
Abstract
Background and Objectives
BI 425809, a novel glycine transporter-1 inhibitor, may ameliorate cognitive deficits in schizophrenia. The objectives of the studies were: to assess absolute bioavailability of oral BI 425809 compared with intravenous (IV) microtracer infusion (study 1), and to determine the mass balance, distribution, metabolism, and excretion of BI 425809 (study 2).
Methods
These were Phase I, open-label, non-randomized, single-period, single-arm studies in healthy males. Study 1 administered a single oral dose of unlabeled BI 425809 25 mg, then an IV microtracer infusion of [14C]-BI 425809 30 µg. In study 2, participants received an oral dose of [14C]-BI 425809 25 mg containing [14C]-labeled (dose: 3.7 megabecquerel (0.41 mSv)) and unlabeled drug. Safety was assessed.
Results
In study 1 (n = 6), the absolute bioavailability of a 25 mg tablet of BI 425809 in a fasted state was 71.64%. The geometric mean dose-normalized maximum plasma concentration was approximately 80% lower after oral administration versus IV dose. In study 2 (n = 6), the total recovery of [14C]-BI 425809 was 96.7%, with ~ 48% of [14C]-radioactivity excreted in urine and ~ 48% excreted in feces. Among the labeled drug in urine, ~ 45% of the amount excreted was composed of BI 425809 (17.4%) and two metabolites (BI 758790, 21.0%; BI 761036, 5.9%). In feces, < 1% of BI 425809 was excreted as unchanged drug. In both studies, BI 425809 was generally well tolerated.
Conclusions
After normalization, the absolute bioavailability of tablet-form BI 425809 was 71.64%. The total recovery of [14C]-BI 425809 25 mg was high (96.7%), with low intraindividual variability and similar amounts excreted in urine and feces.
Clinicaltrials.gov identifiers
NCT03783000 and NCT03654170.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
BI 425809 is a glycine transporter-1 inhibitor being investigated for the treatment of cognitive impairment in schizophrenia. |
Two studies were performed to assess the absolute bioavailability, absorption, distribution, metabolism, and excretion of BI 425809. |
Absolute bioavailability was lower for the oral form of BI 425809 (71.6%) than the IV form, while total recovery was high (96.7%). |
1 Introduction
Schizophrenia is characterized by abnormalities in glutamatergic pathways related to N-methyl-D-aspartate (NMDA) receptor hypofunction, in both the cortical and the hippocampal areas of the brain [1,2,3,4,5]. These abnormalities are associated with negative symptoms (including withdrawal, decreased spontaneous communication, decreased eye contact, dampened or decreased facial expressions, decreased vocal inflection, and reduced spontaneous movement) in schizophrenia, as well as cognitive impairment [6,7,8]. Glycine is an obligatory co-agonist for glutamate signaling at NMDA receptors, and therefore inhibition of glycine transporter-1 (GlyT1) on glial cells may improve NMDA receptor hypoactivation by increasing the concentration of glycine in the synaptic cleft [3]. This may consequently improve negative and cognitive symptoms in patients with schizophrenia (as add-on therapy to antipsychotics) [3, 6, 9, 10].
BI 425809 is a novel, potent and selective GlyT1 inhibitor that has been developed for the symptomatic treatment and cognitive impairment associated with schizophrenia [6, 11]. It has been shown to be generally well tolerated at the anticipated therapeutic dose range of 5–25 mg [11, 12]. Previous studies have shown that BI 425809 increased the levels of glycine in human and rat cerebral spinal fluid (CSF) in a dose-dependent fashion, with multiple dosing leading to accumulation of BI 425809 in plasma and CSF [6]. This demonstrates indirect functional target engagement, indicating that glycine CSF levels can be used to assess GlyT1 inhibition centrally [6]. This report details the findings of two Phase 1 studies; the objective of the first study (NCT03783000) was to investigate the absolute bioavailability of oral BI 425809. The second study (NCT03654170) aimed to determine the basic pharmacokinetics of BI 425809, including mass balance, metabolism, and excretion pathways following a single oral dose of [14C]-BI 425809. The plasma pharmacokinetics were determined for BI 425809 (labeled and unlabeled) and three metabolites (BI 758790 (M530), BI 761036 (M232) and IN79211 (M312)).
2 Methods
2.1 Participants
For both studies, eligible participants were males, 18–65 years of age and healthy (according to the investigator’s assessment, based on a complete medical history including a physical examination, vital signs (blood pressure and pulse rate), a 12-lead electrocardiogram (ECG), and clinical laboratory tests), with a body mass index (BMI) of 18.5–29.9 kg/m2. Key exclusion criteria included abnormal findings from the medical examination, laboratory values outside the reference range, or evidence of concomitant disease judged to be clinically relevant by the investigators. Other exclusion criteria included: smoking (> five cigarettes or one cigar/pipe per day); alcohol (average intake > 24 units per week); drug abuse or having a positive drug result at screening; use of drugs within 30 days prior to the administration of trial medication that might influence the results of the trial; use of cytochrome P450 3A4 (CYP3A4) inhibitors and inducers 1 week prior to administration of the trial medication; chronic or relevant acute infections; and participation in another study where the investigational drug was administered within 60 days prior to the administration of study medication or current participation in another trial involving administration of an investigational drug. Participants were also excluded if they were unable to comply with the dietary regimen.
