allometric scaling on CLL, Q, and V c. The covariate screening revealed only one significant relationship between the CLL and statin co-administration, resulting in a 1.27-fold higher CLL when statins were co-administered. This finding is consistent with the considerable collinearity suggested between DISST and statin co-administration in the TMDD–QSS model with 527 individuals. Similar observations were reported for evolocumab, another anti-PCSK9 monoclonal antibody, where statins significantly increased evolocumab clearance [42]. We propose two hypotheses to explain these findings. The first relates to the upregulation of PCSK9 by statins, as previously reported [43, 44]. However, baseline PCSK9 concentrations, when tested as a covariate, did not reveal a significant relationship with any of the model parameters. The second hypothesis is linked to previous findings that showed that statins have pro-angiogenic effects, via the acceleration of re-endothelialization and the mobilization of endothelial progenitor cells [45, 46]. Indeed, the main catabolic pathway for monoclonal antibodies representing the linear clearance occurs via the reticuloendothelial system [47]. The pro-angiogenic effects of statins could thus result in a significantly higher linear clearance.
The results of the simulations evaluating the impact of these parameter–covariate relationships show, regardless of the analyte (total alirocumab, total or free PCSK9, or PCSK9–alirocumab complex), a notable DISST effect (using the initial TMDD model), and to a lower extent, a statin co-administration effect (using the final TMDD model) on exposures. It is worth noting that healthy volunteers represented 28.5% (150/527) in the initial data set, vs. 5.23% (150/2870) in the expanded data set (i.e., more than 94% of the expanded data set were patients). As previously suggested, the predicted exposures were very similar in patients using the initial TMDD model compared with individuals with statin co-administration in the final TMDD model, regardless of the alirocumab dose and the analyte.
5 Conclusions
This model was well qualified using graphical and numerical quality criteria and with the VPC approach. Therefore, though the available free PCSK9 concentrations were not used as a dependent variable in the present analysis (owing to the application of the QSS approximation) and despite the slight but significant mean prediction error values observed in the numerical quality criteria, the mechanistic model that was developed and qualified in the present analysis correctly integrates the interaction of alirocumab with its target PCSK9, and consequently well describes the behavior of both alirocumab and PCSK9 kinetics.
References
Vogel RA. PCSK9 inhibition: the next statin? J Am Coll Cardiol. 2012;59(25):2354–5.
Lambert G, Sjouke B, Choque B, et al. The PCSK9 decade. J Lipid Res. 2012;53(12):2515–24.
Lambert G, Charlton F, Rye KA, Piper DE. Molecular basis of PCSK9 function. Atherosclerosis. 2009;203(1):1–7.
Seidah NG, Benjannet S, Wickham L, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci. 2003;100(3):928–33.
Zaid A, Roubtsova A, Essalmani R, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology. 2008;48(2):646–54.
Marais AD, Kim JB, Wasserman SM, Lambert G. PCSK9 inhibition in LDL cholesterol reduction: genetics and therapeutic implications of very low plasma lipoprotein levels. Pharmacol Ther. 2015;145:58–66.
Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154–6.
Cohen J, Pertsemlidis A, Kotowski IK, et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37(2):161–5.
Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–72.
Kotowski IK, Pertsemlidis A, Luke A, et al. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet. 2006;78(3):410–22.
Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193(2):445–8.
Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79(3):514–23.
Urban D, Poss J, Bohm M, Laufs U. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J Am Coll Cardiol. 2013;62(16):1401–8.
Bays H, Gaudet D, Weiss R, et al. Alirocumab as add-on to atorvastatin versus other lipid treatment strategies: ODYSSEY OPTIONS I randomized trial. J Clin Endocrinol Metab. 2015;100(8):3140–8.
Cannon CP, Cariou B, Blom D, et al. Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: the ODYSSEY COMBO II randomized controlled trial. Eur Heart J. 2015;36(19):1186–94.
Colhoun HM, Robinson JG, Farnier M, et al. Efficacy and safety of alirocumab, a fully human PCSK9 monoclonal antibody, in high cardiovascular risk patients with poorly controlled hypercholesterolemia on maximally tolerated doses of statins: rationale and design of the ODYSSEY COMBO I and II trials. BMC Cardiovasc Disord. 2014;14:121.
