
Abstract
Background and Objectives
Two phase I drug interaction studies were performed with oral enzalutamide, which is approved for the treatment of metastatic castration-resistant prostate cancer (mCRPC).
Methods
A parallel-treatment design (n = 41) was used to evaluate the effects of a strong cytochrome P450 (CYP) 2C8 inhibitor (oral gemfibrozil 600 mg twice daily) or strong CYP3A4 inhibitor (oral itraconazole 200 mg once daily) on the pharmacokinetics of enzalutamide and its active metabolite N-desmethyl enzalutamide after a single dose of enzalutamide (160 mg). A single-sequence crossover design (n = 14) was used to determine the effects of enzalutamide 160 mg/day on the pharmacokinetics of a single oral dose of sensitive substrates for CYP2C8 (pioglitazone 30 mg), CYP2C9 (warfarin 10 mg), CYP2C19 (omeprazole 20 mg), or CYP3A4 (midazolam 2 mg).
Results
Coadministration of gemfibrozil increased the composite area under the plasma concentration–time curve from time zero to infinity (AUC∞) of enzalutamide plus active metabolite by 2.2-fold, and coadministration of itraconazole increased the composite AUC∞ by 1.3-fold. Enzalutamide did not affect exposure to oral pioglitazone. Enzalutamide reduced the AUC∞ of oral S-warfarin, omeprazole, and midazolam by 56, 70, and 86 %, respectively; therefore, enzalutamide is a moderate inducer of CYP2C9 and CYP2C19 and a strong inducer of CYP3A4.
Conclusions
If a patient requires coadministration of a strong CYP2C8 inhibitor with enzalutamide, then the enzalutamide dose should be reduced to 80 mg/day. It is recommended to avoid concomitant use of enzalutamide with narrow therapeutic index drugs metabolized by CYP2C9, CYP2C19, or CYP3A4, as enzalutamide may decrease their exposure.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Oral enzalutamide is approved for the treatment of patients with metastatic castration-resistant prostate cancer (mCRPC). As mCRPC most commonly occurs in older men, and polypharmacy is prevalent among elderly patients, drug interactions are an important consideration for clinical use of enzalutamide. |
This article describes two phase I drug interaction studies: one investigating the effects of coadministered drugs on the pharmacokinetics of enzalutamide, and one investigating the effects of enzalutamide on the pharmacokinetics of coadministered drugs. |
The results showed that strong cytochrome P450 (CYP) 2C8 inhibitors can increase the composite area under the plasma concentration–time curve from time zero to infinity (AUC∞) of enzalutamide plus its active metabolite by 2.2-fold, and enzalutamide is a moderate inducer of CYP2C9 and CYP2C19 and a strong inducer of CYP3A4. Precautionary measures for mitigating the risks of clinical drug interactions are described within the article. |
1 Introduction
Enzalutamide has been shown to competitively inhibit androgen binding to androgen receptors and inhibit androgen receptor nuclear translocation and interaction with DNA [1]. The efficacy and safety of enzalutamide in patients with metastatic castration-resistant prostate cancer (mCRPC) was assessed in two randomized, placebo-controlled, multicenter phase III clinical trials (AFFIRM and PREVAIL) [2, 3]. The pharmacokinetics of enzalutamide have been studied in patients with mCRPC and in healthy male subjects [4].
Clinical studies have shown that enzalutamide has two major metabolites: N -desmethyl enzalutamide and a carboxylic acid metabolite. N -desmethyl enzalutamide is an active metabolite that is thought to contribute to the clinical effects of enzalutamide because it demonstrates primary and secondary pharmacodynamics of similar potency to enzalutamide and circulates at approximately the same plasma concentrations as enzalutamide. The carboxylic acid metabolite is pharmacologically inactive and circulates at approximately 25 % lower plasma concentrations than enzalutamide [4].
This article summarizes the findings of two clinical drug interaction studies, both of which were informed by the results of prior in vitro experiments (Sect. 4). One of the clinical studies assessed the effects of strong inhibitors of cytochrome P450 (CYP) 2C8 and CYP3A4 on the pharmacokinetics of enzalutamide and its active metabolite. The other study assessed the potential for enzalutamide to affect the pharmacokinetics of orally administered drugs that are sensitive substrates of CYP2C8, CYP2C9, CYP2C19, or CYP3A4. The overall goal of these studies was to determine if potential interactions exist between enzalutamide and other drugs and, if so, to describe measures to mitigate the risk to patients.
2 Methods
2.1 Subjects and Study Drug
Informed consent was obtained from all subjects before study entry. The study with CYP2C8 and CYP3A4 inhibitors was assigned ClinicalTrials.gov registry number NCT01913379 [28], and the study with CYP substrates was assigned NCT01911728 [29]. Both studies were conducted in accordance with the Declaration of Helsinki and with the approval of the appropriate local ethics committees.
To be eligible for the study with CYP2C8 and CYP3A4 inhibitors, subjects were required to be healthy males aged 18–55 years, inclusive. Homozygous carriers for the CYP2C8*3 allele were categorized as poor metabolizers and deemed ineligible.
To be eligible for the study with CYP substrates, individuals were required to be males aged ≥18 years and to have histologically or cytologically confirmed adenocarcinoma of the prostate without neuroendocrine differentiation or small cell features, with ongoing androgen deprivation therapy with a gonadotropin-releasing hormone analogue or orchiectomy. Homozygous carriers of CYP2C8*3, CYP2C9*2, or CYP2C19*2, or heterozygous carriers of CYP2C9*2/CYP2C9*3 or CYP2C19*2/CYP2C19*3 were categorized as poor metabolizers and deemed ineligible. Patients were excluded for any of the following reasons: severe concurrent disease, infection, or co-morbidities; known metastases in the liver or any hepatic disorder that would affect drug metabolism; gastrointestinal disorders that would potentially alter absorption; history of another malignancy within the previous 5 years other than curatively treated non-melanoma skin cancer; and any clinically significant cardiovascular disease.