2.2 Ethical Considerations
The studies were conducted in accordance with the principles of the Declaration of Helsinki [13], the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Harmonized Tripartite Guideline for Good Clinical Practice (GCP) [14], applicable regulatory requirements, and the standard operating procedures of the sponsor company (Boehringer Ingelheim International GmbH). All participants provided a signed and dated written informed consent form in accordance with GCP and local regulatory and legal requirements, prior to admission to the study. The study procedures, protocols, and documents were reviewed and approved by the Independent Ethics Committee of the study center as well as the relevant local authorities.
2.3 Absolute Bioavailability of BI 425809
2.3.1 Study Design
This was an open-label, non-randomized, single-period (12–15 days), single-arm, Phase I study conducted in one center (NCT03783000). Participants were required to attend three study visits: the screening period (Visit 1); the treatment period (Visit 2), including administration of trial medication and extensive pharmacokinetics and safety profiling on Day -1 to Day 2, with participants returning for further pharmacokinetics and safety assessments on Days 4, 6, and 8; and the end-of-trial examination (Visit 3), which occurred between Days 12 and 15 (participants who prematurely discontinued treatment also underwent this examination) (Fig. 1a).

a Study 1 design: absolute bioavailability of BI 425809, and b study 2 design: mass balance recovery of BI 425809. PK pharmacokinetic
2.3.2 Treatments and Administration
The unlabeled tablet formulation of BI 425809, the radiolabeled [14C]-BI 425809 and the unlabeled BI 425809 powder for the intravenous (IV) solution, were manufactured by Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany. The mixture of [14C]-BI 425809 and unlabeled BI 425809, and the solution from this mixture ([14C]-BI 425809) were prepared by PRA Health Sciences, Groningen, the Netherlands. The radioactively labeled mixture was given IV as a comparator to the orally administered tablet form of the study drug; radiolabeling allowed differentiation of the two forms, as the accelerator mass spectrometry (AMS) method used only measures the radiolabeled component (see Sect. 2.3.4). Following an overnight fast (≥ 10 h), each participant received a single oral dose of unlabeled BI 425809 25 mg (a film-coated tablet taken with water) while in a standing or sitting position. Participants remained fasted until at least 4.5 h after oral dosing. Four hours after the intake of the tablet (i.e., at the time of maximum concentration (tmax) of oral BI 425809), an IV microtracer infusion of [14C]-BI 425809 30 µg (a mixture of [14C]-labeled BI 425809 3 µg (radioactive dose per infusion was 0.011 MBq) and unlabeled BI 425809 27 µg) was administered via an intravenous indwelling catheter, kept patent with a saline infusion, while the participants were in a semi-supine position. The infusion duration was 15 min. A second indwelling catheter was placed in the contralateral arm for the collection of blood samples. Blood samples (3 mL) were taken from the antecubital or forearm vein of participants to assess plasma concentrations of unlabeled BI 425809 or [14C]-BI 425809]. Plasma BI 425809 was assessed at 2 h prior to the administration of the oral study drug, and at 1, 2, 3, 4:05, 6, 8, 12, 24, 72, 120, and 168 h after administration. Plasma [14C]-BI 425809] was assessed at 3, 4:05, 4:10, 4:15, 4:30, 5, 6, 7, 8, 12, 16, 24, 72, 120, and 168 h after administration of the oral drug. Once collected, plasma was aliquoted and transferred to a freezer within 60 min and stored on ice at around − 20 °C in an upright position until analysis. Participants were kept under medical surveillance for 24 h after drug administration and adverse events (AEs) were assessed continuously from screening until end-of-trial examination. All study medication was administered under the supervision of the investigating physician or an authorized designated person.
2.3.3 Endpoints and Assessments
The primary pharmacokinetic endpoints were the area under the concentration-time curve (AUC) over the time interval from 0 to infinity in plasma (AUC0–∞) of BI 425809 following oral administration and of [14C]-BI 425809 after IV administration. The maximum measured concentration (Cmax) of oral BI 425809 in plasma following a single oral dose was assessed as a secondary endpoint. Safety and tolerability of the investigational drugs were assessed using clinically relevant findings from physical examinations (AEs), safety laboratory parameters, 12-lead ECG, and vital signs, including pulse rate and blood pressure.
