
Abstract
Background and Objective
Previous clinical trials have suggested that bovine intestinal alkaline phosphatase has renal protective effects in patients with sepsis-associated acute kidney injury. We conducted a first-in-human study to investigate the pharmacokinetics, safety and tolerability of a novel human recombinant alkaline phosphatase (recAP), and we developed a population pharmacokinetic model to support dose selection for future patient studies.
Methods
In a randomized, double-blind, placebo-controlled, phase I trial, healthy volunteers received a single dose of recAP (200, 500, 1000 or 2000 U/kg; n = 33; 3:1 ratio) or multiple doses of recAP (500 or 1000 U/kg; n = 18; 2:1 ratio) via a 1-h intravenous infusion on three consecutive days. Serum recAP concentrations, alkaline phosphatase (AP) activity levels and anti-drug antibodies were measured, and safety parameters were monitored. A population pharmacokinetic model was developed, and simulations were performed to guide dose selection for a phase IIa/b trial.
Results
Peak concentrations of recAP and peak AP activity were reached at the end of the 1-h infusion and showed a rapid decline, with about 10 % of the maximum concentration remaining at 4 h and less than 5 % remaining 24 h post-start. RecAP treatment was generally well tolerated, and anti-drug antibodies could not be detected in the serum up to 2 weeks post-injection after a single dose, or up to 3 weeks post-injection after multiple doses. A four-compartment model best described the pharmacokinetics of recAP administration, with moderate inter-individual variability on the central volume of distribution and elimination rate constant. Simulations showed that 1-h intravenous infusions of 250, 500 and 1000 U/kg recAP once every 24 h for three consecutive days constituted the dosing regimen that best met the criteria for dose selection in patient studies.
Conclusion
RecAP did not raise any safety concerns when administered to healthy volunteers. A population pharmacokinetic model was developed to support dose selection for patient studies.
Trial Registration
2013-002694-21 (EudraCT).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Human recombinant alkaline phosphatase (recAP) is a potential new treatment option for critically ill patients with sepsis-associated acute kidney injury. |
In healthy volunteers, peak concentrations of recAP and peak alkaline phosphatase activity levels were reached at the end of a 1-h infusion and decreased rapidly in a multiphasic manner. Single ascending recAP doses of up to 2000 U/kg and multiple doses of up to 1000 U/kg per day for 3 days did not raise any safety concerns. |
A population pharmacokinetic model was developed and used for simulations to support dose selection for future patient studies. |
1 Introduction
Acute kidney injury (AKI) is a serious complication in critically ill patients, with an incidence of 20–50 % and a mortality of >50 % [1]. While AKI may develop after trauma, cardiovascular surgery or administration of nephrotoxic drugs, sepsis represents the most common cause [1]. Sepsis-associated AKI increases mortality to 70 %, and survivors have an increased risk of developing chronic kidney disease [2, 3]. As yet, no pharmacological interventions have been approved, and treatment is limited to renal replacement therapy (RRT).
Currently, alkaline phosphatase (AP) is one of the limited number of candidate drugs in clinical development to prevent or treat sepsis-associated AKI. This endogenous, membrane-bound enzyme is present in many cells and organs, including the placenta, liver, bone, kidney, germ cells and granulocytes [4]. AP exerts detoxifying effects through dephosphorylation of pro-inflammatory molecules, including endotoxins (lipopolysaccharide [LPS]) involved in sepsis pathogenesis, and adenosine triphosphate (ATP) released from cells during stressful events [5, 6]. The potency of AP in attenuating the innate immune response has been illustrated in several animal models of sepsis, in which exogenous AP reduced inflammatory injury and mortality [7, 8]. Considering the immune-modulating effects of AP, the potential of bovine intestinal AP (biAP) was investigated in critically ill patients with sepsis, with or without AKI, in which biAP treatment significantly improved renal function and prevented further kidney damage [9, 10]. An effect on mortality was not demonstrated in these relatively small studies (n = 36 each).
Administration of bovine-derived material in humans is less than ideal. Repeated administration of animal-sourced AP could provoke immune reactions, and bovine spongiform encephalopathy (BSE)-free sources of bovine-derived enzyme are difficult to obtain. Therefore, a human recombinant AP (recAP) has been developed as a pharmaceutically acceptable alternative by replacing the crown domain of a human intestinal AP with the crown domain of human placental AP [11]. As the renal protective effects of recAP have been confirmed in vitro in renal proximal tubular epithelial cells, and in vivo during LPS-induced AKI in rats [12], therapeutic application of recAP could have potent anti-inflammatory and tissue-protective effects in patients suffering from sepsis-associated AKI. Prior to investigating the potential of recAP in this patient population, it is vital to evaluate the pharmacokinetic characteristics of this enzyme. A translational population pharmacokinetic model has previously been developed, based on preclinical data on recAP and preclinical and clinical data on biAP [13], followed by simulations of several dosing regimens, aiding in dose selection for the first-in-human clinical trial, as reported in this article. Here, we evaluate the tolerability, safety and pharmacokinetics of recAP in healthy volunteers in a two-part, randomized, double-blind, placebo-controlled clinical trial. Using data from this study, the population pharmacokinetic model was further developed, and simulations were performed to support the design and dose selection for patient studies.