In both studies, the enzalutamide dose and drug product presentation were the same as the commercial product (XTANDI®, Astellas Pharma US, Inc., Northbrook, IL, USA, and Medivation Inc., San Francisco, CA, USA): 160 mg as four liquid-filled capsules of enzalutamide 40 mg fully dissolved in caprylocaproyl polyoxylglycerides.
2.2 Pharmacokinetic and Analytical Methods
Pharmacokinetic blood samples were collected with the anticoagulants named in Table 1. To determine the concentrations of the test compounds and their major metabolites (Table 1), plasma samples were analyzed by high-performance liquid chromatography (LC) with detection by tandem mass spectrometry. These methods were validated in accordance with US Food and Drug Administration (FDA) guidance [5]. In all cases, the accuracy and precision were within 15 % over the quantitative ranges shown in Table 1, except at the respective lower limits of quantification, where accuracy and precision were within 20 %. Selectivity tests were performed for coadministered drugs and corresponding major metabolites at clinically relevant concentrations, which were based on the dosing schedule in the clinical protocol. Deuterium-labeled analogs were used as internal standards (IS), and concentrations of the analytes in calibration standards, quality control samples, and study samples were calculated using peak area ratios (analyte/IS). Additionally, the stability of the analytes in frozen human plasma was demonstrated for periods exceeding the storage periods of the samples prior to analysis, as well as under all conditions to which study samples or working solutions were subjected. Details on the bioanalytical methods for enzalutamide and its two metabolites are described elsewhere [6]. Details on the bioanalytical methods for the other test compounds and their respective metabolites are provided below.
To measure pioglitazone and hydroxy-pioglitazone (M-IV), 0.1 mL of plasma was mixed with 20 μL of IS working solution, which contained pioglitazone-d4 and hydroxy-pioglitazone-d4, and an automated solid-phase extraction procedure was used to isolate the analytes. The extracted sample was injected onto a Pursuit XRS 3 diphenyl column (30 × 3 mm; 3 μm) (Agilent Technologies, Santa Clara, CA, USA), and mass spectral (MS) detection was performed with an API 4000 (Applied Biosystems/MDS Sciex, Foster City, CA, USA).
To measure midazolam and 1-hydroxy-midazolam, 0.2 mL of plasma was mixed with 25 μL of IS working solution, which contained midazolam-d4 and 1-hydroxy-midazolam-d4, and a liquid–liquid extraction procedure was used to isolate the analytes. The organic layer was transferred to a clean tube and evaporated under a nitrogen stream at approximately 40 °C. The residue was reconstituted with 250 μL of reconstitution solution. The extracted sample was injected onto a Zorbax Eclipse Plus C18, Rapid Resolution HT column (2.1 × 50 mm; 1.8 μm) (Agilent Technologies), and MS detection was performed with an API 4000.
To measure omeprazole and 5-hydroxy-omeprazole, 0.05 mL of plasma was mixed with 25 μL of IS working solution, which contained omeprazole-d3 and omeprazole sulfone-d3, and the analytes were isolated with protein precipitation extraction using 300 μL of acetonitrile:methanol (65:35). The supernatant was diluted with 200 μL of 0.25 % ammonium hydroxide in water. The final extract was injected into an LC instrument that was configured for column switching between an Aquasil C18 column (100 × 2.1 mm; 5 μm) (Thermo Electron Corp., Madison, WI, USA) and a Javelin Betasil C18 column (20 × 2.1 mm; 5 μm) (Thermo Electron Corp.). MS detection was performed with a Quattro Ultima (Waters Corp., Milford, MA, USA).
To measure R-warfarin, 0.2 mL of plasma was mixed with 20 μL of IS working solution that contained warfarin-d6. Analytes were isolated through liquid–liquid extraction using an organic solvent of methyl tertiary butyl ether and dichloromethane. The extracted organic solvent was evaporated under a nitrogen stream at approximately 45 °C, and the residue was reconstituted with 300 μL of reconstitution solution. The final extract was injected onto a Chiralcel® OD-R column (250 × 4.6 mm; 10 μm) (Daicel Chemical Industries, Inc., Osaka, Japan), and MS detection was performed with an API 3000 (Applied Biosystems/MDS Sciex).
To measure S-warfarin and 7-hydroxy-S-warfarin, 0.2 mL of plasma was mixed with 20 μL of IS working solution that contained warfarin-d6 and 7-hydroxy-warfarin-phenyl-d5. Hydrochloric acid and extraction solvent were added, and the solution was vortexed and centrifuged. The organic layer was transferred to a clean tube, the solution was evaporated, and the residue was reconstituted with 200 μL of reconstitution solution. The final extract was injected into an LC instrument that was configured for column switching between an Eclipse XDB C18 column (150 × 4.6 mm; 5 μm) (Agilent Technologies) and a Chiralcel® OD-R column (250 × 4.6 mm; 10 μm). MS detection was performed with an API 3000.