2.3.4 Bioanalytical Assays
Cold (unlabeled) BI 425809 concentrations in plasma were determined by a validated liquid chromatography coupled to tandem mass spectrometry (LC/MS) assay. This analysis was performed at NUVISAN, Neu-Ulm, Germany. [14C]-BI 425809 was determined by a validated AMS assay performed at TNO, Zeist, The Netherlands.
2.3.5 Statistical Analysis
Descriptive statistics were used to summarize all endpoints; no formal hypothesis testing was performed. The primary pharmacokinetics endpoints were log-transformed and fitted to an analysis of variance (ANOVA) model. Absolute bioavailability was estimated by determination of the ratio of the geometric means (gMean) of BI 425809 25 mg tablet (test) and [14C]-BI 425809 3 µg IV microtracer (reference) for the primary endpoint after dose-normalization using a mixed effects ANOVA model with corresponding two-sided 90% confidence intervals (CIs). As the focus was on estimation, not testing, no acceptance range was specified.
2.4 Mass Balance Recovery of BI 425809
2.4.1 Study Design
This was an open-label, non-randomized, single-period, single-arm, Phase I study conducted in one center in the Netherlands (NCT03654170). On Day 1, participants received the trial medication; blood, urine, and feces samples were collected daily until the morning of Day 15 when release criteria were assessed (Fig. 1b). The release criteria were defined as: ≥ 90% of the administered dose cumulatively recovered in urine and feces combined over the study period; or if < 1% of the dose had been collected (within two separate, consecutive 24-h intervals) in urine and feces; or if concentration of total radioactivity in plasma and blood was < 5% of Cmax of total radioactivity in plasma. If one of these criteria was met, then collection after Day 15 was stopped. If the criteria were not met, the participants returned to the trial site for 24-h collection intervals of urine and feces on Days 21, 28, 35, 42, and 49.
2.4.2 Treatments and Administration
The trial medication was manufactured by Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany. The solution from the mixture was prepared by PRA Health Sciences, Groningen, the Netherlands. Following an overnight fast (≥ 10 h), participants received a single oral dose of [14C]-BI 425809 25 mg, containing a mixture of [14C]-labeled and unlabeled drug, administered in a 12.5 mL solution (2 mg/mL) with 240 mL of water. The planned radioactive dose per participant was 3.7 megabecquerel (MBq; 0.41 mSv). No food was allowed for at least 4 h after medication administration. The trial medication was administered to participants while they were in the sitting position, under the supervision of the investigator and/or authorized designated person. For metabolic profiling, blood was taken from a forearm vein; 20 mL of blood was taken at pre-dose, and all time points apart from at 312 h when 40 mL of blood was drawn. Blood samples were taken 2 h prior to the administration of the oral study drug, and at 1, 4, 8, 24, 48, 96, 120, 168, and 312 h after administration of the drug. For radioactivity analysis and quantification of metabolites, blood samples were taken from the antecubital or forearm vein of the participants. At each time point, 5 mL (for time points 0.5–240 h) or 8 mL (for the pre-dose sample and from time points 264 h to the end) samples were drawn. For metabolic profiling, blood was taken from a forearm vein; 20 mL of blood was taken at pre-dose, and all time points apart from at 312 h when 40 mL of blood was drawn. Three milliliters of blood was also taken for hematocrit measurement 2 h prior to drug administration, then at intervals in the period between 0 and 336 h. Collections were also made on return visits to the trial center at 485 h, 653 h, 821 h, 989 h, and 1157 h for blood cell/plasma ratio. Once collected, plasma was aliquoted and transferred to a freezer within 60 min and stored on ice at around − 20 °C in an upright position until analysis.
Urine collection was made at 2 h prior to drug administration, then at intervals in the period between 0 and 336 h, after which time the participants left the trial center. Collections were also made on return visits to the trial center at 485 h, 653 h, 821 h, 989 h, and 1157 h.
2.4.3 Endpoints and Assessments
The primary pharmacokinetic endpoints were the mass balance recoveries of total [14C]-radioactivity of the administered drug in urine and feces across the whole study, and fraction of [14C]-radioactivity excreted in urine and feces, respectively, as a percentage of the administered oral dose over the time interval from 0 to the last quantifiable time point. Secondary endpoints for total radioactivity and BI 425809 were plasma Cmax and plasma AUC from 0 to the last quantifiable time point (AUC0–tz). Other endpoints measured in whole blood and plasma were AUC0–∞, tmax, percentage AUC0–∞ obtained by extrapolation (%AUCtz–∞), and terminal half-life of the analyte (t1/2). The pharmacokinetic endpoints were determined for unlabeled BI 425809 and three metabolites, BI 758790, BI 761036, and IN 79211 (only in plasma and urine). Safety and tolerability of the investigational drug were assessed using clinically relevant findings from physical examinations (AEs), safety laboratory parameters, 12-lead ECG, and vital signs, including pulse rate and blood pressure.
