
Abstract
Background and Objectives
Carotegrast methyl, a novel prodrug, oral antagonist of α4-integrin, is in development for the treatment of active ulcerative colitis. This randomised, placebo-controlled, double-blind, crossover study evaluated the effect of food on the pharmacokinetics and pharmacodynamics as well as the safety profile after a single dose of carotegrast methyl in healthy male subjects.
Methods
Subjects were randomised to receive a single dose of carotegrast methyl (240, 480 or 960 mg) or placebo in a 6:2 ratio and received the study drug under both fed and fasted conditions separated by an 8-day washout. The pharmacokinetic profiles of carotegrast methyl and its active metabolite, carotegrast, were assessed. The pharmacodynamic profile was evaluated according to a change in the peripheral lymphocyte count. Safety was monitored throughout.
Results
Based on the area under the time curve from zero to the time of the last quantifiable concentration (AUClast), food reduced systemic exposure to both carotegrast methyl and carotegrast by 21–57% and 5–29%, respectively. The fed-to-fasted ratio of least square means for the increase in the lymphocyte count was almost at unity in each dose, indicating no food effect on pharmacodynamics. The time ≥ 90% of maximum effect was prolonged dose dependently, suggesting that a 960 mg-dose can provide a long-lasting effect. Reported adverse events were all mild.
Conclusions
Despite the reduced systemic exposure to both carotegrast methyl and carotegrast, food had no effect on the increase in lymphocyte count. A single administration of carotegrast methyl up to 960 mg was found to be safe.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Carotegrast methyl, a novel α4-integrin antagonist, is an esterified small molecule that enables oral administration. |
Food consumption reduced the systemic exposure of both carotegrast methyl and its active metabolite, carotegrast; however, the reduction noted for carotegrast was minimal at a 960-mg dose. |
A single dose of carotegrast methyl temporarily elevated circulating lymphocyte count; however, this pharmacodynamic effect was not affected by food intake and was prolonged in a dose-dependent manner. |
1 Introduction
Inflammatory bowel disease (IBD) is characterised by a relapsing idiopathic chronic inflammation affecting the gastrointestinal tract. The major types are Crohn’s disease (CD) and ulcerative colitis (UC). The reported prevalence of IBD is highest in Europe (UC, 505 per 100,000; CD, 322 per 100,000) and North America (UC, 286 per 100,000; CD, 319 per 100,000) [1]. Incidence rates of IBD are rapidly increasing worldwide, predominantly in countries that have adopted a Westernised lifestyle, including Japan [2]. The aetiology of IBD is not yet clearly understood, but it is certainly linked to multiple factors involving genetically susceptible individuals and environmental triggers that result in a dysregulated immune response to intestinal microbial flora [3, 4]. Pronounced infiltration of innate and adaptive immune cells into the lamina propria is a particular hallmark of active IBD [4,5,6]. As a result, disease-modifying treatment using an anti-integrin or anti-adhesion agent to inhibit leucocyte trafficking into inflamed tissues has been a focus of attention in the past decade [7]. In the clinical setting, new disease-modifying agents available for IBD treatment are mostly antibody biologics, which are invariably delivered by injection. An orally active drug could increase drug adherence with almost no immunogenicity.
Carotegrast methyl (INN) is a small-molecule α4-integrin antagonist that inhibits leucocyte extravasation into inflammatory sites by blocking the interaction of α4β1 or α4β7 integrins and their counter-receptors, VCAM-1 and MAdCAM-1 [8]. Carotegrast methyl is an esterified prodrug that has limited pharmacological action by itself. It is specifically designed to enhance oral bioavailability as it is hydrolysed to its active carboxylic acid, carotegrast, by carboxylesterase 1 (CES 1) in the liver. Carotegrast is distributed into target tissues through the systemic circulation. It is minimally reabsorbed into the enterohepatic circulation, primarily eliminated via the biliary route and excreted in faeces (unpublished data on file, EA Pharma Co., Ltd., Tokyo, Japan).