2 Materials and Methods
2.1 Phase I Clinical Trial
This two-part, randomized, double-blind, placebo-controlled study was conducted in 51 healthy volunteers (part A: n = 33; part B: n = 18) at a medical screening facility in Zuidlaren, the Netherlands. The local ethics committee approved all protocols, and the study, including amendments, was conducted in accordance with the principles of the Declaration of Helsinki and in compliance with the International Conference on Harmonisation E6 Guideline for Good Clinical Practice (CPMP/ICH/135/95) and the European Union Clinical Trial Directive (2001/20/EC). This study is reported in accordance with the Consolidated Standards of Reporting Trials (CONSORT) guidelines [14]. The trial was registered with the European Clinical Trials Register (EudraCT number 2013-002694-21) and the Dutch Central Committee on Research Involving Human Subjects (CCMO) [reference number NL45925.056.13].
2.1.1 Subjects
All participating healthy volunteers provided written informed consent prior to the start of any study-related procedures. Subjects were eligible for the study if they were aged 18–55 years and considered healthy on the basis of medical screening, including their medical history, a physical examination, routine laboratory testing and a 12-lead electrocardiogram (ECG). The exclusion criteria were pregnancy or lactation, a body mass index (BMI) of <18 or >30 kg/m2, smoking (more than five cigarettes, one cigar or one pipe daily), a history of alcohol abuse or drug addiction, and use of medication (except for acetaminophen). A 4-h fasting period was required prior to screening, admission to the medical screening facility, all visits with clinical laboratory assessments and the follow-up visit. The use of methylxanthine-containing beverages or food (coffee, tea, cola, chocolate, power drinks), grapefruit (juice) and alcohol was not allowed from 48 h prior to entry to the clinical research center and during the stay in the clinic.
2.1.2 Dosing Regimen
To support the dosing rationale for this trial, human pharmacokinetic profiles of recAP were simulated, based on a previously reported translational population pharmacokinetic model for recAP, using various dosing regimens [13]. The simulations, performed in R version 2.12.0 software [15] using the lsoda (deSolve Package 1.8.1) and mvrnorm functions (MASS Package version 7.3-8), were conducted considering the reported population parameters, as well as the full covariance matrix describing inter-individual variability (IIV), for 1000 individuals with a typical weight of 70 kg. Graphical representation of summary statistics of the simulations (medians and 95 % prediction intervals of the simulated IIVs) aided in the selection of optimal dosing regimens. The dosing regimens that were simulated are presented in Supplemental Table 1 in the Electronic Supplementary Material.
2.1.3 Study Design
Part A was a single-ascending-dose study to assess the safety, tolerability and pharmacokinetics of single intravenous doses of recAP in four sequential groups of eight healthy male or female subjects per group (groups 1–4). In each group, subjects were randomly assigned to receive a single dose of recAP (n = 6) or placebo (n = 2) via a 1-h intravenous infusion on day 1. The doses of recAP administered to subjects in groups 1, 2, 3 and 4 were 200, 500, 1000 and 2000 U/kg, respectively (Table 1). Blood samples were drawn pre-dose and at 0.25, 0.5, 0.92, 1.25, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 72, 120 and 192 h after the start of the infusion.
Part B was a multiple-ascending-dose study to assess the safety, tolerability and pharmacokinetics of multiple intravenous doses of recAP in two groups of nine healthy male and female subjects per group (groups 5 and 6). Subjects were randomly assigned to receive recAP (n = 6) or placebo (n = 3) via a 1-h intravenous infusion on days 1, 2 and 3. The doses of recAP or placebo administered to subjects in groups 5 and 6 were 500 and 1000 U/kg, respectively (Table 1). The selection of the dose and dosing regimen administered in part B was based on an interim pharmacokinetic analysis. Blood samples were drawn pre-dose and at 0.5, 0.92, 1.5, 3, 6 and 12 h after the start of the infusion on day 1; pre-dose just before the start of the infusion on day 2; and pre-dose and 0.25, 0.5, 0.92, 1.25, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 72, 120, 168 and 240 h after the start of the infusion on day 3.
2.1.4 Randomization, Blinding and Sample Size
A randomization scheme, assigning the study treatments to subject numbers at random, was generated by an independent biostatistician team, using SAS PROC PLAN (SAS software version 9.1.3) per treatment group. The randomization scheme was then transferred in a sealed envelope to the pharmacist for dispensing purposes and kept in the pharmacy. Subjects who met all eligibility criteria were given a subject number just prior to dosing according to the randomization scheme, and received the treatment assigned by the scheme.