Metabolite radioprofiling was accomplished by high-performance liquid chromatography (HPLC) radiochromatography using fractionation coupled with solid scintillation counting. Metabolites were characterized and identified by LC/MS, including accurate mass determination in conjunction with radioactive detection. Metabolite structural elucidation, conducted in both positive and negative ion mode, was based on CID MS/MS fragmentation and considered tentative unless confirmed with an authentic chemical standard. Radioactive peaks < 20 cpm in peak height were considered to be below the limit of quantitation. Analysis of plasma metabolites was conducted in samples collected up to 168 h after dosing.
2.4.4 Statistical Analysis
No statistical evaluation of the primary and secondary endpoints was performed. Data are reported using descriptive statistics only.
3 Results
3.1 Absolute Bioavailability of BI 425809
3.1.1 Study Population and Disposition
A total of six healthy male participants were included in this trial. All six participants received the trial medication but only four completed the trial according to protocol; although no important protocol violations were observed, one participant missed an ambulatory visit and one was lost to follow-up. Five of the six participants were White, and one was multi-racial. The mean (standard deviation (SD)) age of the participants was 35.0 (14.5) years and mean (SD) BMI was 25.1 (3.6) kg/m2. Two (33.3%) participants were current smokers (> five cigarettes or one cigar/pipe per day) while four (66.7%) had never smoked. Baseline demographic and clinical characteristics are summarized in the Electronic Supplementary Material (ESM), Table 1.
3.1.2 Pharmacokinetic Analysis—Plasma Concentration-Time Profiles
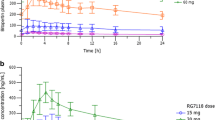
After the single oral administration of a BI 425809 25 mg tablet, the maximum plasma concentration was reached at 3.54 h after dosing (median tmax, range 2.00–6.00 h; Fig. 2a). Following IV administration of [14C]-BI 425809 (4 h after the administration of the oral dose), tmax was reached at 0.25 h (median tmax) after the start of the infusion (range 0.167–0.25 h), i.e., at the end of the 15-min infusion period (Fig. 2b). The concentration-time profiles declined in a biphasic manner after both IV and oral dosing (Fig. 2a, b).

Individual and gMean drug plasma concentration after single a oral administration of BI 425809 25 mg or b intravenous (IV) infusion of labeled [14C]-BI 425809 3 µg. gMean geometric mean
3.1.3 Evaluation of Absolute Bioavailability and Primary Pharmacokinetic Endpoint
The gMean AUC0–∞ following oral dosing of BI 425809 25 mg in the fasted state was 7650 nmol · h/L (geometric coefficient of variation (gCV): 36.5%), and 1.31 nmol · h/L (35.5%) following IV [14C]-BI 425809 administered 4 h later (Table 1). Following dose normalization, AUC0–∞,norm was lower after oral dosing than after IV administration. An ANOVA model, which accounted for the “participant” as a random effect and “formulation” as a fixed effect, was applied to the AUC0–∞,norm. The absolute bioavailability of a 25 mg tablet of BI 425809 in a fasted state was 71.64% (90% CI: 64.3–79.9), thus yielding similar results to the gMean of the ratios calculated for the individual participants. Sensitivity analysis using the same ANOVA model with “participant” as a fixed event also supported the results of the primary analysis.
3.1.4 Secondary and Other Pharmacokinetic Endpoints
The gMean of the dose-normalized maximum plasma concentration (Cmax,norm) was approximately 80% lower after oral administration compared with the IV microtracer infusion. The interindividual variability of Cmax was low to moderate (gCV 34.4–36.8%) and was similar between oral and IV dosing (Table 1).
3.1.5 Safety
Three (50.0%) of the six participants who received BI 425809 reported a least one treatment-emergent AE. These comprised gastrointestinal disorders, nervous system disorders, and eye disorders, all of which were of mild intensity. No serious AEs (SAEs), AEs leading to trial drug discontinuation, AEs of special interest, or other significant AEs were recorded (ESM Table 2).
3.2 Mass Balance Recovery of BI 425809
3.2.1 Study Population and Disposition
A total of six healthy male participants were included in this trial, all of whom received the trial medication. No important protocol violations were observed. Five of the six participants were White and one was multiracial. The mean (SD) age of the participants was 45.7 (17.7) years and mean (SD) BMI was 26.1 (4.0) kg/m2. One participant (16.7%) was a current smoker, while two (33.3%) had never smoked and three (50.0%) were former smokers. Baseline demographic and clinical characteristics are summarized in ESM Table 1. All participants remained at the site until Day 15; however, none of them fulfilled the release criteria. Therefore, the participants left the site but returned once weekly for a 24-h visit including the continuous sampling of urine and feces.
