
Abstract
Background
Imeglimin is a first-in-class novel oral antidiabetic marketed in Japan as TWYMEEG® to treat type 2 diabetes mellitus. Its mode of action is distinct from all other anti-hyperglycemic classes.
Objective
To assess the pharmacokinetic and safety profile of imeglimin in Caucasian and Japanese healthy individuals.
Methods
Two randomized placebo-controlled phase 1 clinical studies were conducted in Caucasian subjects after single (250–8000 mg) and multiple (250–2000 mg twice daily) ascending doses and in Japanese subjects after single (500–6000 mg) and multiple (500–2000 mg twice daily) ascending doses. Imeglimin plasma and urine concentrations were measured.
Results
All imeglimin doses achieved maximal concentration between 1 and 3.5 h in Caucasians, and 1.5 and 3 h in Japanese subjects. The elimination half-lives (t1/2) were dose-independent and means ranged between 9.03 and 20.2 h for Caucasians, and 4.45 and 12 h for Japanese subjects. Dose-normalized area under the plasma concentration-time curve decreased with dose in the 250–8000 mg and in the 500–6000 mg dose range in Caucasians and Japanese, respectively, suggesting a dose-dependent but less than dose-proportional effect in imeglimin exposure. Plasma accumulation was minimal following repeated dosing, and food did not affect the pharmacokinetics in either population. Exposures were generally similar between Caucasian and Japanese subjects with less than 20% difference, although there was a tendency for exposures in Japanese to be slightly higher. Imeglimin had an acceptable safety and tolerability profile, with dose-dependent mild gastrointestinal adverse events.
Conclusion
Imeglimin was safe and well tolerated in these two phases 1 studies, with pharmacokinetics comparable between the two populations.
Clinical Trial Registrations
EudraCT 2005-001946-18 and 2014-004679-21.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Phase 1 studies in healthy Caucasian and Japanese subjects suggest that pharmacokinetic properties after single and repeated administrations of imeglimin, a novel antidiabetic drug, are not sensitive to ethnicity. |
1 Introduction
Despite a growing understanding of its pathophysiology and a substantial portfolio of available drugs, type 2 diabetes mellitus remains a growing epidemic exerting an increasing burden on public health, calling for further efforts from the medical and scientific community to strengthen the therapeutic solutions.
Imeglimin (hydrochloride salt), a recently approved drug for the treatment of type 2 diabetes, is the first in a new chemical class of oral antidiabetic agents, the glimins. Imeglimin provides a novel approach to treat patients due to its unique dual mode of action involving an increase of insulin secretion in response to glucose [1, 2] along with β-cell protection [3], and improvement of insulin sensitivity in muscle and liver [4, 5]. This dual effect is of particular interest to treat type 2 diabetes mellitus as it offers a global approach to the disease pathophysiology that engulfs the potential differences among individuals and ethnicities, such as the prominent decrease in β-cell function and hampered compensation of a gradual increase in insulin resistance in east Asian populations, with a generally higher insulin sensitivity compared to Caucasian [6, 7]. These pathophysiological differences are crucial to optimize the therapeutic strategy [8, 9].
Biochemical and molecular investigations have shown that the effect of imeglimin on insulin secretion was mediated by improvement of glucose-stimulated ATP generation and by an increase in NAD+ synthesis in (rodent) islets [1, 2, 4]. More recently it was suggested that this production of NAD+/cADPR by imeglimin may activate transient receptor potential melastatin 2 (TRPM2) channels in β-cells, leading to the potentiation of glucose-stimulated insulin secretion (GSIS) [10]. In parallel, imeglimin restores mitochondrial function by modulating the respiratory chain complex activities through partial inhibition of Complex I and rescue of Complex III activity, decreasing reactive oxygen species production and preventing cell death by delaying the mitochondrial permeability transition pore’s opening [11,12,13].
Following phase 1 and phase 2 clinical trials demonstrating its efficacy and favorable safety/tolerability profile [14,15,16,17,18], imeglimin completed the TIMES (Trial for Imeglimin Efficacy and Safety) pivotal phase 3 program in Japanese patients with type 2 diabetes, as monotherapy—TIMES 1 [19], as combination therapy with other antidiabetics—TIMES 2 [20], and as an add-on to insulin monotherapy—TIMES 3 [21] at a dose of 1000 mg twice daily (bid). Imeglimin was approved in Japan under the brand name TWYMEEG® in June 2021 [22], constituting the first antidiabetic compound with a new mode of action to be available for clinical use since the advent of SGLT2 (sodium-glucose co-transporter-2) inhibitors.