The products that were administered were a placebo and the active medication (recAP 7036 U/mL; AM-Pharma, Bunnik, the Netherlands). RecAP was diluted with the placebo solution to stock solutions containing 600, 1500, 3000 or 6000 U/mL in order to give each subject an equal amount of the drug product per kilogram of actual body weight. The placebo consisted of 20 mM citrate, 250 mM sorbitol, 2 mM MgCl2 and 50 μM ZnCl2 (pH 7.0). To ensure blinding, the recAP and placebo were indistinguishable in appearance, AP activity results were not communicated to blinded persons during the duration of the study, and the randomization list was accessible only to the pharmacist and the pharmacy assistant. A subject’s treatment assignment was not broken until the end of the study, unless knowledge of the treatment assigned to the subject was necessary to guide medical treatment of the subject.
Considering the exploratory nature of this study, the sample size (with a minimum of six subjects per group) was selected to provide information on safety, tolerability and pharmacokinetics following single and multiple doses of recAP. No prospective calculations of sample statistical power were performed.
2.1.5 Bioanalysis and Safety Assessments
All bioanalytical procedures were performed at the Bioanalytical Laboratory of PRA Early Development Services (EDS, Assen, The Netherlands). Serum recAP concentrations were determined using a validated electrochemiluminescent ligand-binding procedure (ECL) [Meso Scale Discovery, Rockville, MD, USA]. The lower limit of quantitation was 3.91 ng/mL and the upper limit of quantitation was 500 ng/mL, with overall mean accuracy and precision values of ±20.0 % at each concentration of the quality-control samples (10, 66 and 400 ng/mL). Duplicates were not allowed to exceed the coefficient of variation (CV) of 10.0 %. Anti-drug antibodies were determined by an ECL ligand-binding assay, for which a confirmatory cut point of 18.9 % was established during method validation. Any study sample signal reduction equal to or higher than the specificity cut point was confirmed as positive, and any signal reduction lower than the specificity cut point was scored as negative. Duplicates were not allowed to exceed the CV of 20.0 %. The activity of AP was determined by routine laboratory testing, using the Advia 1800 (ALPAMP; Siemens AG Healthcare Sector, Erlangen, Germany).
All adverse events that were reported during the course of the study were recorded. Adverse events were coded using the Medical Dictionary for Regulatory Activities (MedDRA; Version 16.1 [16]) and evaluated by the principal investigator. The intensity of the adverse events was rated as mild, moderate, severe, life threatening or fatal, and the relationship between the adverse events and the study medication was indicated as non-existent, unlikely, possible, likely or definite. Additional safety and tolerability assessments consisted of the results of vital sign assessments, 12-lead ECGs, continuous cardiac monitoring (telemetry), laboratory tests, physical examinations and detection of anti-drug antibodies. All subjects who received at least one dose of the study medication were included in the analysis. Blood sampling for anti-drug antibodies was performed at screening and at days 9 and 15 (part A) or at days 10 and 19 (part B).
The data, represented as mean values and standard deviations or as numbers and percentages, were summarized in tabular or graphical form, and descriptive statistics are given.
2.2 Population Pharmacokinetic Model Development
Population-approach nonlinear mixed-effects modeling was performed with the NONMEM software package (version 7.2.0; Icon Development Solutions, Ellicott City, MD, USA). The location (the typical value or fixed effect) and the spread between individuals (the variability or random effect) were estimated for the model parameters by fitting the model to the data by minimizing the objective function based on the log likelihood (−2× LL). Additive, proportional or combined residual error structures were investigated, while log-normal distribution for the IIV was assumed. Various types of variance–covariance matrices were tested for the IIV.
In order to find the simplest model that adequately described the pharmacokinetic observations, different models were compared with increasing complexity in the structural model and the number of random effects. The likelihood ratio test was used, which compared the difference between −2× log likelihoods of the models (difference in objective function value [∆OFV]) with a Chi squared distribution with degrees of freedom corresponding to the difference in the number of parameters between the two models. Hence, with a difference of at least 6.63 points (P < 0.01), the model with one additional parameter was preferable to its parent model.
Graphical analysis was another tool used to help assessing differences between models. These goodness-of-fit plots included (1) population-predicted (PRED) and individual-predicted (IPRED) versus observed concentrations (DV); (2) conditional weighted residuals with interaction (CWRESI) versus predicted concentrations and versus time, where most observations should be normally distributed around zero and should be within the acceptance criterion of −2 to 2; (3) frequency distributions of CWRESI; (4) frequency distributions of post hoc individual estimates of IIV; and (5) correlation plots with their Pearson’s correlation coefficients for all parameters with IIV [17]. The uncertainty of the parameter estimates was determined by the relative standard error (RSE) [standard error/parameter estimate × 100 %] and was considered acceptable when the RSE was <50 %. The parameter estimates of the IIV were reported as %CV (\( \sqrt {{ \exp }(\omega^{2} ) - 1} \times 100\; \% \)), and shrinkage was considered acceptable when below 35 % [18].