3.2.2 Primary Endpoints—Excretion of BI 425809 and [ 14 C]-BI 425809
The total recovery of [14C]-BI 425809 25 mg to the last quantifiable time point (677 h; fetotal,0–677) ranged from 87.6 to 105%, with a gMean of 96.7% and a low intraindividual variability of 6.10% gCV. The labeled drug was excreted in similar proportions in both urine and feces (Fig. 3a; ESM Table 3). After 120 h, a gMean of 54.0% (gCV: 9.88%) of the labeled drug material had been excreted. After 1 week (168 h), 67.5% (gCV 5.06%) had been eliminated. Two weeks (336 h) after dosing, 86.8% (gCV 3.82%) had been excreted on average. Figure 3a shows the total recovery of [14C]-BI 425809 25 mg in urine (gMean 48.1%; gCV 9.60%) and feces (gMean 48.3%; gCV 14.30%) after 677 h; Fig. 3b, c show the gMean and individual recoveries (n = 6) for the cumulative [14C]-radioactivity excreted after 677 h for urine and feces, respectively.

a Total recovery of [14C]-BI 425809 25 mg in urine and feces; b cumulative [14C]-radioactivity excreted in urine; c cumulative [14C]-radioactivity excreted in feces. a Shows the gMean and gCV; b, c show the gMean plus individual participant data. Fe fraction excreted
3.2.3 Secondary and Other Pharmacokinetics Endpoints
Following rapid absorption, the maximum plasma concentration for BI 425809 was reached with a gMean Cmax of 383 nmol/L (gCV 14.2%) (Table 2); plasma concentrations then declined rapidly in a biphasic manner with a half-life of 50.5 h (gCV 14.0%) (Table 2). Oral BI 425809 was rapidly absorbed with a median tmax of 1.50 h (range 1.00–4.00 h; Table 2). After oral administration, labeled [14C]-BI 425809 appeared almost simultaneously in plasma and in whole blood with a median tmax of 1.51 h (range 1.00–6.00 h) in plasma and 1.26 h (range 1.00–4.00 h) in whole blood (Table 3). Concentrations declined simultaneously in both plasma and whole blood, with similar half-lives of 211 h (gCV 13.4%) and 220 h (gCV 10.4%), respectively (Table 3). The AUC0–∞ for plasma was 70,000 nmol · h/L (gCV 20.2%) and 47,700 nmol · h/L (gCV 20.5%) for whole blood (Table 3). The blood to plasma ratios for Cmax and AUC0–tz delivered results of 0.677 (gCV 3.49%) and 0.662 (gCV 4.11%), respectively.
3.2.4 Metabolites
Based on bioanalysis of unlabeled BI 425809 and its metabolites, the plasma Cmax of BI 758790 was 138 nmol/L (gCV 10.0%) with a median tmax of 10.0 h (range 8.00–24 h) and then declined continuously. BI 761036 and IN 79211 appeared later, with a median tmax of 192 h (144–264 h) and 72 h (72–192 h), respectively (Fig. 4). Cmax values were 55.6 nmol/L (gCV 39.8%) and 2.97 nmol/L (gCV 33.9%) for BI 761036 and IN 79211, respectively. BI 425809, BI 758790, and BI 761036 contributed approximately 45% of the drug-related material represented by the total radioactivity excreted in urine. Parent compound, BI 425809, and the metabolite BI 758790 showed similar cumulative amounts excreted (Ae) in urine up to 366 h (Ae0–366) with a gMean of 3560 nmol and 4050 nmol respectively (Table 4; Fig. 5).

gMean drug concentration time profiles of [14C]-BI 425809 25 mg and its metabolites in plasma. AUC area under curve, AUC0–tz time interval from 0 to the last quantifiable data point, gMean geometric mean

Comparison of [14C]-BI 425809 25 mg and its metabolites excreted in urine. The data shown are from a single participant. Ae0–366 amount excreted up to 366 h
The radioactivity contributions of BI 425809 and its metabolites in male human plasma, urine, and feces, as determined by metabolite radioprofiling, are shown in ESM Tables 4, 5, and 6, respectively. Twenty metabolites were identified collectively from the three matrices. As IN 79211 did not retain the radiolabel, the total number of metabolites identified in this study was 21. In urine, the contribution of BI 425809, BI 758790, and BI 761036 to the total radioprofile (ESM Fig. 5; ~ 41%) was similar to the value determined using exposure data from unlabeled material (45%). In the feces, BI 761036 was not observed, while BI 758790 and BI 425809 accounted for 4.1% (9.7% of total radioactivity) and < 1% of the radioactive dose (< 1% of total radioactivity), respectively (ESM Table 6).
3.2.5 Safety
Five (83.3%) of six participants reported AEs (ESM Table 7), none of which were considered severe or drug related by the investigators. The most frequently reported AEs were nervous system disorders (in four participants) and gastrointestinal disorders (in three participants). Four participants had AEs of mild intensity and one had an AE of moderate intensity (myalgia). No SAEs, AEs leading to trial drug discontinuation, AEs of special interest, or other significant AEs were recorded (ESM Table 7).