Pharmacokinetic analyses were based on plasma concentration–time data for the test compounds and metabolites. For enzalutamide, an additional pharmacokinetic analysis was based on the sum of enzalutamide plus N-desmethyl enzalutamide (i.e., composite of enzalutamide active moieties). Pharmacokinetic parameters were estimated by non-compartmental analysis (NCA) methods in WinNonlin® (Pharsight Corp., Palo Alto, CA, USA) and applicable complimentary software such as SAS® (SAS Institute, Cary, NC, USA). The parameters included area under the plasma concentration–time curve (AUC) from time zero to the last quantifiable concentration, AUC for one 24-h dosing interval at steady state, AUC from time zero to infinity (AUC∞), maximum plasma concentration (C max), time to C max, minimum (pre-dose) plasma concentration (C trough), terminal elimination half-life (t ½), and apparent oral clearance (parent only).
For the study with CYP2C8 and CYP3A4 inhibitors, an additional exposure term was calculated, AUC from time zero to 18 days post-dose (AUC18 d), which corresponded to the AUC from day 4 (day of enzalutamide dosing) to day 22 (day of inhibitor discontinuation) (Sect. 2.3.1). Additionally, a pharmacokinetic analysis based on population pharmacokinetic modeling and simulation was performed to provide more accurate estimates of t ½ and AUC∞. This was necessary because gemfibrozil or itraconazole administration was stopped before complete concentration–time profiles for enzalutamide and metabolites were obtained under inhibitory conditions (Sect. 3.2.1). Pharmacokinetic parameters were estimated using the first-order condition estimation method with interaction in the non-linear mixed-effects modeling software NONMEM® (versions 7.1 and 7.2, ICON Development Solutions, Ellicott City, MD, USA). Simulated concentration–time profiles were generated for each of the 41 subjects in the drug interaction study, and the resulting data were analyzed by NCA methods in WinNonlin®. Further details are provided in Electronic Supplementary Material 1 and 2.
2.3 Study Design
2.3.1 Study with Cytochrome P450 (CYP) 2C8 and CYP3A4 Inhibitors
An open-label, randomized, three-arm parallel-treatment study design was used to determine the effect of multiple-dose gemfibrozil (a strong CYP2C8 inhibitor) or itraconazole (a strong CYP3A4 inhibitor) on the pharmacokinetics of a single oral dose of enzalutamide 160 mg. In arm 1, subjects received enzalutamide on day 1. In arm 2, subjects received gemfibrozil 600 mg orally twice daily on days 1–21; on day 4, enzalutamide was administered with the morning dose of gemfibrozil. Arm 3 was similar to arm 2, except that itraconazole 200 mg orally once daily was used instead of gemfibrozil. In arms 2 and 3, the inhibitor (gemfibrozil or itraconazole) was discontinued on day 22, which was 18 days after the single dose of enzalutamide.
Enzalutamide was administered under fasting conditions (no caloric intake for at least 10 h before dosing) when given alone (arm 1; day 1) or in combination with gemfibrozil (arm 2; day 4) or itraconazole (arm 3; day 4). In arms 2 and 3, the inhibitor was administered first, and enzalutamide was administered within 2 min. On all occasions other than day 4, gemfibrozil was administered at least 30 min prior to intake of food and itraconazole was administered under fed conditions to maximize absorption [7, 8].
The time of enzalutamide dose administration was t = 0 for purposes of pharmacokinetic sample collection times. Due to the long t ½ of enzalutamide, N-desmethyl enzalutamide, and the carboxylic acid metabolite in healthy subjects (approximately 4, 8, and 10 days, respectively [4]), pharmacokinetic samples were collected for 49 days. The sampling schedule was as follows: pre-dose and at 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, and 12 h (day 1), 24 and 36 h (day 2), 48 h (day 3), 72 h (day 4), 120 h (day 6), 168 h (day 8), 264 h (day 12), 336 h (day 15), 432 h (day 19), 504 h (day 22), 600 h (day 26), 672 h (day 29), 768 h (day 33), 840 h (day 36), 936 h (day 40), 1008 h (day 43), 1104 h (day 47), and 1176 h (day 50) post-dose.
2.3.2 Study with CYP2C8, CYP2C9, CYP2C19, and CYP3A4 Substrates
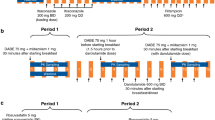
A single-sequence crossover study design was used to determine the effect of enzalutamide on the pharmacokinetics of a single oral dose of pioglitazone (CYP2C8 substrate), S-warfarin (CYP2C9 substrate), omeprazole (CYP2C19 substrate), or midazolam (CYP3A4 substrate). The design was based on validated cocktails and drug interaction trials reported in the literature, with special attention to minimizing undesirable side effects [9–17].
As summarized in Fig. 1, a single oral dose of pioglitazone 30 mg was given on day 1, followed by a 4-day washout. On day 5, a single oral cocktail of warfarin 10 mg (racemic mixture of R- and S-warfarin), omeprazole 20 mg, and midazolam 2 mg was administered, followed by a washout period of 8 days. On days 1 and 5, patients received a single oral dose of enzalutamide placebo-to-match, which was identical to the enzalutamide drug product presentation except that it lacked the drug substance. The purpose of the placebo-to-match was to control for possible effects of excipients (caprylocaproyl polyoxylglycerides) on the absorption of the substrate drugs. Patients received oral doses of enzalutamide 160 mg once daily (i.e., the recommended dose for mCRPC) from days 13 to 97. On day 55, a single oral dose of pioglitazone 30 mg was given. After a 7-day washout (i.e., on day 62), patients received a single oral cocktail of warfarin 10 mg, omeprazole 20 mg, and midazolam 2 mg. Patients experiencing clinical benefit at day 97 were permitted to enroll into an extension study in which they could continue receiving enzalutamide 160 mg once daily.