Imeglimin is absorbed through a passive and active mechanism that can be saturated. It is rapidly and largely distributed to the organs and mainly excreted unchanged in urine. It is poorly metabolized and has no inhibition or induction potential toward CYP450. Imeglimin is a substrate of MATE2-K and also a substrate and an inhibitor of OCT1, OCT2, and MATE1 transporters, but there is no clinically significant interaction when imeglimin is co-administered with either a substrate or an inhibitor of those transporters [23,24,25].
To allow development of imeglimin in Japan and to compare the safety, tolerability, and pharmacokinetic profile of imeglimin in healthy Japanese and Caucasian subjects, two studies were performed in both populations with the same dosing regimen. As the therapeutic dose range was estimated to be between 1000 and 4000 mg/day at the beginning of the development, imeglimin doses within that range were tested in these studies. Doses up to 8000 mg in Caucasian subjects were tested to determine the maximum tolerated dose.
We report here the results of these two phase 1 studies, which described the pharmacokinetic profile of imeglimin after single and repeated oral administration in healthy Caucasian and Japanese subjects.
2 Methods
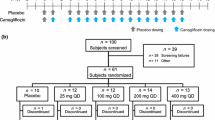
This article describes the results of two phase 1, randomized, double-blind, placebo-controlled studies to assess the safety, tolerability, and pharmacokinetics of imeglimin after single and repeated oral administrations.
The studies were conducted in accordance with the ethical principles of the Declaration of Helsinki (reference numbers: Eth-191/05 and 14/LO/1993). All participants gave written informed consent.
Subjects were followed as in-patients and safety parameters were measured at multiple timepoints including assessment of vital signs, 12-lead ECG, adverse events (AEs), and safety laboratory evaluations (hematology, serum chemistry, and urinalysis). An AE is any untoward medical occurrence in a subject administered a medicinal product and which does not necessarily have a causal relationship with that treatment. A serious adverse event (SAE) or reaction is any untoward medical occurrence that at any dose: results in death, is life-threatening, requires in-patient hospitalization or prolongation of existing hospitalization, results in persistent or significant disability/incapacity, is a congenital anomaly/birth defect, or is a medically important event. The severity of each AE was assessed by the investigator according to the following definitions: mild (awareness of event but easily tolerated), moderate (discomfort enough to cause interference with usual activities), and severe (inability to carry out usual activities).
2.1 Participants
Healthy Japanese and Caucasian men and women aged 18–45 years with a body mass index (BMI) 18.0–29.9 kg/m2 were eligible for inclusion. The estimated glomerular filtration rate (eGFR) was estimated by the Modification of Diet in Renal Disease (MDRD) equation [26]. Japanese subjects had to be born in Japan, had to have four ethnically Japanese grandparents, and had to have lived outside Japan for no longer than 5 years. Subjects were excluded from the studies if they have any history of alcohol abuse or smoke more than five cigarettes per day. During the trials no alcoholic or decaffeinated drinks or smoking were allowed during the period from at least 24 h before admission until discharge. The trials were approved by the Independent Ethics Committee at the Chambers of Physicians (Ethik-Kommission der Ärztekammer) in Berlin and the relevant regulatory authority (Bundesinstitut für Arzneimittel und Medizinprodukte), and by the UK Medicines and Healthcare Products Regulatory Agency and London Harrow Research Ethics Committee.
2.2 Design
2.2.1 Single Ascending-Dose Studies (SADs)
Each cohort consisted of six randomized subjects receiving imeglimin and two or three receiving a placebo. Imeglimin or placebo was administered to Caucasian and Japanese subjects in single oral doses of 250 (Caucasian only), 500, 1000, 1500, 2000, 4000, 6000, and 8000 (Caucasian only) mg on the morning of Day 1 after an overnight fasting period. Blood samples (6 mL) for plasma pharmacokinetic assessments were collected into a lithium heparinate tube on Day 1 at the following time points: pre-dose, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, and 72 h post-dose, except for Caucasians at 1000, 1500, 2000, and 4000 mg where the following timepoints were used: pre-dose, 1, 2, 3, 3.5, 4, 4.5, 5, 6, 8, 12, 16, 24, 36, 48, and 72 h post-dose. Urine samples were collected for each cohort: before drug administration, and at the following intervals 0–4, 4–8, 8–12, 12–24, and 24–48 h after drug administration.