4 Discussion
4.1 Absolute Bioavailability
The first study reported here assessed the absolute bioavailability of a 25 mg tablet of BI 425809 in a fasted state compared with IV microtracer infusion. The ratio of the dose-normalized primary endpoint (AUC0–∞,norm) indicated an absolute bioavailability of 71.64% (90% CI: 64.28–79.84) following a single oral dose. After oral dosing, BI 425809 reached tmax within 3.54 h (median); this was slower than in the mass balance study (tmax 1.50 h) due to the difference in the formulation (tablet vs. solution).
In a previous study, it was found that the bioavailability of BI 425809 from a 25 mg tablet was lower than from a 25 mg solution (adjusted gMean ratios for AUC0–tz and Cmax for BI 425809 administered as a tablet fasted vs. oral solution fasted were 80.5% (90% CI 74.0–87.6) and 50.0% (90% CI 45.1–55.4), respectively) [11]. It was suggested that this was because BI 425809 is a Class II compound (according to the Biopharmaceutics Classification System) [15] of low intrinsic solubility and high permeability, which likely contributed to the low absolute oral bioavailability observed in the study by Moschetti et al. [11]. In addition, the same study observed a moderate increase in
BI 425809 bioavailability from the 25 mg tablet following a high-fat/high-calorie (~ 1000 kcal) meal versus 10-h fasted conditions (adjusted gMean ratios: AUC0–tz 125.9% (90% CI 115.7–137.0); Cmax 142.1% (90% CI 128.3–157.4)) [11]. The administration of BI 425809 with food may increase bile secretion as food increases the dissolution of the tablet, therefore increasing its intrinsic solubility and allowing for greater drug accessibility [12].
Another reason for reduced absolute bioavailability following a single oral dose of BI 425809 25 mg in a tablet form may be elimination via the enzyme CYP3A4 upon first-pass metabolism [11]. The major metabolic pathway for BI 425809 is mediated by CYP3A4, which is the most highly expressed CYP isoform in the liver and intestines, and is important in determining the exposure of oral medications [16, 17]. In the present study, AUC0–∞,norm was lower after oral dosing versus IV infusion, as was gMean Cmax,norm, which was around 80% lower following oral administration of BI 425809 versus IV dosing. These observations could likely be first-pass metabolism via gastrointestinal and hepatic CYP3A4, which would only affect the oral dosing route. In a previous study, the pharmacokinetic interaction of multiple oral doses of the CYP3A4 inhibitor itraconazole and BI 425809 was assessed [18]. It was observed that there was a small increase in Cmax (gMean ratio 116.1% (90% CI 108.0–124.7)) following co-administration of BI 425809 and itraconazole, which most likely reflected the blocking of CYP3A4 in the first-pass metabolism. Overall, the intrinsic solubility and, to a lesser extent, the first-pass metabolism via CYP3A4 might explain the absolute bioavailability of 71.6% of a 25 mg tablet in a fasted state. The interindividual variability of Cmax was low to moderate (gCV 34.4–36.8%) and was similar between oral and IV administration, although it was higher with regard to half-life (Table 1).
In accordance with previous studies investigating its safety and tolerability, oral BI 425809 25 mg was generally well tolerated, with no serious AEs reported [11].
4.2 Mass Balance Recovery
The objective of this study was to determine mass balance, excretion pathways, and metabolism following a single 25 mg oral dose of [14C]-BI 425809 including the basic pharmacokinetics of BI 425809 and [14C]-radioactivity. The total recovery of [14C]-BI 425809 was 96.7%, with a low intra-individual variability of 6.10%. Of the drug-labeled material eliminated, 48.1% was excreted in urine and 48.3% in feces, also with low intraindividual variability. The labeled drug material recovered in urine represents absorbed drug after oral administration, while the labeled drug material excreted in feces represents unabsorbed drug or [14C]-radioactivity excreted via intestinal or biliary excretion. Oral BI 425809 was absorbed rapidly after administration (median tmax 1.50 h), with a plasma gMean Cmax of 383 nmol/L. After drug administration, the labeled drug material appeared nearly simultaneously in whole blood and in plasma, with similar t1/2. However, the levels of [14C]-radioactivity in whole blood were continuously lower than in plasma, which indicates that the greater distribution of [14C]-radioactivity was in plasma. The blood to plasma ratios also indicated a main distribution of the total radioactivity in plasma.
The single 25 mg dose of BI 425809 was generally well tolerated; none of the AEs observed were considered to be serious and none lead to trial drug discontinuation.