Schematic of the phase I fixed-sequence crossover drug interaction study with CYP2C8, CYP2C9, CYP2C19, and CYP3A4 substrates in patients with metastatic castration-resistant prostate cancer. CYP cytochrome P450. aEnzalutamide placebo-to-match capsules were filled with caprylocaproyl polyoxylglycerides and administered under fasting conditions on days 1 and 5. bPatients were instructed to take enzalutamide (160 mg) on days 13–97 as close to the same time each day as possible; enzalutamide could be taken with or without food, except on days 55 and 62, when it was administered under fasting conditions. cPioglitazone (30 mg) was administered under fasting conditions on days 1 and 55. dThe oral drug cocktail, which consisted of warfarin (10 mg), omeprazole (20 mg), and midazolam (2 mg), was administered under fasting conditions on days 5 and 62
Patients were instructed to take enzalutamide as close as possible to the same time each day. Enzalutamide was permitted to be taken with or without food, except on days 55 and 62, when it was required to be taken under fasting conditions. Administration of the substrate drugs, in combination with enzalutamide placebo-to-match (on days 1 and 5) or in combination with enzalutamide 160 mg (on days 55 and 62) occurred under fasting conditions. Enzalutamide or placebo-to-match was always administered first and the substrates were administered within 2 min.
For pioglitazone, time of dosing was t = 0 for the purposes of sample collection times, and pharmacokinetic samples were collected on days 1 and 55 at the following times: pre-dose and post-dose at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 14, 24, 32, 40, 48, 72, and 96 h. For warfarin, omeprazole, and midazolam, time of administration of the drug cocktail was t = 0 for purposes of sample collection times, and pharmacokinetic samples were collected proximal to the day 5 and day 62 doses. Pharmacokinetic samples for warfarin were collected at the following times: pre-dose and post-dose at 1, 2, 3, 4, 6, 12, 24, 36, 48, 72, 96, 120, 144, 168, 192, and 240 h. Pharmacokinetic samples for midazolam were collected at the following times: pre-dose and post-dose at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36, and 48 h. Pharmacokinetic samples for omeprazole were collected at the following times: pre-dose and post-dose at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 16 h. For all substrate drugs, the sampling interval was slightly longer with the second dose (i.e., when administered in combination with enzalutamide) because prior in vitro data suggested that enzalutamide might have possible enzyme inhibitory effects.
2.4 Statistical Analysis
Analysis of variance (ANOVA) tests of natural logarithm-transformed AUC∞ values and C max were performed in SAS® (version 9.1). Point estimates and 90 % confidence intervals (CIs) for the difference in means (test minus reference) from ANOVA were back-transformed using antilogarithms to obtain point estimates and 90 % CI for the ratio (test divided by reference) of the mean AUC∞ and C max values. In the study with CYP2C8 and CYP3A4 inhibitors, ANOVA tests evaluated the effect of gemfibrozil and itraconazole on the AUC∞ and C max of enzalutamide and its active metabolite. Treatment arm was a fixed effect, and linear contrast was applied to compare each of the two test treatments (arms 2 and 3) to the reference treatment (arm 1). In the study with CYP substrates, ANOVA tests evaluated the effect of enzalutamide on the AUC∞ and C max of the substrate drugs and their major metabolites. In each comparison, the test treatment corresponded to the combination with enzalutamide, and the reference treatment corresponded to the combination with enzalutamide placebo-to-match.
3 Results
3.1 Participants
The study with strong CYP2C8 and CYP3A4 inhibitors enrolled 41 healthy male subjects, and the study with CYP substrates enrolled 14 male patients with mCRPC. Demographics and baseline characteristics for both studies are summarized in Table 2.
3.2 Pharmacokinetics
3.2.1 Study with Strong CYP2C8 and CYP3A4 Inhibitors
As evidenced in Fig. 2, gemfibrozil decreased the rates of elimination of enzalutamide and formation of N -desmethyl enzalutamide while increasing the rate of formation of the carboxylic acid metabolite; these rates changed suddenly when gemfibrozil was discontinued on day 22. Given the apparent changes in pharmacokinetics of N-desmethyl enzalutamide after discontinuation of gemfibrozil, extrapolation of the observed concentration–time data in the terminal phase could not be used to estimate the magnitude of the effect of gemfibrozil on AUC∞. To address this issue, pharmacokinetic models were used to simulate concentration–time profiles for enzalutamide and metabolites for enzalutamide administered alone and enzalutamide coadministered with continuous gemfibrozil (i.e., no discontinuation on day 22) (Electronic Supplementary Material 1) [18]. Simulated concentration–time data for each of the 41 subjects in the study were then analyzed by NCA methods to estimate AUC∞ values. As AUC18 d and C max were defined by plasma concentration–time data prior to gemfibrozil discontinuation on day 22, these parameters were estimated by NCA analysis of observed data.

Mean concentration–time profiles for a enzalutamide, b N-desmethyl enzalutamide, and c carboxylic acid metabolite after a single oral dose of enzalutamide in healthy male subjects. Subjects received enzalutamide alone (n = 13), enzalutamide plus gemfibrozil (strong CYP2C8 inhibitor) (n = 13), and enzalutamide plus itraconazole (strong CYP3A4 inhibitor) (n = 14). Enzalutamide (160 mg) was administered under fasted conditions on day 4. Gemfibrozil (600 mg twice daily) was administered at least 30 min prior to food intake on days 1–21. Itraconazole (200 mg once daily) was administered under fed conditions on days 1–21. CYP cytochrome P450
As indicated by the geometric mean ratios (GMRs; Table 3), gemfibrozil had the following effects on enzalutamide and the active metabolite: for enzalutamide, AUC18 d and AUC∞ increased by 2.53-fold and 4.26-fold, respectively, while C max decreased by 18 %; for N-desmethyl enzalutamide, AUC18 d, AUC∞, and C max decreased by 67, 25, and 44 %, respectively; and for the composite sum of enzalutamide plus N-desmethyl enzalutamide, AUC18 d and AUC∞ increased by 1.39-fold and 2.17-fold, respectively, while C max decreased by 16 %. Notably, the estimated magnitude of the effect of gemfibrozil on the sum of exposure to active moieties (enzalutamide plus N-desmethyl enzalutamide) was smaller for the AUC term based on observed data (AUC18 d) than for the AUC term based on modeling and simulation (AUC∞).