Carotegrast methyl shares the same mechanisms of action as natalizumab, which is approved for the treatment of CD in the USA but is associated with a risk of progressive multifocal leucoencephalopathy (PML), a rare opportunistic brain infection caused by John Cunningham virus (JCV) [9]. Three risk factors of developing natalizumab-associated PML have been identified: the presence of JCV antibodies in serum, prior use of an immunosuppressant, and treatment duration, especially extending beyond 2 years [10, 11]. Due to these safety concerns, carotegrast methyl has entered clinical development as short-term induction therapy in patients with IBD.
A first-in-human study, conducted in the Netherlands, found that a single dose of carotegrast methyl up to 160 mg was safe and tolerable in healthy male subjects [12]. Thereafter, two phase 1 studies were conducted in Japan that found that both single and multiple doses of carotegrast methyl of up to 320 mg for 6 days were safe and tolerable in healthy male subjects (multiple-dose study: unpublished data on file, EA Pharma Co., Ltd., Tokyo, Japan). In a single-dose study [13], we also investigated the effects of food on pharmacokinetics following an 80-mg dose of carotegrast methyl, the results of which suggested that food may interfere with drug absorption. Next, we conducted a phase 2 trial in patients with CD and found that multiple doses of carotegrast methyl up to 240 mg for 8 weeks were safe and tolerable; however, it failed to show efficacy in regard to the primary endpoint, namely a change in the CD activity index when compared with the placebo groups (unpublished data on file, EA Pharma Co., Ltd., Tokyo, Japan).
Taking these studies into consideration, we expected that an administration of more than 240 mg of carotegrast methyl three times daily would be required to demonstrate efficacy in patients with IBD. In this study, we aimed to assess the effect of food on the pharmacokinetics and pharmacodynamics of carotegrast methyl and carotegrast as well as the safety profile after a single oral administration of carotegrast methyl up to 960 mg in a dose-escalating manner in healthy male subjects.
2 Methods
2.1 Study Participants
Subjects were non-smoking, Japanese males aged 20–39 years, with a body mass index ≥ 18.5 and < 25.0 kg/m2. Eligible subjects had normal vital signs, laboratory tests, and ECG, no clinically significant medical history and were taking no medications or supplements that could interfere with procedures or compromise subject safety. Subjects were ineligible if their white blood cell (WBC) count was ≤ 3.0 × 103/µL and/or ≥ 8.5 × 103/µL. Other exclusion criteria included neurological symptoms and serious infectious diseases, including opportunistic infections within 1 year prior to administration of the drug.
2.2 Study Design
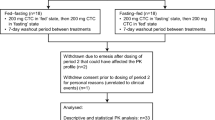
This was a single-centre, randomised, placebo-controlled, double-blind, two-period, crossover, food-effect study conducted at the CPC clinic (Kagoshima, Japan). Subjects were randomly assigned to receive 240 mg, 480 mg or 960 mg of the active drug (n = 6 per group) or the corresponding placebo (n = 2 per group), and were assigned to one of two sequences: fed–fasted or fasted–fed, according to the ascending order of screening numbers. Subjects received a single oral dose of the study drug once under fasted conditions (without breakfast) and once under fed conditions (with breakfast), separated by an 8-day washout period as described in Fig. 1. All subjects were admitted to the study centre on day − 2 for baseline assessments prior to the first treatment period and discharged on day 3. They were readmitted on day 7 for another baseline assessment prior to commencing the second treatment period and discharged on day 11, visiting on day 15 for follow-up examinations. For baseline assessments, subjects under fasted conditions did not take breakfast on 2 days: either days − 1 and 1 or days 8 and 9. In previous clinical phase 1 and phase 2 studies, a single dose of carotegrast methyl of up to 320 mg in healthy subjects and multiple doses of 240 mg of carotegrast methyl three times daily for 8 weeks in patients with active CD, respectively, were both found to be safe (unpublished data on file, EA Pharma Co., Ltd., Tokyo, Japan). Therefore, we commenced with the 240 mg, 480 mg and corresponding placebo administration, whilst the subsequent administration of the 960 mg and placebo took place following a safety evaluation by the investigator. The investigator discontinued participation if a subject showed any central nervous system-related neurological symptoms, such as motor and/or sensory paresis, cognitive impairment, aphasia or visual impairment, or if their total WBC count was ≤ 3.0 × 103/µL.

