
Abstract
Background and Objectives
Exogenous human chorionic gonadotropin (hCG) acts on the final phase of the follicle maturation. Choriomon®, a highly purified hCG formulation, is approved in many European and extra-European countries for the induction of ovulation after stimulation of follicular development. The present study compares hCG bioavailability of Choriomon® (Test product) versus a recombinant hCG preparation (Ovitrelle®; Reference product).
Methods
In this randomized, two-way cross-over study, 26 healthy women received a single dose of Choriomon® (10,000 IU) and Ovitrelle® (250 µg; 6500 IU) by subcutaneous injection. hCG was determined in serum up to 192 h post-dose. Dose-normalized peak concentration (Cmax) and area under the concentration-time curve up to the time of the last quantifiable concentration (AUC0–t) and extrapolated to infinity (AUC0–∞) were calculated and compared between the two treatments.
Results
Serum hCG concentrations increased rapidly with a very similar pharmacokinetic curve for the two products. The test/reference geometric means ratio (GMR) for AUC0–t and AUC0–∞ corresponded to 121.31 and 119.81%, and the upper limits of the 90% confidence intervals (CIs) (130.21% and 128.51%, for AUC0–t and AUC0–∞, respectively) exceeded the 125% bioequivalence threshold. Cmax GMR was 146.89%, indicating a rate of hCG absorption approximately 50% greater for the test product (90% CI 132.30–163.10). Half-life (t1/2) was very similar (36.77 ± 5.11 h and 38.63 ± 6.08 h), whereas time to achieve Cmax (tmax) significantly differed, with median values of 16 h and 24 h for Choriomon® and Ovitrelle®, respectively, (p = 0.0023).
Conclusions
The differences between Choriomon® and Ovitrelle® pharmacokinetic parameters can be ascribed to the different raw source of the products and are reflected in the approved dose regimens of the two hCG formulations. The observed lack of bioequivalence between the two compounds at the given doses is not clinically relevant, as results from Phase III studies indicated similar clinical efficacy and safety. The safety data are in line with the known safety profile of the two products.
Clinicaltrials.gov registration no
NCT03735030.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In healthy women, a single subcutaneous administration of highly purified hCG (HP-hCG) resulted in a slightly greater exposure than recombinant hCG (r-hCG) administration. |
The observed higher rate of absorption of HP-hCG compared to r-hCG does not affect the clinical efficacy or safety in the induction of ovulation and/or assisted reproductive technology outcome, as demonstrated in previous comparative Phase III studies. |
1 Introduction
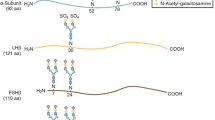
Human chorionic gonadotropin (hCG) is a dimeric glycoprotein hormone normally produced by the trophoblasts as early as 6 days post-conception, which stimulates the corpus luteum and early feto-placental endocrine function [1,2,3]. In women, exogenous hCG stimulates estradiol and progesterone production, and acts on the final phase of the follicle maturation, promoting oocyte maturation, follicular rupture, and E2 and progesterone secretion through sustaining the corpus luteum function [4].
A study demonstrated that hCG of urinary origin can be self-administered by subcutaneous (s.c.) injection without reducing the chances of retrieving mature oocytes and altering clinical outcome, pregnancy rates and development [5]. Stelling et al. [6] confirmed that s.c. administration of purified hCG (10,000 IU) achieves serum and follicular fluid levels adequate to obtain the desired clinical effects with less patient inconvenience than intramuscular (i.m.) administration. In a study by Mannaerts at al. [7], i.m. and s.c. injections of 10,000 IU hCG were bioequivalent with respect to hCG extent of absorption. Cmax values were similar between the two administration routes but bioequivalence could not be proven due to intersubject variability. In particular, after s.c. injection, serum hCG increased from 0.4 to 0.5 IU/L at baseline to a mean peak concentration of 339 IU/L (Cmax), which was reached at approximately 20 h post-dose. Elimination half-life (t1/2) was on average 32–33 h, irrespective of the i.m. or s.c. treatment regimen.