2.2.2 Multiple Ascending-Dose Studies (MADs)
At doses of 250 (Caucasian only), 500, 1000, 1500, and 2000 mg, subjects received a first dose on Day 1 followed by repeated twice-daily administration from Day 4 to Day 9, with a last morning administration on Day 10. On Day 10 blood and urine samples were collected at the same timepoints as Day 1 (SAD studies) for each group to assess the pharmacokinetic parameters at steady state.
2.2.3 Food Effect
Food effect was assessed after administration in a fasting condition on Day 1 and after a high-fat breakfast on Day 4 at 1000 mg for Caucasian and at 1500 mg for Japanese. Blood and urine samples for imeglimin assays were collected for 12 h post-dose at Day 4 and Day 1, following the timepoints of the SAD studies.
2.3 Bioanalytical Method
The measurement of imeglimin in plasma and urine samples was performed by using a validated liquid chromatography with tandem mass spectrometry (LC–MS/MS) method [24]. Plasma samples were centrifuged within 30 min at about 4 °C and 1500g for 10 min and stored at − 20 °C. Imeglimin is a hydrochloride salt. Stock solutions have not been corrected for salt content, so imeglimin plasma concentrations were expressed as hydrochloride salt. Imeglimin hydrochloride salt plasma concentrations can be converted to imeglimin free-base concentrations by multiplying by a 0.810 factor.
Pharmacokinetic parameters were calculated according to standard non-compartmental methods [27] using KINETICA version 4.1.1 (Innaphase, Philadelphia, PA, 2002): time to reach peak plasma concentration (tmax), peak plasma concentration (Cmax), area under the plasma concentration-time curve (AUC) from time zero to last measurable concentration (AUC0–last), AUC from time zero to infinity (AUC0–∞), AUC from time zero to 12 h (AUC0–12h), elimination half-life (t1/2), mean residence time (MRT), apparent clearance (CL/F), volume of distribution (Vz/F), renal clearance (CLR), amount of imeglimin excreted unchanged in urine over the 12- and 48-h period (Ae0–12h and Ae0–48h), and excreted fraction in urine over the 12- and 48-h period (fe0–12h and fe0–48h %).
2.4 Statistical Analysis
Plasma and urine pharmacokinetic parameters were summarized using descriptive statistics according to treatment group. An analysis of variance (ANOVA) model was applied to estimate the food effect. Dose proportionality was assessed graphically using a regression analysis between log(AUC) and log(dose) for Japanese and Caucasian subjects separately based on the power model (Eqs. 1, 2):
The slope (β) and intercept (α) are estimated for AUC0–∞ after single administration and AUC0–12 after multiple administrations (bid) of imeglimin.
3 Results
3.1 Demographics
The key demographic and baseline characteristics of participants are summarised in Table 1.
3.2 Pharmacokinetics
3.2.1 Pharmacokinetic Parameters After SAD in Healthy Caucasian and Japanese Volunteers
Mean concentration–time profiles after single administration in Japanese and/or in Caucasian subjects from 500 to 6000 mg are presented in Fig. 1A, B. For single doses of imeglimin from 250 to 8000 mg in Caucasian subjects and from 500 to 6000 mg in Japanese subjects, imeglimin was readily absorbed; the median tmax was within 1–3.5 h and within 1.5–3 h after administration, respectively (Table 2).

Mean plasma concentration–time profiles, linear plot (A) and semi-log plot (B) after a single dose of imeglimin in Japanese and Caucasian healthy individuals (n = 6)
Caucasians achieved a mean Cmax of imeglimin ranging from 488 to 4086 ng/mL within the 250–8000 mg dose range with an interindividual variability ranging from 15.5 to 35.4%. Over the entire range of doses tested, the mean AUC0–∞ ranged from 3582 to 37,335 ng⋅h/mL with an interindividual variability ranging from 11.8 to 34.0% (Table 2). Japanese subjects achieved a mean Cmax of imeglimin ranging from 1007 to 4194 ng/mL within the 500–6000 mg dose range with an interindividual variability ranging from 20.2 to 40.3%. The mean AUC0–∞ ranged from 6631 to 35,963 ng⋅h/mL with an interindividual variability ranging from 10.8 to 31.6% (Table 2). Over the range of 500–2000 mg, geometric mean Cmax and AUC0–∞ were comparable in Japanese and Caucasians. Exposures were generally within 20%, although there was a tendency for exposures in Japanese to be slightly higher, particularly for Cmax. Similarly, after higher single doses (4000 and 6000 mg), exposure in Japanese tended to be slightly higher (about 15–20%) than that in Caucasians.