Farnier M, Jones P, Severance R, Averna M, et al. Efficacy and safety of adding alirocumab to rosuvastatin versus adding ezetimibe or doubling the rosuvastatin dose in high cardiovascular-risk patients: the ODYSSEY OPTIONS II randomized trial. Atherosclerosis. 2016;244:138–46.
Kastelein JJ, Ginsberg HN, Langslet G, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J. 2015;36(43):2996–3003.
Kastelein JJ, Robinson JG, Farnier M, et al. Efficacy and safety of alirocumab in patients with heterozygous familial hypercholesterolemia not adequately controlled with current lipid-lowering therapy: design and rationale of the ODYSSEY FH studies. Cardiovasc Drugs Ther. 2014;28(3):281–9.
Kereiakes DJ, Robinson JG, Cannon CP, et al. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: the ODYSSEY COMBO I study. Am Heart J. 2015;169(6):906–15.e13.
Moriarty PM, Jacobson TA, Bruckert E, et al. Efficacy and safety of alirocumab, a monoclonal antibody to PCSK9, in statin-intolerant patients: design and rationale of ODYSSEY ALTERNATIVE, a randomized phase 3 trial. J Clin Lipidol. 2014;8(6):554–61.
Moriarty PM, Thompson PD, Cannon CP, et al. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: the ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol. 2015;9(6):758–69.
Robinson JG, Colhoun HM, Bays HE, et al. Efficacy and safety of alirocumab as add-on therapy in high-cardiovascular-risk patients with hypercholesterolemia not adequately controlled with atorvastatin (20 or 40 mg) or rosuvastatin (10 or 20 mg): design and rationale of the ODYSSEY OPTIONS studies. Clin Cardiol. 2014;37(10):597–604.
Robinson JG, Farnier M, Krempf M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1489–99.
Roth EM, McKenney JM. ODYSSEY MONO: effect of alirocumab 75 mg subcutaneously every 2 weeks as monotherapy versus ezetimibe over 24 weeks. Future Cardiol. 2015;11(1):27–37.
Stein EA, Raal F. Reduction of low-density lipoprotein cholesterol by monoclonal antibody inhibition of PCSK9. Annu Rev Med. 2014;65:417–31.
Lambert G, Petrides F, Chatelais M, et al. Elevated plasma PCSK9 level is equally detrimental for patients with nonfamilial hypercholesterolemia and heterozygous familial hypercholesterolemia, irrespective of low-density lipoprotein receptor defects. J Am Coll Cardiol. 2014;63(22):2365–73.
Praluent® (alirocumab). Prescribing information (USA). 2015. Available from: http://products.sanofi.us/praluent/praluent.pdf. Accessed 9 Sept 2016.
Praluent® (alirocumab). Summary of product characteristics (EC). 2015. Available from: http://ec.europa.eu/health/documents/community-register/2015/20150923132812/anx_132812_en.pdf. Accessed 9 Sept 2016.
Lunven C, Paehler T, Poitiers F, et al. A randomized study of the relative pharmacokinetics, pharmacodynamics, and safety of alirocumab, a fully human monoclonal antibody to PCSK9, after single subcutaneous administration at three different injection sites in healthy subjects. Cardiovasc Ther. 2014;32(6):297–301.
Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507–32.
Stein EA, Mellis S, Yancopoulos GD, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366(12):1108–18.
Toth PP, Hamon SC, Jones SR, et al. Effect of alirocumab on specific lipoprotein non-high-density lipoprotein cholesterol and subfractions as measured by the vertical auto profile method: analysis of 3 randomized trials versus placebo. Lipids Health Dis. 2016;15(1):28.
Koren MJ, Kereiakes D, Pourfarzib R, et al. Effect of PCSK9 inhibition by alirocumab on lipoprotein particle concentrations determined by nuclear magnetic resonance spectroscopy. J Am Heart Assoc. 2015;4(11). doi:10.1161/JAHA.115.002224 (pii: e002224).