Choriomon® (or Gonasi® HP) is a highly purified hCG preparation (HP-hCG) extracted from the urine of pregnant women, approved in many European and extra-European countries, and indicated in women for the induction of ovulation after stimulation of follicular development with human menopausal gonadotropin or follicle stimulating hormone (FSH) [8]. Ovitrelle® [9], a recombinant hCG formulation (r-hCG), is the bench-market product for this indication in Europe, and was previously compared to Choriomon® in a Phase III randomized controlled study assessing the efficacy and safety of the two hCG preparations in women undergoing in vitro fertilization (IVF) [10]. Results of the study demonstrated that Choriomon® 10,000 IU was not inferior to Ovitrelle® 250 µg, both administered subcutaneously, with respect to the number of retrieved oocytes, oocyte quality, embryo quality, hormonal assessments, pregnancy rates and follow-up parameters [10]. Safety was very good for both treatments.
The present study, which was carried out to respond to a specific request from the Danish Competent Authorities, compares hCG serum bioavailability after a single s.c. dose of HP-hCG and r-hCG.
2 Methods
2.1 Study Design and Procedures
The study was open-label, randomized, two-way cross-over and was designed according to the EMA guideline on bioequivalence investigations [11]. The study (Clinicaltrial.gov No. NCT03735030) was approved by the Canton Ticino Ethics Committee, Switzerland, and the Swiss Federal Health Authority, and was performed in accordance with the Helsinki Declaration and Good Clinical Practice at CROSS Research S.A., Clinical Phase I Unit, Arzo (Switzerland), between November 2018 and August 2019. All subjects received a detailed study description and gave their written informed consent before enrolment.
The test product was HP-hCG, 5000 IU/vial, lyophilized powder for injection (Choriomon®, IBSA Institut Biochimique S.A., Switzerland) [8]. Before administration, the test hCG solution was prepared by dissolving the powder (two vials) in the 1 mL solvent provided. The reference product was Ovitrelle®, 250 µg hCG/0.5 mL (equivalent to 6500 IU hCG) pre-filled pen (Merck Serono GmbH, Germany), prepared and used according to Ovitrelle® information leaflet [9].
Subjects received a single s.c. dose of the test (10,000 IU hCG) and reference (6500 IU hCG) products in two periods under fasting conditions, with a wash-out interval of at least 21 days between the two administrations (Fig. 1). All s.c. injections were performed in the umbilical region. The randomization list was computer-generated using SAS® version 9.3 PLAN procedure.

Study design. hCG human chorionic gonadotropin, PK pharmacokinetics, Test Test product: Choriomon®, R randomization, Ref Reference product: Ovitrelle®
2.2 Subjects
Healthy women aged 20–45 years, with a body mass index of 18.5.0–30.0 kg/m2 and a normal menstrual cycle were enrolled. Participants were on a combined oral contraceptive pill for at least 2 months before screening and at least 16 continuous days before treatment.
All subjects were in good physical health, as assessed by a full physical examination, electrocardiogram (ECG), vital signs and clinical laboratory assays. Pituitary downregulation (luteinizing hormone < 5 IU/L; FSH < 4 IU/L) and endogenous hCG values < 1.2 IU/L were confirmed before randomization. All subjects had a negative or not clinically significant Papanicolaou smear test result at screening or within the 12 preceding months. No subjects had a history of drug/alcohol/caffeine/tobacco abuse. Exclusion criteria included: history/presence of any disease, in particular polycystic ovary disease, ovarian cysts, primary ovarian failure, early menopause or abnormal bleeding of undetermined origin. History of hypersensitivity or allergic reactions to the active principle and/or formulations' ingredients was also ruled out. No medications were allowed for 2 weeks before the study. Subjects taking gonadotropin preparations within the 6 months before the study were excluded. Subjects were also not enrolled if they had participated in other clinical trials or donated blood in the past 3 months.