AUC0–∞ plots versus dose showed less than dose-proportional relationships (Fig. 2A). The slope and the intercept estimated by regression analysis were similar between Japanese and Caucasian subjects with slopes of 0.672 and 0.664 and intercepts of 106 and 94.9, respectively. The slope estimates were lower than 1, suggesting a less than proportional AUC increase along with a dose increase. AUC/dose versus dose plots indicated a clear decrease in AUC/dose along with a dose increase in the lower dose range of 250–2000 mg (Fig. 2B).

Relationship between AUC0–∞ and dose and comparison between Japanese and Caucasian after single-dose administration of imeglimin. AUC0–∞ (A) and AUC0–∞/dose (B) were plotted against dose and the regression lines obtained with power model were transformed and overlaid. AUC0–∞ area under the plasma concentration-time curve from time zero to infinity
In both populations, imeglimin appeared to be distributed with a rapid initial phase occurring until 24 h followed by a slower terminal elimination phase (Fig. 1B). The pharmacokinetic characteristics of imeglimin were defined using population pharmacokinetic analysis. The imeglimin population pharmacokinetics were described by a two-compartment model with first-order absorption with a lag time and first-order elimination from the central compartment [28]. In the entire range of tested doses, the mean t1/2 varied from 9.03 to 20.2 h in Caucasians and from 4.45 to 12.0 h in Japanese with a high variability (up to 163%) (Table 2), making t1/2 quite difficult to interpret. The MRT is less variable and is a better reflection of the rate of elimination of imeglimin. The mean apparent MRT ranged from 7.69 and 12.5 h in Caucasians with a variability up to 43.9% and from 6.75 to 12.7 h in Japanese with a variability up to 72% (Table 2). Overall, the MRT was shorter than the t1/2, reflecting that imeglimin was mainly eliminated within 12 h of administration, and only a small proportion underwent the slower, terminal, elimination phase.
In Caucasians, the CL/F and the Vz/F after a single dose increased with dose (from 69.8 L/h at 250 mg to 214 L/h at 8000 mg and from 1036 L at 250 mg to 2792 L at 8000 mg, respectively), reflecting a decrease in bioavailability when the dose increased (Table 2).
After single doses of up to 1500 mg in Japanese subjects, apparent clearance was similar: CL/F ranged from 75.4 to 89.4 L/h, probably due to the interindividual variability since at higher doses CL/F increased with dose (ranged from 137 to 167 L/h). Vz/F generally increased with increasing single doses and ranged from 485 to 2760 L (Table 2). In Caucasian subjects this reflects the decrease in bioavailability at higher doses.
Similar to plasma exposure, the amount of imeglimin excreted in urine after a single administration increased with dose. Considering that imeglimin is not metabolized and is excreted unchanged in urine, the excreted fraction in urine reflects the absorbed fraction [24]. The Ae0–48h increased along with the dose but non-dose-proportionally in both populations. The fraction absorbed decreased from 34.6% at 500 mg to 16.4% at 6000 mg in Japanese subjects and from 54.9% at 250 mg to 12% at 8000 mg in Caucasians. While fe0–48h decreased with dose, CLR was generally dose independent. Overall, CLR ranged between 26.3 and 37.4 L/h in Caucasians and between 21.5 and 29.1 L/h in Japanese subjects (Table 2).
There was no relevant food effect on the pharmacokinetic parameters of imeglimin (single 1000 mg dose) in Caucasians with only slight decreases of AUC0–12h (3%) and Cmax (8%), but a delay of 2.75 h in tmax in fed conditions (Table 3). Similarly, exposures after a single 1500 mg dose under fed and fasted conditions were comparable in Japanese subjects: Cmax and AUC0–12h were similar in both conditions with a slight delay (1 h) in tmax under fed conditions (Table 3).
3.2.2 Pharmacokinetic Parameters Following MAD in Healthy Caucasian and Japanese Volunteers
At all doses, mean concentration-time profiles after repeated administration (7-day) were similar to those after single doses in both populations (Tables 2, 4). The AUC0–12h were comparable to AUC0–∞, confirming that the pharmacokinetics of imeglimin is independent of time in both populations. Based on visual inspection of pre-dose plasma concentrations, steady state was reached after 5 days of treatment in both populations.