A covariate analysis was performed on age, actual body weight, sex, height, lean body mass and BMI. The covariate in a variable cluster that showed the highest correlation with the empirical Bayes estimates of the parameters, which was also clinically relevant, was implemented in the model. Continuous covariates were centered on their median values. Power functions, exponentials and linear functions were explored for the covariate relationships. Covariate analysis was performed by forward inclusion (ΔOFV > 3.84), followed by backward elimination (ΔOFV > 6.63).
The first-order method with conditional estimation and interaction (FOCEI) was used for the maximum likelihood parameter estimation. Additionally, R version 2.12.0 software was used for the graphical presentation, evaluation of goodness of fit, covariate selection and model evaluation. The analyses closely followed the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines for performing and reporting population pharmacokinetic analyses [19, 20].
2.3 Simulations for Phase IIa/b Dose Selection
All simulations for the patient study were performed in R software, using the lsoda (deSolve Package 1.8.1) and mvrnorm functions (MASS Package version 7.3-8). The target used for the simulations was a serum recAP concentration of >290 U/L (170 ng/mL), maintained for at least 48 h and preferably for 5–7 days. The dosing rationale for the patient studies was primarily based on trough concentration (C min) values and on exposure values shown to be associated with objective clinical benefit in sepsis-associated AKI patients treated with biAP in previous clinical studies [9, 21]. The simulations were performed considering the estimated population parameters, as well as the full covariance matrix describing IIV, in 1000 individuals. To simulate the dose in units per kilogram, individual weights were drawn from a normal distribution (with resampling) based on the mean and standard deviation of the population of included subjects. Graphical representation of the summary statistics of the simulations (the median and 95 % prediction interval values for the simulated IIV) aided in the selection of optimal dosing regimens. The dosing regimens that were simulated are presented in Supplemental Table 2.
3 Results
3.1 Phase I Clinical Trial
3.1.1 Subjects
Fifty-one subjects were included in the study: 33 subjects (16 female) were assigned to part A, the single-ascending-dose part of the study; 18 subjects (8 female) were assigned to part B, the multiple-ascending-dose part of the study. All subjects complied with the inclusion and exclusion criteria. The dosing schedules and baseline characteristics are presented in Tables 1 and 2, respectively. There were no clinically significant findings with regard to medical history or previous medication. No subjects withdrew or dropped out during the course of the study. A total of 50 subjects completed the study per protocol in the period of September–December 2013. One subject in group 1 (Table 1) received less than the planned recAP dose of 200 U/kg because of leakage of the infusion set during administration, and that subject was replaced. The safety data for that subject, but not the pharmacokinetic data, were considered evaluable for analysis. All other subjects received the study medication as planned and had measurable serum recAP concentrations.
3.1.2 Pharmacokinetic Single-Ascending-Dose Study
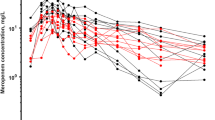
The pharmacokinetics were evaluated in healthy volunteers during a 1-h intravenous infusion of single-ascending-doses recAP (200, 500, 1000 or 2000 U/kg) in groups of eight subjects (six active, two placebo) [Table 1]. The maximum serum concentration values for recAP were typically reached at the end of the infusion (Fig. 1a). The mean recAP serum concentration profiles displayed an increase in serum concentrations following administration of increasing doses of recAP and ranged from 1715 to 26,253 ng/mL. At the end of the infusion, the recAP serum concentrations decreased rapidly in a multiphasic manner, with 10 % of the maximum concentration remaining at 4 h and <5 % remaining at 24 h after the start of the infusion. RecAP concentrations were detectable for up to 120 h post-dose in the 200 U/kg treatment group and for up to 192 h post-dose (the last pharmacokinetic sampling time point, on day 9) in the other three treatment groups.