2.3 Blood Sampling
Blood samples for hCG determination were collected from a forearm vein at − 1, − 0.5, and − 0.1 h pre-dose and 0.5, 1, 1.5, 2, 4, 6, 8, 12, 16, 24, 30, 36, 48, 72, 96, 120, 144, 168 and 192 h post-dose using an indwelling catheter with switch valve. After each sampling the cannula was rinsed with about 1 mL of sterile saline solution containing 20 IU/mL Na-heparin. At each collection time, the first 2 mL of blood were discarded to avoid contamination of the sample with heparin. The remaining 8 mL were collected from the catheter and transferred with a syringe into polypropylene tubes. The samples were kept at room temperature for 30–60 min and centrifuged at 2500 × g for 10 min. Serum samples were transferred into polyethylene tubes and stored frozen at ≤ − 20 °C until analyses.
2.4 Bioanalytical Assay
Serum hCG concentrations were determined at Ardena Bioanalysis B.V., the Netherlands, using two independent fluoroimmunoassays (DELFIA® hCG solid-phase, time-resolved; AutoDelfia hCG kit B007-101, PerkinElmer) with lower-upper quantification limits of 5–500 IU/L for Choriomon® and 2–500 IU/L for Ovitrelle®, independently developed and validated according to EMA guidance document requirements [12], using appropriate reference standards for the two molecules. This was needed due to the different nature of the two molecules (extractive vs. recombinant). The methods adhered to the regulatory requirements for selectivity, sensitivity, precision, accuracy and stability.
Calibration standards and quality control (QC) samples were prepared using the reference standards Choriomon® and Ovitrelle®. For the preparation of the calibration and the QC samples, respectively, a surrogate matrix (PBS containing 0.5% BSA) and blank human serum with low hCG levels (hCG level < 20% of the LLOQ, matrix number M-7290) were used. Calibrators (5.0 (LLOQ), 10.0, 20.0, 50.0, 100.0, 200.0, 500.0 (ULOQ) IU/L for Choriomon® and 2.0 (LLOQ), 5.0, 10.0, 20.0, 50.0, 100.0, 200.0, 500.0 (ULOQ) IU/L for Ovitrelle®) were prepared freshly on the day of analysis.
Choriomon® QC samples at the levels (IU/L) low (12.5), medium 1 (50.0) and high (400.0) and Ovitrelle® QC samples at the levels (IU/L) low (6.0), medium 1 (50.0) and high (400.0) were prepared as a batch, and aliquots were stored at ≤ − 18 °C. Sample volume was 25.0 µL per well.
The samples were analysed using a SynergyTM 2 reader (BioTek Instruments, Inc., USA) with time-resolved fluorescence, and Gen5TM secure software (BioTek Instruments, Inc., USA) was used for data processing. Each calibrator, QC and study sample was measured in duplicate. The coefficient of variation (CV%) of duplicate measurements had to be ≤ 20.0% (calculated as: CV% = 100 * (SD duplicates/mean of the duplicates)), otherwise the result was rejected. The mean of the duplicate measurements was reported for calibrators, QC samples and study samples. Concentrations were calculated using a 4 Parameter Logistic (4-PL) nonlinear regression model.
2.5 Pharmacokinetic Parameters
Serum hCG pharmacokinetic parameters Cmax (maximum serum concentration), tmax (time to achieve Cmax), AUC0-t (area under concentration-time curve from time 0 to the last quantifiable concentration time t), AUC0–∞ (AUC extrapolated to infinity) and half-life (t½) were determined or calculated with the validated software Phoenix WinNonLin® 6.3, Certara, Inc., using a non-compartmental analysis and the linear trapezoidal rule. Pharmacokinetic parameters Cmax and AUCs were dose-normalized to account for the different doses used (10,000 IU vs. 6500 IU for Choriomon® and Ovitrelle®). No baseline-correction or correction for potency was performed.
The primary study outcome measures were serum dose-normalized Cmax and AUC0–t.