4.2.1 Metabolites
Twenty-one plasma metabolites of BI 425809 were identified in human males in the metabolite radioprofiling study. Using HPLC radio chromatography with fractionation, coupled with solid scintillation counting, BI 425809 was found to be the most abundant circulating component at 41.0%, followed by metabolites BI 758790, BI 761036, and BI 764352, which accounted for 34.3%, 11.9%, and 6.6% of total plasma radioactivity, respectively (ESM Table 4). The remaining identified metabolites accounted for only 0–1.5% each of the total plasma reactivity.
In the present study, the plasma and urine concentrations of BI 758790, BI 761036, and IN 79211 were followed by absolute quantitation via liquid chromatography-tandem mass spectrometry (LC-MS/S) (Table 2). Based on the single-dose pharmacokinetics of BI 758790, the exposure (AUC0–∞ and Cmax) and half-life were similar to BI 425809. The single-dose pharmacokinetics of BI 761036 indicated markedly higher AUC0–∞, but lower Cmax of this metabolite in comparison to BI 425809 or BI 758790. This higher exposure was explained by a markedly higher half-life of BI 761036 in comparison to the half-life of BI 425809 and BI 758790 (243 h vs. ~ 50 h). IN 79211 exposure level (Cmax and AUC0–∞) was markedly lower than for BI 425809 and BI 758790, even though its half-life was higher than the other two species (127 h vs. ~ 50 h). The plasma exposure AUC0–∞ of BI 425809, BI 758790, BI 761036, and IN 79211 accounted for 71% of the total radioactivity detected. Based on comprehensive metabolite profiling studies, the remaining 29% of radioactivity is composed of minor metabolites, each accounting for a small percentage of remaining radioactivity. Overall, BI 425809, BI 761036, and BI 758790 contributed approximately 45% of the drug-related radioactive material excreted in urine (Table 4). In the feces, < 1% of BI 425809 was excreted as unchanged drug, and BI 758790 accounted for ~ 10% of the total excreted radioactivity.
4.3 Study Limitations
Interpretation of the data presented in this study is limited by the fact that the number of participants in both trials was small (absolute bioavailability, n = 6 with four completions; mass balance recovery, n = 6 with six completions). This led to results that showed large coefficients of variation for the pharmacokinetic endpoints and therefore may slightly reduce confidence in their dependability.
5 Conclusions
The AUC0–∞,norm and Cmax norm were both higher following IV infusion of [14C]-BI 425809 30 µg compared with the oral dose. The absolute bioavailability of BI 425809 25 mg given as a tablet in a fasted state was 71.64% (gMean). In terms of mass balance recovery, upon administration of [14C]-BI 425809 25 mg as an oral solution, 96.7% (gMean) of the dose was recovered in excreta, with approximately 48% excreted in both urine and feces. BI 425809 was metabolized to two major human metabolites, BI 758790 and BI 761036, which accounted for 18% and 33% of total radioactivity in the plasma, respectively. Although the pharmacokinetics of BI 758790 was similar to BI 425809, the pharmacokinetics of BI 761036 was markedly different as indicated by higher exposures driven by its slow elimination. In both studies, BI 425809 was well tolerated by the healthy male participants.
Change history
14 March 2022
A Correction to this paper has been published: https://doi.org/10.1007/s40261-022-01134-w
References
Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry. 2001;158(9):1367–77.
Hu N-W, Ondrejcak T, Rowan MJ. Glutamate receptors in preclinical research on Alzheimer’s disease: update on recent advances. Pharmacol Biochem Behav. 2012;100(4):855–62.
Hashimoto K. Glycine transport inhibitors for the treatment of schizophrenia. Open Med Chem J. 2010;4:10–9.
Lin CH, Lane HY, Tsai GE. Glutamate signaling in the pathophysiology and therapy of schizophrenia. Pharmacol Biochem Behav. 2012;100(4):665–77.
Balu DT. The NMDA receptor and schizophrenia: from pathophysiology to treatment. Adv Pharmacol (San Diego, Calif). 2016;76:351–82.
Rosenbrock H, Desch M, Kleiner O, Dorner-Ciossek C, Schmid B, Keller S, et al. Evaluation of pharmacokinetics and pharmacodynamics of BI 425809, a novel GlyT1 inhibitor: translational studies. Clin Transl Sci. 2018;11(6):616–23.
Rubio MD, Drummond JB, Meador-Woodruff JH. Glutamate receptor abnormalities in schizophrenia: implications for innovative treatments. Biomol Ther (Seoul). 2012;20(1):1–18.
Lakhan S, Caro M, Hadzimichalis N. NMDA receptor activity in neuropsychiatric disorders. Front Psychiatry. 2013;4:52.
Lane H-Y, Chang L-H, Liu Y-C, Chiu C-C, Tsai G. Sarcosine or D-serine add-on treatment for acute exacerbation of schizophrenia: a randomized, double-blind, placebo-controlled study. Arch Gen Psychiatry. 2005;62:1196–204.