After repeated administrations, there was a dose-dependent increase in imeglimin exposure; Cmax and AUC0–12h ranged from 572 to 2313 ng/mL and 3927 to 17,513 ng⋅h/mL, respectively, within the 250–2000 mg bid dose range in Caucasians, and from 994 to 2267 ng/mL and 6288 to 16,672 ng⋅h/mL, respectively, within the 500–2000 mg bid dose range in Japanese subjects.
The plots of AUC0–12h at steady state versus dose also showed less than dose-proportional relationships (Fig. 3A). The slopes and the intercepts estimated by regression analysis were similar to the estimates for AUC0–∞ after single doses for Caucasian and Japanese subjects, with slopes of 0.6957 and 0.7291 and intercepts of 87.333 and 72.796, respectively. The slope estimates were lower than 1, again suggesting less than proportional AUC increase along with a dose increase. AUC0–12/dose versus dose plots suggested a decrease in AUC0–12h/dose along with a dose increase in the dose range of 250–2000 mg bid (Fig. 3B).

Relationship between AUC0–12 and dose and comparison between Japanese and Caucasian after multiple-dose administrations of imeglimin. AUC0–12 (A) and AUC0–12/dose (B) were plotted against dose. AUC0–12 area under the plasma concentration-time curve from time zero to 12 h
At steady state, t1/2 remained similar to those calculated after single doses, ranging from 9.54 to 15.9 h in Caucasians, from 10.1 to 13.6 h in Japanese, and were unaffected by the dose (Table 4). Similarly, MRT ranged from 8.92 to 16.5 h in Caucasians, from 9.16 to 10.8 h in Japanese subjects, and were unaffected by the dose (Table 4). In both populations, the apparent clearance and volume of distribution at steady state were similar to those calculated after single doses.
Similar to plasma exposure, the imeglimin amount excreted in urine after repeated doses increased in a dose-dependent manner. In Japanese subjects, the fraction excreted was similar at 500 and 1000 mg bid (34.3 and 33.7% of dose, respectively) and then decreased to 23.9 and 27.4% after 1500 mg bid and 2000 mg bid. CLR,ss ranged between 22.3 and 33.9 L/h (Table 4). The absorbed fraction of imeglimin decreased with dose slightly less after repeated administration than after a single dose (data not shown). In Caucasians, urinary excretion of imeglimin tended to decrease with dose from 56.7 to 28.7% of administered dose after repeated administration. Mean CLR,ss ranged between 32.2 and 39.5 L/h.
There was no relevant accumulation after 6 days of bid administration within the dose range of 250–2000 mg bid in Caucasians and within the dose range of 500–2000 mg bid in Japanese subjects. Ratios of Cmax ranged from 1.15 to 1.38 in Caucasians and from 1.00 to 1.43 in Japanese subjects; ratios of AUC0–12h ranged from 1.27 to 1.57 in Caucasians and from 1.08 to 1.57 in Japanese subjects (Online Supplementary Material (OSM) Table 1).
3.3 Safety and Tolerability
3.3.1 Overall Safety Following Repeated Dosing
Overall, the safety and tolerability of imeglimin was acceptable and consistent between Japanese and Caucasian subjects, with no difference between imeglimin treatment and placebo in the incidence of subjects presenting with treatment-emergent adverse events (TEAEs) (Tables 5, 6).
In the Caucasian population, the most commonly observed TEAEs following repeated administrations were from investigations with miscellaneous changes in laboratory values (ECG or hematology or biochemistry) followed by central nervous system and gastrointestinal disorders (OSM Table 2). The gastrointestinal TEAEs (abdominal pain or discomfort) mainly occurred at the beginning of the treatment. In the Japanese population, the most commonly reported TEAEs were also from the gastrointestinal tract and central nervous system (headache). Most of the TEAEs were of mild intensity.
One subject experienced a serious event of transient asystolia a few minutes after the first administration of the 2000 mg bid dose and recovered spontaneously without abnormal findings in vital signs or ECG. This event was considered as severe and unlikely related to the study drug by the investigator.
3.3.2 Maximal Tolerated Dose (MTD) Following Single Administration
For the setting of the maximum tolerated dose (MTD), only safety from single doses above 4000 mg is reported.
Overall, single administration of imeglimin was well tolerated up to a dose of 6000 mg in both Japanese and Caucasian populations, with a maximum 50% of the subjects presenting with mild or moderate TEAEs with imeglimin or placebo (Table 6).