2.6 Safety
Safety of the investigational products was assessed by physical examination, electrocardiogram (ECG), routine laboratory tests and vital sign check, performed at screening (before study enrolment) and final visit. Untoward medical occurrences (adverse events) in the subjects were recorded throughout the study. Adverse events (AEs) were coded by System Organ Class and Preferred Term, using the Medical Dictionary for Regulatory Activities (MedDRA) version 22.0. All adverse events were classified according to the categories serious/non-serious, expected/unexpected, and mild, moderate and severe. In addition, the study investigator always indicated whether a causal relationship existed between the specified event and the study product/treatment.
2.7 Sample Size
Twenty-six (26) healthy women were randomized to have 24 subjects in the pharmacokinetic analysis. Although no formal bioequivalence proof was required, sample size estimation was performed based on the bioequivalence test: for Cmax and AUC, when the sample size in each sequence group is 12 a crossover design will have 80% power to reject the null hypotheses that the ratio of the test mean to the standard mean is below 0.800 and above 1.250; i.e., that the test and standard are not equivalent, in favour of the alternative hypothesis that the two treatment means are equivalent, assuming that the expected means ratio is 1.000, the Crossover ANOVA, mean square error (MSE) (ln scale) is 0.250 (the SD difference, sd (ln scale) is 0.354), and that data are analysed in the natural log scale using t tests (5% level) for means differences. The expected mean ratios and parameters variability were hypothesized because no information from previous comparisons was available.
2.8 Statistical Analyses
Study data were summarized by descriptive statistics. The statistical analyses were performed using SAS® version 9.3 (TS1M1) and Phoenix WinNonLin® 6.3.
For the comparisons, dose-normalized Cmax, AUC0–t and AUC0–∞ were analysed using analysis of variance (ANOVA), after neperian logarithmic transformation. Before analysis, the pharmacokinetic parameters were dose-normalized using the respective test and reference batch assayed contents.
Bioavailability was compared based upon the 90% confidence interval (CI) and each parameter geometric means test/reference ratio (GMR). The statistical analysis included treatment, period, sequence and subject-within-sequence as fixed effects. tmax was compared between treatments using the non-parametric Wilcoxon signed-rank test.
3 Results
3.1 Subjects
Twenty-six (26) healthy women aged 20–45 years, with a BMI of 23.63 ± 2.44 kg/m2, were randomized, completed the study per protocol, and were included in the safety evaluation. Twenty-four (24) women were included in the pharmacokinetic analysis and two were excluded due to analytical sample swap.
3.2 Pharmacokinetics
Serum hCG concentration-time profiles for the test and reference products are shown in Fig. 2. No baseline-correction for pharmacokinetic parameters was performed because no endogenous hCG was detected. Main serum hCG pharmacokinetic parameters are presented in Table 1.

Mean (±SD) serum hCG original concentrations (IU/L) vs. time profiles. Logarithmic/linear scale (N = 24). h hours, hCG human chorionic gonadotropin, IU/L international units/litre, Test Test product: Choriomon®, Ref Reference product: Ovitrelle®
Serum hCG levels increased rapidly after a single dose of the test and reference hCG preparations and the shape of the two pharmacokinetic curves was very similar. Half-life (t1/2) of the two products was also very similar, with values of 36.77 ± 5.11 and 38.63 ± 6.08 h for Choriomon® and Ovitrelle®.
Results of the exploratory statistical analysis on dose-normalized pharmacokinetic parameters are presented in Table 2. The ratio of geometric means (GMR) corresponded to 121.31 and 119.81% for AUC0–t and AUC0–∞, indicating that hCG extent of exposure was approximately 20% greater for the test than the reference hCG preparation. Accordingly, the 90% CI for the two parameters exceeded the prespecified upper limit for bioequivalence (125%) (Table 2). The rate of absorption was approximately 50% higher with Choriomon® than with Ovitrelle®, as indicated by a GMR of 146.89% for Cmax. Also, Cmax 90% CI was completely above the pre-specified range (Table 2). No period or sequence effects were observed. On the other hand, treatment had an effect on the three parameters (p ≤ 0.0002).