Gaudet D, Kereiakes DJ, McKenney JM, et al. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am J Cardiol. 2014;114(5):711–5.
McKenney JM, Koren MJ, Kereiakes DJ, et al. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59(25):2344–53.
Roth EM, McKenney JM, Hanotin C, et al. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367(20):1891–900.
Stein EA, Gipe D, Bergeron J, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380(9836):29–36.
Roth EM, Taskinen MR, Ginsberg HN, et al. Monotherapy with the PCSK9 inhibitor alirocumab versus ezetimibe in patients with hypercholesterolemia: results of a 24 week, double-blind, randomized phase 3 trial. Int J Cardiol. 2014;176(1):55–61.
Holford N, Heo YA, Anderson B. A pharmacokinetic standard for babies and adults. J Pharm Sci. 2013;102(9):2941–52.
Sutjandra L, Rodriguez RD, Doshi S, et al. Population pharmacokinetic meta-analysis of denosumab in healthy subjects and postmenopausal women with osteopenia or osteoporosis. Clin Pharmacokinet. 2011;50(12):793–807.
Gibbs JP, Doshi S, Kuchimanchi M, et al. Impact of target-mediated elimination on the dose and regimen of evolocumab, a human monoclonal antibody against proprotein convertase subtilisin/kexin type 9 (PCSK9). J Clin Pharmacol. 2016. doi:10.1002/jcph.840 (Epub ahead of print).
Mayne J, Dewpura T, Raymond A, et al. Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis. 2008;11(7):22.
Guo YL, Zhang W, Li JJ. PCSK9 and lipid lowering drugs. Clin Chim Acta. 2014;1(437):66–71.
Walter DH, Rittig K, Bahlmann FH, et al. Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation. 2002;105(25):3017–24.
Walter DH, Zeiher AM, Dimmeler S. Effects of statins on endothelium and their contribution to neovascularization by mobilization of endothelial progenitor cells. Coron Artery Dis. 2004;15(5):235–42.
Shi S. Biologics: an update and challenge of their pharmacokinetics. Curr Drug Metab. 2014;15(3):271–90.
Mager DE, Krzyzanski W. Quasi-equilibrium pharmacokinetic model for drugs exhibiting target-mediated drug disposition. Pharm Res. 2005;22(10):1589–96.
Gibiansky L, Gibiansky E, Kakkar T, Ma P. Approximations of the target-mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Pharmacodyn. 2008;35(5):573–91.
Acknowledgements
All authors contributed to the concept/design and data analysis/interpretation of this study, and critically reviewed the manuscript and approved it for submission. The authors take full responsibility for the content of this article. The authors acknowledge Bill Sasiela and Jay Edelberg for their contributions to the manuscript, and Grace Shim, PhD, an employee of Prime (New York, NY), for medical writing assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Sanofi and Regeneron Pharmaceuticals, Inc. Grace Shim, PhD, an employee of Prime (New York, NY), provided assistance with the writing, formatting, proofreading, and collation of the author comments and this assistance was funded by Sanofi and Regeneron Pharmaceuticals, Inc. Open access publication of this article was sponsored by Sanofi and Regeneron Pharmaceuticals, Inc.
Conflict of interest
ND., J.M.M., A.B., F.H., and D.F. are full-time employees of, and stockholders in, Sanofi. S.K. is an expert scientist in pharmacokinetics at the Université de Montpellier and INSERM 1194–Institut de Recherche en Cancérologie de Montpellier (IRCM), consulting for Sanofi. L.L. was a student at the Université de Montpellier, hosted by Sanofi at the time of these analyses.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study. Additional informed consent was obtained from all individual participants for whom identifying information is included in this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Djebli, N., Martinez, JM., Lohan, L. et al. Target-Mediated Drug Disposition Population Pharmacokinetics Model of Alirocumab in Healthy Volunteers and Patients: Pooled Analysis of Randomized Phase I/II/III Studies. Clin Pharmacokinet 56, 1155–1171 (2017). https://doi.org/10.1007/s40262-016-0505-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0505-1