The only TEAEs reported in both populations by at least two subjects were from the gastrointestinal tract (nausea, diarrhea, and vomiting) at a dose of 6000 mg, from cardiac disorders (ventricular extrasystole) at a dose of 4000 mg, or from general disorders (fatigue) with the placebo (OSM Table 3). The dose of 8000 mg was not well tolerated as 100% of Caucasian subjects presented with gastrointestinal TEAEs (diarrhea, nausea, vomiting, or abdominal pain/discomfort), starting 1–3 h after drug intake, mostly of mild intensity and resolving within the day without any treatment. There was no biochemistry abnormality observed during an unscheduled test performed 2 h post-dose at the height of the symptoms. Based on these gastrointestinal symptoms, 6000 mg was considered to be the maximum tolerated dose in both populations and the dose of 8000 mg was not administered to the Japanese cohort.
There was no meaningful change in lactate concentrations between imeglimin and placebo during the course of the treatment. In the Caucasian population, laboratory biochemistry assessment performed 2 h post the 8000 mg dose, at the time gastrointestinal symptoms were the most marked, did not evidence any change in lactate concentrations.
4 Discussion
Imeglimin is currently marketed in Japan as TWYMEEG®. The safety, tolerability, and pharmacokinetics of SADs and MADs of imeglimin were investigated in Caucasian and Japanese volunteers. Japanese and Caucasian individuals received high single doses of imeglimin (> 4000 mg) or placebo to assess the maximum tolerated dose in both populations. Based on the general known pharmacokinetic characteristics of imeglimin, rather good absorption, no hepatic metabolism, weakly bound to plasma protein and eliminated unchanged into urines [24], no major impact of ethnicity on its pharmacokinetic profile was expected. The results of these phase 1 studies in Caucasian and Japanese individuals confirmed this expectation. However, although exposures were generally within 20%, there was a tendency for slightly higher exposure in Japanese individuals, particularly for Cmax. Similarly, exposures in Japanese subjects tended to be slightly higher (about 15–20%) than in Caucasians after higher single doses (4000 mg and 6000 mg). Several covariates such as age, body weight, and creatine clearance have been identified as key covariates of imeglimin clearance as well as dose of imeglimin relative bioavailability during the building of a population pharmacokinetic model [28]. We therefore looked at whether one or all of these covariates could be responsible for the slightly higher exposure observed in Japanese subjects. The estimated glomerular filtration rate values at screening in the 1500 and 2000 mg groups were similar and most values were > 90 mL/min/1.73 m2, so unlikely to explain the results between the two populations. Similarly, age ranged from 25.7 to 35.3 years across populations and is also unlikely to explain the small exposure difference. However, the Japanese cohort’s body weight was on average 21% lower than the Caucasian body weight (58.3 vs. 73.6 kg). The lower body weight resulted in a slightly lower CL/F and slightly higher exposure, but this was not considered clinically relevant.
The Ae0–48h after single doses of 500–6000 mg increased in a dose-dependent manner. The average difference in Ae0–48h between Japanese and Caucasians across doses was 13%.
CLR ranged between 21.5 and 39.5 L/h across single and repeated dose levels, indicating active renal secretion by the kidney. CLR was similar between both populations and was dose independent.
Single and twice-daily repeated oral doses of up to 2000 mg were well tolerated in both Japanese and Caucasian subjects. The maximal tolerated dose was 6000 mg in both populations, with 17 and 33% of subjects reporting gastrointestinal AEs in Caucasian and Japanese cohorts, respectively. At a dose of 8000 mg, 100% of the Caucasian subjects complained of nausea, vomiting, abdominal discomfort, or diarrhea without biological abnormalities including absence of lactate increase. Overall, imeglimin is well tolerated in the therapeutic dose range of 1000–1500 mg bid [17, 19, 22].
5 Conclusions
The results of these studies evaluating single and multiple administration of imeglimin in Japanese and Caucasian subjects suggest that it is unlikely that ethnic diversity between these two groups would cause a difference in imeglimin exposure and pharmacokinetic parameters. The safety data obtained in these studies indicate that treatment with imeglimin at doses up to 6000 mg was generally safe and well tolerated, and that safety parameters were similar between Caucasian and Japanese subjects at each dose level studied.
Overall, these data showed that ethnic factors do not have a major impact on the safety, tolerability, and pharmacokinetics of imeglimin in Caucasian or Japanese subjects.