The tmax ranged from 12 to 36 h (median 16 h) for the test and from 12 to 48 h (median 24 h) for the reference product, and significantly differed between treatments (p = 0.0023).
3.3 Safety
The investigational products administered by s.c. injection were well tolerated. No subject withdrew from the study for an adverse event. Fatigue, experienced by one participant, was the only adverse event judged related to Choriomon®. “Aggressive behaviour”, which was deemed by the investigator as related to treatment, was reported for the same subject with Ovitrelle®. The other adverse events related to the reference product were headache (two subjects) and nausea, breast tenderness and hyperhidrosis (one subject each). All adverse events were mild or moderate and resolved before study end. No adverse reactions at the injection site or significant effects on laboratory parameters, vital signs, body weight or ECG were observed.
4 Discussion
The present study, which compared the pharmacokinetics of Choriomon® and Ovitrelle®, administered by s.c. injection at the doses of 10,000 and 250 µg (6500 IU) in women, showed that the serum concentration profiles of the two preparations have a similar shape. However, serum hCG bioavailability was approximately 20% greater with HP-hCG than with r-hCG, based on the results obtained with dose-normalized AUC parameters. In addition, HP-hCG administration resulted in an approximately 50% higher rate of absorption than r-hCG, with median tmax values of 16 h and 24 h for Choriomon® and Ovitrelle®, respectively. This result is not unexpected, considering the different source of raw material (human derived vs. recombinant) and the possible different affinity of the two products for the antibodies used in the immunoassay [13]. The observed differences in the rate and extent of hCG absorption are indeed reflected in the different approved dose regimens for the two hCG formulations, i.e., 5000–10,000 IU for Choriomon® versus 250 µg (6500 IU) for Ovitrelle®.
These differences do not appear to be clinically relevant, as clearly confirmed by the previous study by Bellavia et al. [10], in which the two products, administered by s.c. injection, were equally safe and equally effective in inducing the final follicular maturation in women undergoing controlled ovarian stimulation for IVF treatment.
The doses used in the study are those already approved for the respective formulations and previously compared in the Phase III trial [10]. Choriomon® is approved at dosages of 5000 and 10,000 IU. For the clinical study, the 10,000 IU dose was selected to test its safety in the worst conditions. The previous study [10] demonstrated that even at the highest dose, Choriomon® was as safe as Ovitrelle®. Moreover, in the Choriomon® group more oocytes were collected but since the study had a “non-inferiority” design, no conclusions about the superiority of the test drug could be made and only equivalence was claimed. The study presented in this article was performed as a complement of information.
To note, elimination half-life (t1/2) values were similar for the two investigational products. According to these results, the products are completely cleared 7–8 days after the injection, suggesting that results of a serum pregnancy test (beta-hCG test), usually performed 2 weeks after injections, will not be affected.
Serum hCG concentrations and pharmacokinetic data of this study are in line with the results of published data, indicating a quite high inter-subject variability in serum hCG concentrations and derived pharmacokinetic parameters. In the study by Mannaerts et al. [7], dose-normalized Cmax (tmax) and AUC0–∞ after a single s.c. injection of 10,000 IU urine-extracted hCG corresponded to 0.03 ± 0.01 1/L (20.03 ± 8.3 h) and 3.05 ± 0.50.53 h/L, respectively. Serum hCG was eliminated with a t1/2 of 32.28 ± 3.80 h.
In a more recent study [14], a single s.c. injection of 250 µg r-hCG administered to healthy Caucasian women resulted in a peak concentration of 158.06 ± 24.1 IU/L, achieved at a median tmax of 22h (9.0–48.0 h). Elimination t1/2 was 35.7 ± 11.2 h.