Serum human recombinant alkaline phosphatase (recAP) concentrations and alkaline phosphatase (AP) enzyme activity in healthy volunteers. Subjects received a 1-h intravenous infusion of single ascending doses of recAP (200, 500, 1000 or 2000 U/kg) (a) or multiple ascending doses of recAP (500 and 1000 U/kg) on days 1, 2 and 3 (b, c). Serum recAP concentrations (upper graphs) and AP enzyme activity levels (lower graphs) were determined at several time points after the infusions. The data are expressed as geometric means and standard deviations (n = 6 per group)
The serum AP activity levels in recAP-treated subjects followed a pattern similar to that of the serum recAP concentrations; after reaching a maximum, at the end of the infusion, the AP activity decreased rapidly, followed by a slower decline (Fig. 1a), and the mean AP activity–time profiles displayed an increase in AP activity following administration of increasing doses of recAP. In comparison with baseline levels (49–53 U/L), serum AP levels dose-dependently increased approximately 35- to 500-fold at the end of the infusion and were still 2.5–20 times higher after 24 h. At 192 h post-dose, the AP activity in most subjects had returned to values near those observed at baseline. In each placebo-treated subject, the endogenous AP activity varied only minimally over time (not shown).
3.1.3 Pharmacokinetic Multiple-Ascending-Dose Study
Next, the pharmacokinetics were evaluated in a multiple-ascending-dose study. Healthy volunteers received multiple doses of recAP (500 and 1000 U/kg, n = 6 per group) or placebo via a 1-h intravenous infusion on days 1, 2 and 3 (Table 1). On days 1 and 3, the maximal serum concentrations of recAP were typically reached at the end of the infusion (Fig. 1b, c). In just one subject (in group 6) on day 1, the maximal serum concentration was reached 0.5 h after the end of the infusion. For both multiple-dose levels, the maximum concentrations of recAP were similar between days 1 and 3. RecAP was detectable up to 240 h (the last pharmacokinetic sampling time point) post-dose in both groups.
Like serum recAP concentrations, serum AP activity increased to a maximum at the end of the recAP infusion on both days, and the mean values were similar between days 1 and 3 for both multiple-dose levels (Fig. 1b, c). Directly after the infusion, 200- and 460-fold increases in AP activity levels were observed in the lower-dose and higher-dose groups, respectively, in comparison with baseline (58 U/L in both groups). Serum AP levels were still 5- to 12-fold higher than baseline levels 24 h after the start of the first infusion, and they further increased 8- to 20-fold 24 h after the third infusion. After an initial rapid decline in serum AP activity during the first 4 h post-infusion, a slower decline ensued, which was similar to what was observed after single-dose administration. At the last pharmacokinetic measurement (240 h post-dose), AP activity had returned to values near to those observed at baseline in most subjects. AP activity in placebo-treated subjects varied only minimally over time (not shown). When the serum recAP concentrations and AP activity in all subjects at all dose levels and time points evaluated in the study were compared, AP activity increased with increasing concentrations of recAP in a linear relationship.
3.1.4 Safety Assessments
Single intravenous infusions of recAP in the range of 200–2000 U/kg, as well as daily intravenous infusions of 500 and 1000 U/kg recAP for three consecutive days, were well tolerated by healthy male and female subjects and did not lead to any safety concerns. No serious adverse events were reported, and there were no discontinuations due to adverse events in any parts of the study. All drug-related treatment-emergent adverse events are presented in Supplemental Table 3. The most common events were headache (3/33) and postural dizziness (2/33) after the single infusion, and infusion-site reactions (3/18) and local swelling (2/18) after multiple infusions. None of the subjects demonstrated evidence of the presence of serum antibodies against recAP up to 2 weeks after single-dose administration or up to 3 weeks after multiple-dose administration. There were no significant clinical laboratory findings, 12-lead ECG findings, continuous cardiac monitoring findings or physical examination findings during the study. With respect to vital signs, one subject (in group 4) showed a postural drop in blood pressure (from 113/66 mmHg in the supine position to 82/49 mmHg in the standing position) 3 h post-dose.
3.2 Pharmacokinetic Modeling
One-, two- and three-compartment models proved inferior to a four-compartment model as the structural model. Introducing a fifth compartment did not significantly improve the model. For the four-compartment model, a proportional error structure was preferred over an additive error structure or a combined proportional and additive error structure. After investigation of IIV (represented as η in the model) on different parameters, the best model included an η on the central volume of distribution (V1) and elimination rate constant (K10), with a parameter describing the covariance between the η values. Finally, a covariate analysis was performed, based on the available covariates. On the basis of the correlation plots, the best candidates for covariates on either V1 or K10 were selected and tested in the models, but none improved the model fit and they were therefore not taken forward in the model development. The parameter estimates are listed in Table 3. The relatively low RSE values (<21 %) indicate accurate parameter estimation. The IIV, represented as %CV, is within the acceptance criterion, and the shrinkage was well below 35 %. The goodness-of-fit plots and individual pharmacokinetic profiles are represented in Fig. 2. The observed versus population-predicted values indicate that the model is structurally sound, and inclusion of IIV improves the fit, as seen in the observations versus individual predictions. A bias was observed in the plot representing CWRESI as a function of time and PRED. The model slightly overpredicts the concentrations at 1.5 and 2 h. Most observations, however, lie within the acceptance criterion (−2 to 2), indicating that the model describes the data accurately. The visual predictive check shows that 95 % of the data lie within the 95 % prediction interval, indicating that the model describes the variability accurately (Fig. 3).