Itraconazole appeared to have only a small impact on the elimination of enzalutamide and the rates of formation of N -desmethyl enzalutamide and the carboxylic acid metabolite (Fig. 2); therefore, all pharmacokinetic parameters for assessing the itraconazole drug interaction were based on observed data. As indicated by the GMR values (Table 3), itraconazole had the following effects on enzalutamide and the active metabolite: for enzalutamide, AUC18 d and AUC∞ increased 1.34-fold and 1.41-fold, respectively, while C max decreased by 2 %; for N-desmethyl enzalutamide, AUC18 d decreased by 4 %, AUC∞ increased 1.21-fold, and C max decreased by 14 %; and for the sum of enzalutamide plus N-desmethyl enzalutamide, AUC18 d and AUC∞ increased 1.14-fold and 1.28-fold, respectively, while C max decreased by 3 %.
3.2.2 Study with CYP2C8, CYP2C9, CYP2C19, and CYP3A4 Substrates
The pharmacokinetic parameters for enzalutamide and its major metabolites (Table 4) confirmed that plasma exposures in this study were similar to those observed in other studies in which enzalutamide was administered at 160 mg once daily to steady state [4]. The mean C trough values for enzalutamide, N-desmethyl enzalutamide, the carboxylic acid metabolite, and the sum of enzalutamide plus N-desmethyl enzalutamide were 12.0, 10.6, 6.32, and 23.0 μg/mL, respectively.
The mean plasma concentration–time profiles of the orally administered CYP substrates before and after enzalutamide at steady state are shown in Fig. 3. The pharmacokinetic parameters and comparative statistics for the CYP substrates and their major metabolites are shown in Tables 5 and 6, respectively. A forest plot showing the effects of multiple doses of enzalutamide on exposure parameters for the orally administered CYP substrates pioglitazone, S-warfarin, omeprazole, and midazolam is presented in Fig. 4.

Mean plasma concentration–time profiles (n = 14) after single-dose oral administration of CYP substrates alone or in the presence of enzalutamide 160 mg. Insets show data on logarithmic scale. CYP cytochrome P450

Forest plot summarizing the effects of enzalutamide on exposures to other drugs. AUC area under the plasma concentration–time curve, CI confidence interval, C max maximum plasma concentration, CYP cytochrome P450, PK pharmacokinetic parameters
Enzalutamide increased the AUC∞ for pioglitazone (CYP2C8 substrate) by 20 % and decreased the AUC∞ for S-warfarin (CYP2C9 substrate), omeprazole (CYP2C19 substrate), and midazolam (CYP3A4 substrate) by 56, 70, and 86 %, respectively. Enzalutamide also reduced the AUC∞ for hydroxy-pioglitazone (M-IV) by 37 %, 7-hydroxy-S-warfarin by 53 %, R-warfarin by 45 %, 5-hydroxy-omeprazole by 54 %, and 1-hydroxy-midazolam by 76 %.
3.3 Safety
No deaths, serious adverse events, or adverse events resulting in discontinuation occurred during the healthy subject study with CYP2C8 and CYP3A4 inhibitors. Thirteen subjects (three in arm 1, six in arm 2, and four in arm 3) experienced at least one treatment-emergent adverse event (TEAE). All events were categorized as National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) grade 1, with the exception of grade 2 flatulence in one subject (arm 2) that was attributed to a possible relationship to gemfibrozil. Four additional subjects experienced at least one TEAE that was attributed a possible relationship to the study drug. All TEAEs resolved by the end of the study.
In the patient study with CYP substrates, the most frequent TEAEs (i.e., in at least three of 14 patients, ≥21.4 %) were nausea, constipation, dizziness, arthropod bite, fatigue, and hot flush. The majority of reported TEAEs were NCI-CTCAE grade 1 or 2. One patient experienced a single and transient episode of generalized tonic–clonic seizure that was assessed as probably related to enzalutamide and led to discontinuation of study treatment with enzalutamide. No clinically significant changes were noted for safety laboratory tests or electrocardiograms.
4 Discussion
Clinical pharmacology studies of factors that affect enzalutamide pharmacokinetics typically measure plasma concentrations of the parent drug and its two major human metabolites, N-desmethyl enzalutamide (active metabolite) and the carboxylic acid metabolite (inactive metabolite). Pharmacokinetics data on the carboxylic acid metabolite are used to understand mechanistic underpinnings of changes in exposure to active moieties, but changes in exposure to this metabolite are not considered clinically important. In contrast, exposure to N-desmethyl enzalutamide is considered clinically important. N -desmethyl enzalutamide is thought to contribute to the clinical effects of enzalutamide with regard to efficacy and safety, because its molecular structure is similar to enzalutamide [4], it has similar potency to enzalutamide in all primary and secondary pharmacodynamic endpoints, it circulates at approximately the same plasma concentrations as enzalutamide, and an exposure–response analysis of enzalutamide and N-desmethyl enzalutamide plasma concentration data from a phase III clinical trial (AFFIRM) [2] did not produce results that would suggest differences in the clinical efficacy or safety profiles of the two molecules [30]. For these reasons, when interpreting clinical relevance (both in terms of efficacy and safety), conclusions are based on the sum of enzalutamide plus N-desmethyl enzalutamide, which corresponds to exposure to enzalutamide active moieties.