In the same study, also comparing a single 250 µg dose of r-hCG with a single 5000 IU dose of urinary hCG (u-hCG) in healthy Japanese women, r-hCG s.c. injection resulted in an 11% lower Cmax but a 19% higher AUC0–∞ compared with u-hCG.
This gives additional evidence that administration of the two preparations can result in different pharmacokinetic profiles although no difference in efficacy between r-hCG and u-hCG has been observed [10, 15, 16]. Notably, all women enrolled in this study had a normal menstrual cycle and were pituitary suppressed using their usual combined oral hormonal contraceptives reducing the levels of endogenous FSH < 4 IU/L and LH < 5IU/L, which is considered a marker of an appropriate gonadotropin suppression [17]. Injections were performed in the umbilical region because this area has an easily accessible layer of tissue and is one of the most common injection sites for subcutaneous administration. A possible limitation of the study was that the two assays for the determination of the two hCG preparations had slightly different lower limits of quantifications (2 IU for Choriomon® vs. 5 IU for Ovitrelle®) because the two hCG preparations are different (human-derived vs. recombinant) and consequently two different analytical assays had to be developed and validated using appropriate reference standards. Considering that the results of this study are comparable to previously published data with different preparations, and that the aim of this study was to descriptively compare the bioavailability of the two formulations, we do not believe that the different lower limits of quantification have an impact on the study results.
5 Conclusions
The present study cannot exclude a difference in the serum pharmacokinetic profile of HP-hCG and r-hCG. Yet, the observed higher rate of absorption of HP-hCG compared to r-hCG does not affect the clinical efficacy or safety in the induction of ovulation and/or assisted reproductive technology outcome, as demonstrated in previous comparative Phase III studies. The safety data collected during this study are in line with the known safety profile of the two products and did not raise any safety concerns.
References
Muyan M, Boime I. Secretion of chorionic gonadotropin from human trophoblasts. Placenta. 1997;18:237–41. https://doi.org/10.1016/s0143-4004(97)80056-2.
Pierce JG, Parsons TF. Glycoprotein hormones: structure and function. Annu Rev Biochem. 1981;50:465–95. https://doi.org/10.1146/annurev.bi.50.070181.002341.
Fournier T, Guibourdenche J, Evain-Brion D. Review: hCGs; different sources of production, different glycoforms and functions. Placenta. 2015;29:S60–5. https://doi.org/10.1016/j.placenta.2015.02.002.
Gemzell C. Induction of ovulation with human gonadotropins. Recent Prog Horm Res. 1965;21:179–204.
Wikland M, Borg J, Forsberg AS, Jakobsson AH, Svalander P, Waldenström P. Human chorionic gonadotrophin self-administered by the subcutaneous route to induce oocyte maturation in an in-vitro fertilization and embryo transfer programme. Hum Reprod. 1995;10(7):1667–70. https://doi.org/10.1093/oxfordjournals.humrep.a136152.
Stelling JR, Chapman ET, Frankfurter D, Harris DH, Oskowitz SP, Reindollar RH. Subcutaneous versus intramuscular administration of human chorionic gonadotropin during an in vitro fertilization cycle. Fertil Steril. 2003;79(4):881–5. https://doi.org/10.1016/s0015-0282(02)04918-x.
Mannaerts BM, Geurts TB, Odink J. A randomized three-way cross-over study in healthy pituitary-suppressed women to compare the bioavailability of human chorionic gonadotrophin (Pregnyl) after intramuscular and subcutaneous administration. Hum Reprod. 1998;13(6):1461–4. https://doi.org/10.1093/humrep/13.6.1461.
Choriomon® 5000 IU. IBSA Institut Biochimique S.A., Switzerland. https://compendium.ch/fr/product/index/1211588-choriomon-5000-ie-c-solv-spritze. Accessed 29 Sept 2021.
Ovitrelle 250 micrograms/0.5 ml prefilled syringe. Summary of product characteristics. https://www.medicines.org.uk/emc/product/5527. Accessed 29 Sept 2021.