Goodness-of-fit plots for human recombinant alkaline phosphatase concentrations. Left graphs observed (DV) versus population-predicted (PRED) and individual-predicted (IPRED) concentrations, showing the lines of unity (black lines) and regression lines (red lines). Right graphs conditional weighted residuals with interaction (CWRESI) versus IPRED concentrations and time after dose (TAD), showing the Loess fits through the data (red lines)

Visual predictive checks of the pharmacokinetic model after 500 U/kg human recombinant alkaline phosphatase (recAP) administered once or administered once daily on three consecutive days (normalized to the same dosing regimen if needed). The black lines are the median predicted human recAP concentrations over time, with the 95 % prediction intervals shown in gray. The dots are the observations after single doses of 200 U/kg (red), 500 U/kg (green), 1000 U/kg (orange) or 2000 U/kg (blue) [left graph], or after multiple doses of 500 U/kg (blue) or 1000 U/kg (red) [right graph]
3.3 Simulations for Phase IIa/b Dose Selection
Simulations were performed for a wide range of dosing regimens. These included multiple doses and continuous infusions, as shown in Supplemental Table 2, and were selected on the basis of the knowledge from the current study, taking into account that no dose-limiting toxicity was observed in healthy volunteers after single-dose administration of up to 2000 U/kg and after multiple-dose administration of up to three daily doses of 1000 U/kg. The dosing rationale for the patient studies was primarily based on C min values and exposure shown to be associated with objective clinical benefit in sepsis-associated AKI patients treated with biAP in previous clinical studies [9, 21], which indicated that clinical benefit occurred above serum recAP concentrations of 290 U/L (170 ng/mL). Therefore, the lowest predicted (trough) concentrations should stay above this level for at least 48 h and preferably for 5–7 days. On the basis of predictive simulations, three recAP dose levels—all 1-h intravenous infusions administered once every 24 h for three consecutive days—were selected, such that a low-dose group (250 U/kg) achieves borderline exposure and C min values, while the medium-dose and high-dose groups (500 and 1000 U/kg, respectively) should achieve substantially higher exposure and C min values. The simulation of the high dose is presented in Fig. 4. The three dosing regimens were also selected because the simulated serum recAP concentration–time profiles per dose group were sufficiently differentiated, which allows attribution of a potential observed treatment effect to a specific dose cohort.

Prediction of human recombinant alkaline phosphatase (recAP) concentrations over time after intravenous infusion of the high dose (1000 U/kg, administered once every 24 h for three consecutive days) selected for patient studies. The solid line is the median predicted recAP concentration over time, with the 95 % prediction interval shown in gray. The dashed line is the target trough concentration of 170 ng/mL
4 Discussion
Sepsis-associated AKI is a common complication observed in the intensive care unit and is accompanied by high mortality rates, while survivors have an increased risk of developing end-stage renal disease [22]. Two previous clinical trials suggested that biAP had renal protective effects in critically ill patients with sepsis-associated AKI [9, 10]. On the basis of these results, recAP was developed to replace the bovine-derived enzyme [11]. This randomized, double-blind, placebo-controlled, first-in-human trial demonstrated that single ascending recAP doses of up to 2000 U/kg and multiple recAP doses of up to 1000 U/kg per day for 3 days were well tolerated by healthy volunteers and that recAP displays a good safety profile. Following recAP infusion, no anti-drug antibodies were detectable, no serious adverse events occurred and recAP could be administered sequentially without an increase in either the frequency or the severity of any treatment-emergent adverse events, in comparison with single recAP infusions.
The results from the biAP trial showed that a loading dose was required to significantly raise serum AP activity levels, followed by a continuous infusion over 24 h, to maintain relatively stable serum AP levels [21]. In order to improve the treatment regimen, recAP was developed by replacing the crown domain of a human intestinal AP with the crown domain of human placental AP [11], which has a half-life of approximately 6–7 days in humans [23]. This is expected to improve enzyme stability while preserving or enhancing catalytic function. Indeed, our results demonstrated that a 1-h recAP infusion resulted in an apparent terminal half-life of 48 h, and that 24 h after a single administration of 2000 U/kg recAP, or after 3 days of recAP dosing of 1000 U/kg, serum AP activity levels were still up to 20-fold higher than at baseline. The improved bioavailability of recAP could be an important feature of a potential new treatment option for sepsis-associated AKI, a disease with a complex pathogenesis in which multiple pathological processes—including inflammation, hemodynamic instability, microvascular dysfunction and unbalanced renal bioenergetics [24]—should be targeted for a longer period of time.