Study design. All subjects were admitted to the trial centre twice from day − 2 (evening) until day 3 (morning) and from day 7 (evening) until day 11 (morning). The subjects allocated into group A did not take breakfast on both day 8 and day 9 (fed–fasted sequence), similarly, the subjects allocated into group B did not take breakfast both day − 1 and day 1 (fasted–fed sequence). The orange arrow indicates a single administration of the study drug. The administration of 240 mg and 480 mg of the study drug was conducted in parallel. The blue arrow indicates safety examination performed to determine further progress of the study
Subjects were randomised based on a permuted block method (block size 8) and assigned by the investigator. Computer-generated randomisation sequences were prepared at BELLSYSTEM24, Inc. (Tokyo, Japan). Blinding was achieved by providing a placebo tablet identical in weight and appearance to the carotegrast methyl tablet, and subjects, investigators and data analysts were masked to treatment allocation. All food provided by the study centre consisted of standardised low-fat meals (approx. energy, 2050 kcal; protein, 75 g; fat, 54 g; carbohydrate, 310 g; sodium, 8.5 g per day with breakfast). When subjects were allocated to the fasted group, breakfast was removed from the standardised meals and only lunch and dinner were provided (approx. energy 1480 kcal; protein, 58 g; fat, 32 g; carbohydrate, 233 g; sodium, 6.4 g/day). No special dietary meals were provided and no additional food or drink (in particular, alcohol, caffeine and products containing grapefruit) was permitted during admission. To exclude any potential daily fluctuation in lymphocyte counts caused by different nutritional intakes, the following breakfast was provided at 8 a.m. on days − 1 and 1, and days 8 and 9: three bread rolls, one portion each of butter and strawberry jam, one boiled egg, one can of apple juice (160 g), and salt (energy, 574 kcal; protein, 17.5 g; fat, 21.2 g; carbohydrate, 79.0 g; sodium, 2.2 g). Subjects were required to finish meals at least 30 min before dosing and were not allowed to take any additional food or drink other than water until 4 h post-dose. Under fasted conditions, subjects received the study drug following an overnight fast of at least 13 h. Subjects were not allowed to lie down for at least 2 h post-dose.
2.3 Sample Collection and Analytical Methods
In order to quantify plasma carotegrast methyl and carotegrast concentrations, 2-mL venous blood samples were collected via an intravenous cannula at 0 (just before dosing), 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, 15 (days 1 and 9), 24 (days 2 and 10), 48 (days 3 and 11) h after dosing. Blood was collected in a blood collection tube containing an anticoagulant (VENOJECT® II, VP-H052K, sodium heparin, Terumo Corporation, Tokyo, Japan), and blood samples were centrifuged at 1200×g for 10 min at 4 °C. Thereafter, the plasma samples were stored at ≤ − 20 °C within 4 h of collection until analysis. Lymphocyte count was used as the pharmacodynamic marker and 2-mL venous blood samples were collected at 0, 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, 15 (days − 1 and 1; days 8 and 9), 24 (day 2 and day 10), 48 (day 3 and day 11) h after dosing. These collection points (on days − 1 and 1; days 8 and 9) were set to observe daily fluctuations in lymphocyte count. The pharmacodynamic analysis was conducted at CPC Clinic using a SP-3000 automated haematology analyser (Sysmex Corporation, Hyogo, Japan). Accumulated urine samples were collected pre-dose, every 4 h on days 1 and 9 and every 8 h on days 2 and 10. Urine samples were stored under acidic conditions (pH 3.0) at − 20 °C with a 5% 1 M sodium dihydrogen phosphate buffer to maintain the stability of the carotegrast methyl until analysis. Plasma and urinary concentrations of carotegrast methyl and carotegrast were measured using a liquid chromatography–tandem mass spectrometry (LC–MS/MS) assay at the Toray Research Center, Inc. (Kanagawa, Japan). The method utilised was developed and validated according to the recommendations of the US Food and Drug Administration (FDA) guidance on bioanalytical method validation [14]. A linear analytic range was used with a 1/x2 weighting between 0.5 and 500 ng/mL for the plasma concentration of analytes (carotegrast methyl and carotegrast) based on an r value greater than 0.999. Inter-day and intra-assay accuracy for the plasma concentration of analytes with low (1 ng/mL), medium (10 ng/mL) and high (400 ng/mL) quality control samples were within the recommended range (± 15%): 107.0–114.0% for carotegrast methyl and 103.7–113.9% for carotegrast. Inter-day and intra-assay precision (% coefficient of variation) for the plasma concentration was ≤ 2.5% for carotegrast methyl and ≤ 5.4% for carotegrast. The absolute recovery ranged between 83.7% and 98.8% for carotegrast methyl and 84.9% and 101.1% for carotegrast.