Bellavia M, de Geyter C, Streuli I, Ibecheole V, Birkhäuser MH, Cometti BPS, de Ziegler D. Randomized controlled trial comparing highly purified (HP-hCG) and recombinant (r-hCG) for triggering ovulation in ART. Gynecol Endocr. 2013;29(2):93–7. https://doi.org/10.3109/09513590.2012.730577.
Guidance on the investigation of bioequivalence. CPMP/EWP/QWP/1401/98 Rev. 1/Corr **, 20 January 2010
EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2**. Guideline on bioanalytical method validation. 21 July 2011
Gronowski AM, Grenache DG. Characterization of the hCG variants recognized by different hCG immunoassays: an important step toward standardization of hCG measurements. Clin Chem. 2009;55(8):1447–9. https://doi.org/10.1373/clinchem.2009.129205 (Epub 2009 Jun 18).
Bagchus W, Wolna P, Uhl W. Single-dose pharmacokinetic study comparing the pharmacokinetics of recombinant human chorionic gonadotropin in healthy Japanese and Caucasian women and recombinant human chorionic gonadotropin and urinary human chorionic gonadotropin in healthy Japanese women. Reprod Med Biol. 2017;17(1):52–8. https://doi.org/10.1002/rmb2.12066.
Youssef MA, Abou-Setta AM, Lam WS. Recombinant versus urinary human chorionic gonadotrophin for final oocyte maturation triggering in IVF and ICSI cycles. Cochrane Database Syst Rev. 2016;4(4):CD003719. https://doi.org/10.1002/14651858.CD003719.pub4.
Chang P, Kenley S, Burns DG, Currie K, DeVane G, O’Dea L. Recombinant human chorionic gonadotropin (rhCG) in assisted reproductive technology: results of a clinical trial comparing two doses of rhCG (Ovidrel) to urinary hCG (Profasi) for induction of final follicular maturation in in vitro fertilization-embryo transfer. Fertil Steril. 2001;76(1):67–74. https://doi.org/10.1016/s0015-0282(01)01851-9.
Dissanayake S. Assessing the bioequivalence of analogues of endogenous substances ('endogenous drugs’): considerations to optimize study design. Br J Clin Pharmacol. 2010;69(3):238–44. https://doi.org/10.1111/j.1365-2125.2009.03585.x.
Acknowledgements
The authors acknowledge CROSS Research S.A., Switzerland for study coordination and Ardena Bioanalysis B.V., the Netherlands, for serum hCG determination.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The study was funded by IBSA Institut Biochimique S.A., Switzerland.
Conflict of interest
B.C. is an employee of IBSA Institut Biochimique S.A., M.R. and C.L. are employees of CROSS Research S.A., Switzerland, which was contracted by IBSA Institut Biochimique S.A. for the conduction of this study and received financial support for its services. The authors declare that they have no other relationships or activities that could appear to have influenced the submitted work.
Ethics approval
The study was approved by the Canton Ticino Ethics Committee, Switzerland, and the Swiss Federal Health Authority, and was performed in accordance with the Helsinki Declaration and Good Clinical Practice.
Consent to participate
All subjects received a detailed study description and gave their written informed consent before enrolment.
Consent for publication
Not applicable.
Availability of data and material
The study is registered on Clinicaltrials.gov (no. NCT03735030). The data supporting the study fundings are available from the author (B.C.) upon reasonable request.
Code availability
Not applicable.
Authors’ contributions
All authors made substantial contributions to the conception and design of the study, the acquisition of data, the analysis and interpretation of data, drafting of the article or critical revision for important intellectual content and final approval of the submitted version.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Radicioni, M., Leuratti, C. & Cometti, B. Randomized Pharmacokinetic Study of a Highly Purified Human Chorionic Gonadotropin and of a Recombinant Human Chorionic Gonadotropin Following Single Subcutaneous Administration in Healthy Women. Clin Drug Investig 42, 199–206 (2022). https://doi.org/10.1007/s40261-022-01118-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-022-01118-w