Enzalutamide is indicated for the treatment of mCRPC, which most commonly occurs in older men [19]. Given that polypharmacy is prevalent in elderly populations [20–22], drug interactions were recognized as an important issue to investigate early in the enzalutamide development program. As recommended by FDA guidance [23], the investigation of enzalutamide drug interactions began with in vitro studies.
In vitro testing was performed to inform clinical investigations of enzalutamide as a potential victim of drug interactions. A study with recombinant CYP enzymes showed that enzalutamide is metabolized by CYP2C8 and CYP3A4, both of which play a role in the formation of N -desmethyl enzalutamide [30].
To assess the in vivo effects of a strong CYP2C8 inhibitor on enzalutamide pharmacokinetics, a single oral dose of enzalutamide was given concomitantly with gemfibrozil dosed to steady state. An in vivo drug interaction was evident from the plasma concentration–time plots (Fig. 2), which showed that concomitant gemfibrozil altered the rates of enzalutamide elimination and metabolite formation, and that discontinuation of gemfibrozil caused an abrupt change in the elimination and metabolite formation rates. Such changes upon discontinuation of the inhibitor were not wholly unexpected, as similar changes have been reported in other drug interaction studies [18]. Given the change in pharmacokinetics, extrapolation of the observed concentration–time data in the terminal phase could not be used to estimate the AUC∞ for the sum of enzalutamide plus N-desmethyl enzalutamide. If the AUC term from observed data (AUC18 d) had been used to assess changes in the composite sum of exposure to active moieties, then the magnitude of the drug interaction effect would have been underestimated. To protect against this bias, pharmacokinetic modeling and simulation were used to predict the plasma concentration–time profiles with continued administration of gemfibrozil. The final analysis showed that the composite AUC∞ of enzalutamide plus N -desmethyl enzalutamide increased by 2.2-fold with minimal effect on C max. Thus, in vivo inhibition by a strong CYP2C8 inhibitor resulted in an approximate doubling of exposure to enzalutamide active moieties. In patients taking 160 mg once daily (i.e., the recommended dose of enzalutamide), an approximate doubling of exposure to active moieties would result in plasma concentrations higher than those associated with the maximum tolerated dose of 240 mg/day [24]. In a dose-escalation study in patients with mCRPC, no seizures were reported at ≤240 mg, whereas three seizures were reported at higher doses, one each at 360, 480, and 600 mg [24]; therefore, patients taking enzalutamide 160 mg once daily concomitantly with a strong CYP2C8 inhibitor may be at an increased risk of seizure. To mitigate the risks to patients, it is recommended to reduce the dose of enzalutamide to 80 mg once daily during concomitant use with a strong CYP2C8 inhibitor (e.g., gemfibrozil).
To assess the in vivo effects of a strong CYP3A4 inhibitor on enzalutamide pharmacokinetics, a single oral dose of enzalutamide was given concomitantly with itraconazole dosed to steady state. A slight in vivo drug interaction was evident from the plasma concentration–time plots (Fig. 2), which showed that itraconazole was associated with a small decrease in the rate of enzalutamide elimination and small changes in the rates of metabolite formation. The calculated pharmacokinetic parameters showed that the composite AUC∞ of enzalutamide plus N -desmethyl enzalutamide increased by 1.3-fold with no effect on C max. As this change in exposure is small and not clinically meaningful, no dose adjustments are warranted for concomitant use of enzalutamide with CYP3A4 inhibitors.
Taken together, data from the gemfibrozil and itraconazole treatments indicate that CYP2C8 is a more important contributor to the in vivo metabolism of enzalutamide than CYP3A4.
In vitro testing was additionally used to inform clinical evaluations of enzalutamide as a potential perpetrator of drug interactions. This in vitro testing focused on the possible effects of enzalutamide as an inhibitor or inducer of CYP enzymes. Due to the low aqueous solubility of enzalutamide (≤2.0 μg/mL) [4], concentrations greater than the plasma C max (approximately 17 μg/mL) were difficult to achieve in these in vitro test systems. Inhibition was investigated through in vitro studies with human hepatic microsomes, which showed that enzalutamide, N-desmethyl enzalutamide, and the carboxylic acid metabolite may act as inhibitors of CYP2C8 and CYP2C19, with lesser inhibitory effects on CYP2B6 and CYP2C9. The potential for enzalutamide to inhibit CYP1A2 and CYP2D6 could not be fully excluded, as the highest concentration tested was approximately 2.4-fold of the clinical C max. An in vitro induction study with human primary hepatocyte cultures showed that enzalutamide may cause increases in messenger RNA and enzymatic activity for CYP3A4 [30], suggesting that enzalutamide could have the potential to induce CYP3A4; induction effects on CYP1A2 and CYP2B6 were inconclusive as the highest concentration of enzalutamide tested was approximately 7 % of the C max in patients taking enzalutamide 160 mg once daily (Table 4).
Based on the totality of the in vitro inhibition and induction data, the clinical investigation plan was to evaluate the effects of enzalutamide on the pharmacokinetics of sensitive substrates of CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. First priority was assigned to CYP2C8, CYP2C9, CYP2C19, and CYP3A4, and the results for these four enzymes are summarized in this report.