A linear analytic range was used with a 1/x2 weighting between 2.11 and 526 ng/mL for urinary analyte concentration based on an r value ≥ 0.997. Inter-day and intra-assay accuracy for urinary analyte concentration with low (5.26 ng/mL), medium (52.6 ng/mL) and high (421 ng/mL) quality control samples was within the FDA criteria [14] (± 15%): 98.1–110.6% for carotegrast methyl and 100.9–109.3% for carotegrast. Inter-day and intra-assay precision for each urinary analyte was ≤ 3.5% for both carotegrast methyl and carotegrast. The absolute recovery ranged between 78.3% and 86.8% for carotegrast methyl and 89.7% and 93.0% for carotegrast.
2.4 Pharmacokinetic Analysis
The main pharmacokinetic parameters assessed for carotegrast methyl and carotegrast were the maximum plasma concentration (Cmax) and the area under the plasma concentration–time curve from zero to the time of the last quantifiable concentration (AUClast); other secondary parameters are referred to in the footnote of Table 1. Descriptive pharmacokinetic analyses were conducted using a non-compartmental method with WinNonlin Professional Version 5.0.1 (Pharsight Corporation, St. Louis, MO, USA). The linear trapezoidal rule was used to estimate the AUC. Plasma drug concentrations below the limit of quantification were treated as zero. We did not impute any missing data and included outliers in the study data.
2.5 Safety Assessments
Safety variables included adverse event (AE) monitoring, physical examinations, clinical laboratory tests, electrocardiogram (ECG) and monitoring of vital signs and neurological symptoms. AEs were classified using the Medical Dictionary for Regulatory Activities (MedDRA), version 10.1. All safety assessments were conducted at screening and on days − 2, 1, 3, 7, 9, 11 and 15, and AEs were monitored throughout the study.
2.6 Data Analysis and Statistics
Descriptive statistics were used for all pharmacokinetic, pharmacodynamic, safety and demographic parameters. All statistical analyses were performed using SAS 8.2 (SAS Institute Inc., NC USA) at BELLSYSTEM24, Inc. (Tokyo, Japan). Statistical tests for significance were two‐sided, with the significance level was set at α = 0.05.
The natural logarithmic (ln) transformations of the pharmacokinetic parameters, except for time to reach Cmax (Tmax), were applied for all statistical inferences. The pharmacokinetic dose-proportionality in regard to Cmax and AUClast was analysed using a power model and was considered to have been demonstrated if the corresponding 95% confidence intervals (CIs) were within the predefined range of 0.7–1.3 [15]. The pharmacokinetic dose-proportionality in regard to terminal half-life (t1/2) and t1/2 from Tmax to 6 h (\(t_{{ 1/ 2\,[T_{ \rm{max} } - 6 {\text{h}}}]}\)) was analysed using an analysis of variance (ANOVA). The variability of the median Tmax was assessed using a Kruskal–Wallis test. All of these parameters were assessed under fed and fasted conditions, respectively.
The effect of food on Cmax and AUClast was assessed using a CI approach and it was considered to have no effect if a fed-to-fasted ratio of geometric least square means (LSMs) and the corresponding 90% CIs were within the predefined range of 0.8–1.25. All pharmacokinetic parameters, except for median Tmax, were analysed using an ANOVA with fixed factors of subject, dose, food and period. The evaluation of any food effect on Tmax was undertaken using a Wilcoxon’s signed-rank test.
The pharmacodynamic analyses were conducted by assessing changes from a time-matched baseline corresponding to the blood collection points for the lymphocyte count. The percentage change from the time-matched baseline at each measuring point was calculated using the following equation:
where X = the day after dosing of carotegrast methyl. The statistical significance of a change in the lymphocyte count between carotegrast methyl and placebo was assessed using a t-test.