On the basis of the pharmacokinetic observations, a four-compartment population pharmacokinetic model was developed with IIV on V1 and K10. The apparent terminal half-life (48 h) and volume of distribution (3.5 L) are comparable to those in the previously developed translational two-compartment pharmacokinetic model (41 h and 4.2 L, respectively) used to support the design of this trial [13]. The discrepancy in the model structure could be explained by the experimental design of this trial, as the sampling was more intensive, with a longer follow-up period. Nevertheless, our model accurately described the data and was used to support the selection of optimal dosing regimens to investigate the potential of recAP in patients. The simulations aimed to reach trough recAP concentrations above 290 U/L (170 ng/mL), maintained for at least 48 h and preferably for 5–7 days. This resulted in selection of three recAP doses, ranging from 250 to 1000 U/kg, administered once every 24 h for three consecutive days. The dosing rationale was primarily based on the results of previous clinical studies conducted with biAP [9, 21]. As could be expected from the enhanced stability by design of the chimeric enzyme, the data from this trial showed that AP activity increased with increasing concentrations of recAP in a linear relationship. Once the optimal dose of recAP is determined, therapeutic drug monitoring allows exposure to be monitored, as AP activity can be quantified by routine laboratory chemistry.
Patients suffering from sepsis-associated AKI not only have impaired renal function, but also have critical illness-related symptoms such as hypotension, edema and increased vascular permeability, all of which might influence recAP pharmacokinetics. In addition, impaired liver function could affect recAP clearance, as circulating AP is normally cleared through uptake by the hepatic asialoglycoprotein receptor [13]. Previously, we demonstrated in vivo that recAP radioactivity and AP enzyme activity levels were increased during endotoxemia-induced AKI in rats, in comparison with placebo, whereas the organ distribution of recAP was not critically affected [13]. Currently, the safety and efficacy of recAP are being investigated in patients with sepsis-associated AKI (ClinicalTrials.gov study ID: NCT02182440). In that trial, recAP pharmacokinetics will also be assessed in patients receiving one of three doses of recAP (250, 500 or 1000 U/kg). Even if pharmacokinetic derangements affect the recAP dosing requirement, the highest dose selected for that patient study significantly exceeds the target C min of 290 U/L. Whether recAP administration will result in sufficient exposure in the target organs of the heterogeneous patient population, and will thereby exert renal protective effects, will be revealed by that large, multicenter trial.
5 Conclusion
In this study, we investigated the pharmacokinetics, safety and tolerability of recAP, a potential new biological treatment option for critically ill patients with sepsis-associated AKI. In healthy volunteers, peak concentrations of recAP and AP activity levels were reached at the end of the 1-h infusion and decreased rapidly in a multiphasic manner. Single ascending recAP doses of up to 2000 U/kg and multiple doses of up to 1000 U/kg three times daily did not cause serious adverse events. A population pharmacokinetic model was developed and was used to simulate different dosing regimens. From these simulations, a more informed dose was selected for the patient studies. These results pave the way to investigation of the potential of recAP as a new treatment option for sepsis-associated AKI in patient studies.
References
Case J, Khan S, Khalid R, Khan A. Epidemiology of acute kidney injury in the intensive care unit. Crit Care Res Prac. 2013;2013:479730.
Oppert M, Engel C, Brunkhorst FM, Bogatsch H, Reinhart K, Frei U, et al. Acute renal failure in patients with severe sepsis and septic shock—a significant independent risk factor for mortality: results from the German Prevalence Study. Nephrol Dial Transpl. 2008;23(3):904–9.
Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int. 2011;79(12):1361–9.
Millan JL. Alkaline phosphatases: structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic Signalg. 2006;2(2):335–41.
Bentala H, Verweij WR, Huizinga-Van der Vlag A, van Loenen-Weemaes AM, Meijer DK, Poelstra K. Removal of phosphate from lipid A as a strategy to detoxify lipopolysaccharide. Shock. 2002;18(6):561–6.
Picher M, Burch LH, Hirsh AJ, Spychala J, Boucher RC. Ecto 5′-nucleotidase and nonspecific alkaline phosphatase: two AMP-hydrolyzing ectoenzymes with distinct roles in human airways. J Biol Chem. 2003;278(15):13468–79.
Beumer C, Wulferink M, Raaben W, Fiechter D, Brands R, Seinen W. Calf intestinal alkaline phosphatase, a novel therapeutic drug for lipopolysaccharide (LPS)-mediated diseases, attenuates LPS toxicity in mice and piglets. J Pharmacol Exp Ther. 2003;307(2):737–44.
Su F, Brands R, Wang Z, Verdant C, Bruhn A, Cai Y, et al. Beneficial effects of alkaline phosphatase in septic shock. Crit Care Med. 2006;34(8):2182–7.