References
Hallakou-Bozec S, Kergoat M, Fouqueray P, Bolze S, Moller DE. Imeglimin amplifies glucose-stimulated insulin release from diabetic islets via a distinct mechanism of action. PLoS ONE. 2021;16(2): e0241651.
Pacini G, Mari A, Fouqueray P, Bolze S, Roden M. Imeglimin increases glucose-dependent insulin secretion and improves β-cell function in patients with type 2 diabetes. Diabetes Obes Metab. 2015;17(6):541–5.
Hallakou-Bozec S, Kergoat M, Moller DE, Bolze S. Imeglimin preserves islet beta-cell mass in type 2 diabetic ZDF rats. Endocrinol Diabetes Metab. 2021;4(2): e00193.
Hallakou-Bozec S, Vial G, Kergoat M, Fouqueray P, Bolze S, Borel AL, et al. Mechanism of action of Imeglimin: a novel therapeutic agent for type 2 diabetes. Diabetes Obes Metab. 2021;23(3):664–73.
Vuylsteke V, Chastain LM, Maggu GA, Brown C. Imeglimin: a potential new multi-target drug for type 2 diabetes. Drugs R&D. 2015;15(3):227–32.
Koh-Banerjee P, Wang Y, Hu FB, Spiegelman D, Willett WC, Rimm EB. Changes in body weight and body fat distribution as risk factors for clinical diabetes in US men. Am J Epidemiol. 2004;159(12):1150–9.
Wang Y, Rimm EB, Stampfer MJ, Willett WC, Hu FB. Comparison of abdominal adiposity and overall obesity in predicting risk of type 2 diabetes among men. Am J Clin Nutr. 2005;81(3):555–63.
Kim YG, Hahn S, Oh TJ, Kwak SH, Park KS, Cho YM. Differences in the glucose-lowering efficacy of dipeptidyl peptidase-4 inhibitors between Asians and non-Asians: a systematic review and meta-analysis. Diabetologia. 2013;56(4):696–708.
Kim YG, Hahn S, Oh TJ, Park KS, Cho YM. Differences in the HbA1c-lowering efficacy of glucagon-like peptide-1 analogues between Asians and non-Asians: a systematic review and meta-analysis. Diabetes Obes Metab. 2014;16(10):900–9.
Funazaki S, Yoshida M, Yamada H, Kakei M, Kawakami M, Nagashima S, et al. A novel mechanism of imeglimin-mediated insulin secretion via the cADPR-TRP channel pathway. J Diabetes Investig. 2022;13(1):34–41.
Vial G, Chauvin MA, Bendridi N, Durand A, Meugnier E, Madec AM, et al. Imeglimin normalizes glucose tolerance and insulin sensitivity and improves mitochondrial function in liver of a high-fat, high-sucrose diet mice model. Diabetes. 2015;64(6):2254–64.
Vial G, Lamarche F, Cottet-Rousselle C, Hallakou-Bozec S, Borel AL, Fontaine E. The mechanism by which imeglimin inhibits gluconeogenesis in rat liver cells. Endocrinol Diabetes Metab. 2021;4(2): e00211.
Detaille D, Vial G, Borel AL, Cottet-Rouselle C, Hallakou-Bozec S, Bolze S, et al. Imeglimin prevents human endothelial cell death by inhibiting mitochondrial permeability transition without inhibiting mitochondrial respiration. Cell Death Discov. 2016;2:15072.
Dubourg J, Ueki K, Grouin JM, Fouqueray P. Efficacy and safety of imeglimin in Japanese patients with type 2 diabetes: A 24-week, randomized, double-blind, placebo-controlled, dose-ranging phase 2b trial. Diabetes Obes Metab. 2021;23(3):800–10.
Fouqueray P, Pirags V, Inzucchi SE, Bailey CJ, Schernthaner G, Diamant M, et al. The efficacy and safety of imeglimin as add-on therapy in patients with type 2 diabetes inadequately controlled with metformin monotherapy. Diabetes Care. 2013;36(3):565–8.
Fouqueray P, Pirags V, Diamant M, Schernthaner G, Lebovitz HE, Inzucchi SE, et al. The efficacy and safety of imeglimin as add-on therapy in patients with type 2 diabetes inadequately controlled with sitagliptin monotherapy. Diabetes Care. 2014;37(7):1924–30.
Fouqueray P, Bolze S, Pirags V, Bailey CJ, Schernthaner G, Inzucchi SE, et al. Dose ranging-study to determine the optimum dose for imeglimin, a novel treatment for type 2 diabetes. Boston: ADA; 2015.