The effects of enzalutamide on the pharmacokinetics of a single oral dose of sensitive substrates for CYP2C8 (pioglitazone), CYP2C9 (S-warfarin), CYP2C19 (omeprazole), and CYP3A4 (midazolam) were determined in a single-sequence crossover design study in which patients with mCRPC received enzalutamide at the recommended dose of 160 mg once daily. The results showed no clinically meaningful changes in exposure to the CYP2C8 substrate; however, enzalutamide decreased exposure to the CYP2C9, CYP2C19, and CYP3A4 substrates by 56, 70, and 86 %, respectively. Based on the magnitude of the induction effects, enzalutamide is considered a moderate inducer of CYP2C9 and CYP2C19 and a strong inducer of CYP3A4 [23].
Given that enzalutamide is an inducer of CYP2C9, CYP2C19, and CYP3A4, and that co-induction of these enzymes commonly occurs via activation of the nuclear pregnane X receptor, enzalutamide may also induce uridine 5′-diphospho-glucuronosyltransferase (UGT) 1A1 [25]. Induction of UGT1A1 could explain the 37–76 % decrease in exposure to the hydroxy-metabolites of the CYP substrates (Table 6), as many of these metabolites are further metabolized via glucuronidation that is mediated by UGT enzymes [26, 27].
To mitigate the risks to patients, it is recommended to avoid concomitant use of enzalutamide with substrates of CYP2C9 (such as phenytoin and warfarin), CYP2C19 (such as S-mephenytoin), or CYP3A4 [such as alfentanil, ergotamine, dihydroergotamine, cyclosporine (ciclosporin), fentanyl, pimozide, quinidine, sirolimus, and tacrolimus] that have a narrow therapeutic index. The full induction potential of enzalutamide may not occur until approximately 1 month after the start of treatment, when steady-state plasma concentrations of enzalutamide are reached, although some induction effects may be apparent earlier. Patients taking drugs that are substrates of CYP2C9, CYP2C19, and CYP3A4 should be evaluated for possible loss of pharmacological effects (or increase in effects in cases where active metabolites are formed) during the first month of enzalutamide treatment, and dose adjustment should be considered as appropriate. In consideration of the long t ½ of enzalutamide (5.8 days in patients [4]), effects on enzymes may persist for 1 month or longer after stopping enzalutamide. A gradual dose reduction of the concomitant drug may be necessary when stopping enzalutamide treatment.
5 Conclusion
In the drug interaction study with CYP2C8 and CYP3A4 inhibitors, coadministration of gemfibrozil (strong CYP2C8 inhibitor) increased the composite AUC∞ of enzalutamide plus N-desmethyl enzalutamide by 2.2-fold, and coadministration of itraconazole (strong CYP3A4 inhibitor) increased the composite AUC∞ by 1.3-fold. Based on these findings, it is recommended that if a patient requires coadministration of a strong CYP2C8 inhibitor with enzalutamide, then the enzalutamide dose should be reduced to 80 mg once daily. In the drug interaction study with CYP2C8, CYP2C9, CYP2C19, and CYP3A4 substrates, enzalutamide increased the AUC∞ of oral pioglitazone by 20 % and reduced the AUC∞ of oral S-warfarin, omeprazole, and midazolam by 56, 70, and 86 %, respectively; therefore, enzalutamide is a moderate inducer of CYP2C9 and CYP2C19 and a strong inducer of CYP3A4. Enzalutamide did not affect exposure to oral pioglitazone (CYP2C8 substrate) to a significant extent. Based on these findings, it is recommended to avoid concomitant use of enzalutamide with administered narrow therapeutic index drugs that are metabolized by CYP2C9, CYP2C19, or CYP3A4 as enzalutamide may decrease their exposure.
References
Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–90.
Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. AFFIRM Investigators. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–97.
Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate before chemotherapy. N Engl J Med. 2014;371(5):424–33.
Gibbons JA, Ouatas T, Krauwinkel W, Ohtsu Y, van der Walt JS, Beddo V, et al. Clinical pharmacokinetics of enzalutamide. Clin Pharmacokinet. 2015 [Epub ahead of print].
U.S. Department of Health and Human Services. Guidance for industry: bioanalytical method validation, 2001. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107. Accessed 13 Feb 2015.
Bennett D, Gibbons JA, Mol R, Ohtsu Y, Williard C. Validation of a method for quantifying enzalutamide and its major metabolites in human plasma by LC–MS/MS. Bioanalysis. 2014;6(6):737–44.
Gemfibrozil tablets [prescribing information], 2014. http://labeling.pfizer.com/ShowLabeling.aspx?id=636. Accessed 13 Feb 2015.
Barone JA, Koh JG, Bierman RH, Colaizzi JL, Swanson KA, Gaffar MC, et al. Food interaction and steady-state pharmacokinetics of itraconazole capsules in healthy male volunteers. Antimicrob Agents Chemother. 1993;37(4):778–84.
Chainuvati S, Nafziger AN, Leeder JS, Gaedigk A, Kearns GL, Sellers E, et al. Combined phenotypic assessment of cytochrome p450 1A2, 2C9, 2C19, 2D6, and 3A, N-acetyltransferase-2, and xanthine oxidase activities with the “Cooperstown 5 + 1 cocktail”. Clin Pharmacol Ther. 2003;74(5):437–47.
Goh BC, Reddy NJ, Dandamudi UB, Laubscher KH, Peckham T, Hodge JP, et al. An evaluation of the drug interaction potential of pazopanib, an oral vascular endothelial growth factor receptor tyrosine kinase inhibitor, using a modified Cooperstown 5 + 1 cocktail in patients with advanced solid tumors. Clin Pharmacol Ther. 2010;88(5):652–9.
Jaakkola T, Backman JT, Neuvonen M, Laitila J, Neuvonen PJ. Effect of rifampicin on the pharmacokinetics of pioglitazone. Br J Clin Pharmacol. 2006;61(1):70–8.