Pickkers P, Heemskerk S, Schouten J, Laterre PF, Vincent JL, Beishuizen A, et al. Alkaline phosphatase for treatment of sepsis-induced acute kidney injury: a prospective randomized double-blind placebo-controlled trial. Crit Care. 2012;16(1):R14.
Heemskerk S, Masereeuw R, Moesker O, Bouw MP, van der Hoeven JG, Peters WH, et al. Alkaline phosphatase treatment improves renal function in severe sepsis or septic shock patients. Crit Care Med. 2009;37(2):417–23, e1.
Kiffer-Moreira T, Sheen CR, Gasque KC, Bolean M, Ciancaglini P, van Elsas A, et al. Catalytic signature of a heat-stable, chimeric human alkaline phosphatase with therapeutic potential. PLoS One. 2014;9(2):e89374.
Peters E, Geraci S, Heemskerk S, Wilmer MJ, Bilos A, Kraenzlin B, et al. Alkaline phosphatase protects against renal inflammation through dephosphorylation of lipopolysaccharide and adenosine triphosphate. Br J Pharmacol. 2015;172(20):4932–45.
Peters E, Stevens J, Arend J, Guan Z, Raaben W, Laverman P, et al. Biodistribution and translational pharmacokinetic modeling of a human recombinant alkaline phosphatase. Int J Pharm. 2015;495(1):122–31.
Kanji S, Hayes M, Ling A, Shamseer L, Chant C, Edwards DJ, et al. Reporting guidelines for clinical pharmacokinetic studies: the ClinPK statement. Clin Pharmacokinet. 2015;54(7):783–95.
R Development Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2010.
International Conference on Harmonisation. Introductory guide: MedDRA version 16.1. McLean: MedDRA Maintenance and Support Services Organization; 2013. http://www.meddra.org/sites/default/files/guidance/file/intguide_16_1_english.pdf. Accessed 3 Mar 2016.
Pearson K. Note on regression and inheritance in the case of two parents. Proc R Soc Lond. 1895;58(58):240–2.
Savic RM, Karlsson MO. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11(3):558–69.
US Food and Drug Administration [FDA] Center for Drug Evaluation and Research. Guidance for industry: population pharmacokinetics. Rockville: FDA; 1999. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133184.pdf. Accessed 1 Mar 2016.
European Medicines Agency [EMA] Committee for Medicinal Products for Human Use. Guideline on reporting the results of population pharmacokinetic analyses. London: European Medicines Agency; 2007. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003067.pdf. Accessed 3 Mar 2016.
Pickkers P, Snellen F, Rogiers P, Bakker J, Jorens P, Meulenbelt J, et al. Clinical pharmacology of exogenously administered alkaline phosphatase. Eur J Clin Pharmacol. 2009;65(4):393–402.
Lameire NH, Bagga A, Cruz D, De Maeseneer J, Endre Z, Kellum JA, et al. Acute kidney injury: an increasing global concern. Lancet. 2013;382(9887):170–9.
McComb RB, Bowers GN Jr, Posen S. Isoenzymes. In: Alkaline phosphatase. New York: Plenum Press; 1979. p. 415.
Gomez H, Ince C, De Backer D, Pickkers P, Payen D, Hotchkiss J, et al. A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock. 2014;41(1):3–11.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Compliance with Ethical Standards
All protocols were approved by the local ethics committee, and the study, including amendments, was conducted in accordance with the principles of the Declaration of Helsinki and in compliance with the International Conference on Harmonisation E6 Guideline for Good Clinical Practice (CPMP/ICH/135/95) and the European Union Clinical Trial Directive (2001/20/EC). All healthy volunteers who participated in the study provided written informed consent before the start of any study-related procedures.
Funding
This study was fully funded by AM-Pharma.
Conflict of interest
Peter Pickkers received consultancy fees from AM-Pharma for the design of the phase IIa/b trial of recAP. Andrea van Elsas is an inventor on patent applications relating to recAP and received advisory fees and stock options from AM-Pharma. Jacques Arend is an employee of AM-Pharma. Renger Tiessen is an employee of the contract research organization (CRO) sponsored to conduct this research. The remaining authors declare that they have no relevant financial interests. Jacques Arend has full control of all primary data from the study and agrees to allow the journal to review the data if requested.
Additional information
E. Peters, J. A. A. C. Heuberger and R. Tiessen contributed equally to this work.
J. Stevens and P. Pickkers share senior authorship of this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Peters, E., Heuberger, J.A.A.C., Tiessen, R. et al. Pharmacokinetic Modeling and Dose Selection in a Randomized, Double-Blind, Placebo-Controlled Trial of a Human Recombinant Alkaline Phosphatase in Healthy Volunteers. Clin Pharmacokinet 55, 1227–1237 (2016). https://doi.org/10.1007/s40262-016-0399-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0399-y