Field JR, Walker AG, Conn PJ. Targeting glutamate synapses in schizophrenia. Trends Mol Med. 2011;17(12):689–98.
Moschetti V, Desch M, Goetz S, Liesenfeld K-H, Rosenbrock H, Kammerer KP, et al. Safety, tolerability and pharmacokinetics of oral BI 425809, a glycine transporter 1 Inhibitor, in healthy male volunteers: a partially randomised, single-blind, placebo-controlled, first-in-human study. Eur J Drug Metab Pharmacokinet. 2018;43(2):239–49.
Moschetti V, Schlecker C, Wind S, Goetz S, Schmitt H, Schultz A, et al. Multiple rising doses of oral BI 425809, a GlyT1 inhibitor, in young and elderly healthy volunteers: a randomised, double-blind, phase I study investigating safety and pharmacokinetics. Clin Drug Investig. 2018;38(8):737–50.
World Medical A. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–4.
International Conference on Harmonisation of technical requirements for registration of pharmaceuticals for human u. ICH harmonized tripartite guideline: guideline for Good Clinical Practice. J Postgrad Med. 2001;47(1):45–50.
Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20.
Danielson PB. The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans. Curr Drug Metab. 2002;3(6):561–97.
Desch M, Goettel M, Goetz S, Liesenfeld K-H, Chan T, Zhou J, et al. Effects of the potent cytochrome p450 3A4 inhibitor, itraconazole, on the pharmacokinetics of BI 425809, a new glycine transporter 1 (GlyT1) inhibitor. Clin Pharmacol Ther. 2017;101:S52.
Desch M. Effects of the potent cytochrome P450 3A4 inhibotor, itraconazole, on the pharmacokinetics of BI 425809, a new glycine transporter 1 (GLYT 1) inhibitor. Clin Pharmacol Ther. 2017;101(S1):S5–99.
Acknowledgements
Editorial support in the form of initial preparation of the outline based on input from all authors, and collation and incorporation of author feedback to develop subsequent drafts, assembling tables and figures, copyediting, and referencing was provided by Lesley Blogg, PhD and Lisa Auker, PhD, of Fishawack Communications Ltd, UK, and was funded by Boehringer Ingelheim International GmbH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
These studies were funded by Boehringer Ingelheim International GmbH (study numbers: 1346-0015 and 1346-0016; Clinicaltrials.gov identifiers: NCT03783000 and NCT03654170).
Conflicts of interest
UB, MD, YS, SRM, and CS are employees of Boehringer Ingelheim Pharma GmbH; GW, AMT, PL, and TSC are employees of Boehringer Ingelheim Pharmaceuticals Inc.
Ethics approval
The studies were conducted in accordance with the principles of the Declaration of Helsinki, the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Harmonized Tripartite Guideline for Good Clinical Practice (GCP), applicable regulatory requirements, and the standard operating procedures of the sponsor company (Boehringer Ingelheim International GmbH). The study procedures, protocols, and documents were reviewed and approved by the Independent Ethics Committee of the study center as well as the relevant local authorities.
Consent to participate
All participants provided a signed and dated written informed consent form in accordance with GCP and local regulatory and legal requirements, prior to admission to the study.
Consent to publish
Not applicable.
Availability of data and material
All publications reporting BI clinical trial data need to include this data disclosure statement to fulfil ICMJE requirements: “To ensure independent interpretation of clinical study results, Boehringer Ingelheim grants all external authors access to relevant material, including participant-level clinical study data, as needed by them to fulfill their role and obligations as authors under the ICMJE criteria. Clinical study documents and participant clinical study data are available to be shared on request after publication of the primary manuscript in a peer-reviewed journal, and if regulatory activities are complete and other criteria met as per the BI Policy on Transparency and Publication of Clinical Study Data (see Medical & Clinical Trials | Clinical Research | MyStudyWindow). Bona fide, qualified scientific and medical researchers are eligible to request access to the clinical study data with corresponding documentation describing the structure and content of the datasets. Upon approval, and governed by a Legal Agreement, data are shared in a secured data-access system for a limited period of 1 year, which may be extended upon request. Prior to providing access, clinical study documents and data will be examined, and, if necessary, redacted and de-identified, to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants. Researchers should use the https://vivli.org/ link to request access to study data and visit Medical & Clinical Trials | Clinical Research | MyStudyWindow for further information”.
Code availability
Not applicable.
Additional information
The original online version of this article was revised due to a retrospective Open Access order.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Burkard, U., Desch, M., Shatillo, Y. et al. The Absolute Bioavailability, Absorption, Distribution, Metabolism, and Excretion of BI 425809 Administered as an Oral Dose or an Oral Dose with an Intravenous Microtracer Dose of [14C]-BI 425809 in Healthy Males. Clin Drug Investig 42, 87–99 (2022). https://doi.org/10.1007/s40261-021-01111-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-021-01111-9