Krishna G, Moton A, Ma L, Savant I, Martinho M, Seiberling M, et al. Effects of oral posaconazole on the pharmacokinetic properties of oral and intravenous midazolam: a phase I, randomized, open-label, crossover study in healthy volunteers. Clin Ther. 2009;31(2):286–98.
Ma JD, Nafziger AN, Villano SA, Gaedigk A, Bertino JS Jr. Maribavir pharmacokinetics and the effects of multiple-dose maribavir on cytochrome P450 (CYP) 1A2, CYP 2C9, CYP 2C19, CYP 2D6, CYP 3A, N-acetyltransferase-2, and xanthine oxidase activities in healthy adults. Antimicrob Agents Chemother. 2006;50(4):1130–5.
Padhi D, Salfi M, Emery M. Cinacalcet does not affect the activity of cytochrome P450 3A enzymes, a metabolic pathway for common immunosuppressive agents: a randomized, open-label, crossover, single-centre study in healthy volunteers. Drugs R D. 2008;9(5):335–43.
Shord SS, Chan LN, Camp JR, Vasquez EM, Jeong HY, Molokie RE, et al. Effects of oral clotrimazole troches on the pharmacokinetics of oral and intravenous midazolam. Br J Clin Pharmacol. 2010;69(2):160–6.
Turpault S, Brian W, Van Horn R, Santoni A, Poitiers F, Donazzolo Y, et al. Pharmacokinetic assessment of a five-probe cocktail for CYPs 1A2, 2C9, 2C19, 2D6 and 3A. Br J Clin Pharmacol. 2009;68(6):928–35.
Yeh RF, Gaver VE, Patterson KB, Rezk NL, Baxter-Meheux F, Blake MJ, et al. Lopinavir/ritonavir induces the hepatic activity of cytochrome P450 enzymes CYP2C9, CYP2C19, and CYP1A2 but inhibits the hepatic and intestinal activity of CYP3A as measured by a phenotyping drug cocktail in healthy volunteers. J Acquir Immune Defic Syndr. 2006;42(1):52–60.
Lehr T, Staab A, Trommeshauser D, Schaefer HG, Kloft C. Semi-mechanistic population pharmacokinetic drug–drug interaction modelling of a long half-life substrate and itraconazole. Clin Pharmacokinet. 2010;49(1):53–66.
American Cancer Society. Cancer facts and figures 2014. http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2014/index. Accessed 13 Feb 2015.
Hajjar ER, Cafiero AC, Hanlon JT. Polypharmacy in elderly patients. Am J Geriatr Pharmacother. 2007;5(4):345–51.
Haider SI, Johnell K, Thorsland M, Fastbom J. Trends in polypharmacy and potential drug-drug interactions across educational groups in elderly patients in Sweden for the period 1992–2002. Int J Clin Pharmacol Ther. 2007;45(12):643–53.
Husson N, Watfa G, Laurain MC, Perret-Guillaume C, Niemier JY, Miget P, et al. Characteristics of polymedicated (≥4) elderly: a survey in a community-dwelling population aged 60 years and over. J Nutr Health Aging. 2014;18(1):87–91.
U.S. Department of Health and Human Services. Guidance for industry. Drug interaction studies - study design, data analysis, implications for dosing, and labeling recommendations. 2012. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf. Accessed 13 Feb 2015.
Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, et al; Prostate Cancer Foundation/Department of Defense Prostate Cancer Clinical Trials Consortium. Antitumour activity of enzalutamide in castration-resistant prostate cancer: a phase 1–2 study. Lancet. 2010;24;375(9724):1437–46.
Tolson AH, Wang H. Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv Drug Deliv Rev. 2010;62:1238–49.
Jones DR, Moran JH, Miller GP. Warfarin and UDP-glucuronosyltransferases: writing a new chapter of metabolism. Drug Metab Rev. 2010;42(1):55–61.
Seo KA, Bae SK, Choi YK, Choi CS, Liu KH, Shin JG. Metabolism of 1′- and 4-hydroxymidazolam by glucuronide conjugation is largely mediated by UDP-glucuronosyltransferases 1A4, 2B4, and 2B7. Drug Metab Dispos. 2010;38(11):2007–13.
Astellas Pharma Inc. (Astellas Pharma Europe B.V.) Drug–drug interaction study with MDV3100 (ASP9785) and gemfibrozil and itraconazole. [ClinicalTrials.gov identifier NCT01913379]. US National Institutes of Health, ClinicalTrials.gov. http://www.clinicaltrials.gov. Accessed 23 Apr 2015.
Astellas Pharma Inc. (Astellas Pharma Europe B.V.) Drug–drug interaction study with MDV3100 and a cocktail of substrates. [ClinicalTrials.gov identifier NCT01911728]. US National Institutes of Health, ClinicalTrials.gov. http://www.clinicaltrials.gov. Accessed 23 Apr 2015.
Data on file, Medivation, 2012.
Acknowledgments
Jacqueline Gibbons and Joyce Mordenti are employees of Medivation. Jan Noukens is an employee of Kinesis. All other authors are employees of Astellas. The research described here was funded by Medivation and Astellas, and individuals from these organizations were involved in the design and conduct of the studies, collection of the data, and analysis and interpretation of the data. Copy editing assistance was provided by Shannon Davis of Infusion Communications and funded by Medivation. The authors take full responsibility for the content of the paper.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gibbons, J.A., de Vries, M., Krauwinkel, W. et al. Pharmacokinetic Drug Interaction Studies with Enzalutamide. Clin Pharmacokinet 54, 1057–1069 (2015). https://doi.org/10.1007/s40262-015-0283-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-015-0283-1