Pirags V, Lebovitz H, Fouqueray P. Imeglimin, a novel glimin oral antidiabetic, exhibits a good efficacy and safety profile in type 2 diabetic patients. Diabetes Obes Metab. 2012;14(9):852–8.
Dubourg J, Fouqueray P, Thang C, Grouin JM, Ueki K. Efficacy and safety of imeglimin monotherapy versus placebo in japanese patients with type 2 diabetes (TIMES 1): a double-blind, randomized, placebo-controlled, parallel-group, multicenter phase 3 trial. Diabetes Care. 2021;44(4):952–9.
Dubourg J, Fouqueray P, Quinslot D, Grouin JM, Kaku K. Long-term safety and efficacy of imeglimin as monotherapy or in combination with existing antidiabetic agents in Japanese patients with type 2 diabetes (TIMES 2): a 52-week open-label, multicenter phase 3 trial. Diabetes Obes Metab. 2022;24(4):609–19.
Reilhac C, Dubourg J, Thang C, Grouin JM, Fouqueray P, Watada H. Efficacy and safety of imeglimin add-on to insulin monotherapy in Japanese patients with type 2 diabetes (TIMES 3): a randomized, double-blind, placebo-controlled phase 3 trial with a 36-week open-label extension period. Diabetes Obes Metab. 2022;24(5):838–48.
SA P. Poxel and Sumitomo Dainippon Pharma announce the approval of TWYMEEG® (Imeglimin hydrochloride) for the treatment of type 2 diabetes in Japan. 2021. https://www.poxelpharma.com/en_us/news-media/press-releases/detail/187/poxel-and-sumitomo-dainippon-pharma-announce-the-approval. Accessed 20 Jun 2022.
Fouqueray P, Perrimond-Dauchy S, Bolze S. Imeglimin does not induce clinically relevant pharmacokinetic interactions when combined with either metformin or sitagliptin in healthy subjects. Clin Pharmacokinet. 2020;59(10):1261–71.
Clemence C, Fouqueray P, Sebastien B. In vitro investigation, pharmacokinetics, and disposition of imeglimin, a novel oral antidiabetic drug, in preclinical species and humans. Drug Metab Dispos. 2020;48(12):1330–46.
Chevalier C, Perrimond-Dauchy S, Dubourg J, Fouqueray P, Bolze S. Lack of drug–drug interaction between cimetidine, a renal transporter inhibitor, and imeglimin, a novel oral antidiabetic drug, in healthy volunteers. Eur J Drug Metab Pharmacokinet. 2020;45(6):725–33.
Matsuo S, Imai E, Horio M, Yasuda Y, Tomita K, Nitta K, et al. Revised equations for estimated GFR from serum creatinine in Japan. Am J Kidney Dis. 2009;53(6):982–92.
Gabrielsson J, Weiner D. Non-compartmental analysis. Methods Mol Biol. 2012;929:377–89.
Tomita Y, Hansson E, Mazuir F, Wellhagen GJ, Ooi QX, Mezzalana E, et al. Imeglimin population pharmacokinetics and dose adjustment predictions for renal impairment in Japanese and Western patients with type 2 diabetes. Clin Transl Sci. 2022;15(4):1014–26.
Acknowledgements
The sponsor, Poxel, would like to acknowledge Sandrine Perrimond-Dauchy and Julie Dubourg for their contribution to the studies reported in this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Consent for publication
Not applicable.
Code availability
Not applicable.
Funding
Studies were funded by Poxel SA.
Conflict of interest
Pascale Fouqueray, Clémence Chevalier, and Sébastien Bolze are employees and shareholders of Poxel.
Author contributions
All authors were involved in the design and the conduct of the study, analysis of the results, as well as the preparation of the manuscript.
Ethics approval
The studies were conducted in accordance with the ethical principles of the Declaration of Helsinki. The studies were approved by the Independent Ethics Committee at the Chambers of Physicians (Ethik-Kommission der Ärztekammer), Berlin and by London—Harrow Research Ethics Committee (reference numbers: Eth-191/05 and 14/LO/1993)
Informed consent
All participants gave written informed consent.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Fouqueray, P., Chevalier, C. & Bolze, S. Pharmacokinetics of Imeglimin in Caucasian and Japanese Healthy Subjects. Clin Drug Investig 42, 721–732 (2022). https://doi.org/10.1007/s40261-022-01181-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-022-01181-3