2.7 Post Hoc Analyses
In order to clarify the effect of food on the pharmacodynamics of carotegrast methyl, we conducted post hoc analyses on the food effect on the area under the effect curve (AUEC), which was measured as a change in lymphocyte count from baseline, and pharmacokinetic-pharmacodynamic relationship. The food effect on the AUEC was analysed using the same CI approach as the effect on pharmacokinetic parameters. The pharmacokinetic-pharmacodynamic relationship between carotegrast plasma concentration and the percentage change in the lymphocyte count was assessed using a maximum effect (Emax) model. The time in which the change in the lymphocyte count was equal to 90% or greater than Emax (time ≥ 90% of Emax) at each dose was analysed using a non-compartmental method.
3 Results
3.1 Study Participants
This study was conducted between January and March 2008 at the CPC clinic (Kagoshima, Japan). Fifty subjects were screened and 24 were accepted for inclusion in the study. A total of 24 subjects received at least one dose of the study drug, therefore all of them were included in the pharmacokinetic, pharmacodynamic and safety analyses. However, one of the six subjects allocated to the 480-mg dose group was withdrawn on day 8 as he met the discontinuation criteria (WBC count ≤ 3.0 × 103/µL). As a result, only five subjects were included in the analyses for the 480-mg dose group under fed conditions (Supplemental Material, Fig. S1). Baseline demographics were generally similar across all dosing groups (Supplemental Material, Table S1).
3.2 Pharmacokinetics and Food Effect
The main pharmacokinetic parameters with geometric means of the analytes are summarised in Table 1 (arithmetic means of the pharmacokinetic parameters are available in Supplemental Material, Table S2) with the concentration–time profiles of carotegrast methyl and carotegrast under fasted and fed conditions shown in Fig. 2 (logarithmic scale; Supplemental Material, Fig. S2). The systemic exposure to carotegrast, based on AUClast, was higher than that of carotegrast methyl (4.6- to 6.1-fold higher under fasted conditions and 5.5- to 7.8-fold higher under fed conditions), reflecting the metabolism of the parent drug to its active metabolites. The fed-to-fasted LSM ratios of carotegrast methyl and carotegrast for Cmax and AUClast and the corresponding 90% CIs were all outside the 0.8–1.25 range across all dose groups, indicating the effect of food on these pharmacokinetic parameters (Table 2). The ANOVA with food as a fixed factor also demonstrated significant differences in carotegrast methyl Cmax (p = 0.002), AUClast (p = 0.011) and t1/2 (\(_{T_{ \rm{max} } - 6 {\text{h}}}\)) (p = 0.017). However, it only demonstrated significance in regard to carotegrast pharmacokinetic parameters for Cmax (p = 0.030) (Supplemental Material, Table S3). Wilcoxson’s signed-rank test confirmed no food effect on both carotegrast methyl and carotegrast median Tmax across all dose groups (data not shown).

Carotegrast methyl and carotegrast plasma concentration–time curves under fasted and fed conditions. Note that the concentration of carotegrast (active metabolite, bottom) was much higher than that of carotegrast methyl (prodrug, top) and that the plasma concentrations showed decrease under fed conditions (red) compared to fasted conditions (blue) at each dose. Data are presented as the mean (+ standard deviation) of the concentration
The power model analysis could not establish the dose proportionality for carotegrast methyl and carotegrast Cmax and AUClast, regardless of fed or fasted conditions at all doses tested as the 95% CIs did not fall within the predefined window. The systemic exposure of both carotegrast methyl and carotegrast showed an increase in a sub-proportional manner (Supplemental Material, Table S4). The Kruskal-Wallis test showed no variability in median Tmax among dose groups under either fed (carotegrast methyl, p = 0.882; carotegrast p = 0.726) or fasted (carotegrast methyl, p = 0.683; carotegrast p = 1.000) conditions. ANOVA showed no statistical significance among the three dose groups for carotegrast methyl t1/2 and t1/2 (\(_{T_{ \rm{max} } - 6 {\text{h}}}\)) or for carotegrast t1/2, t1/2 (\(_{T_{ \rm{max} } - 6 {\text{h}}}\)) and under fed or fasted conditions, thus indicating no dose-proportionality of these pharmacokinetic parameters (Supplemental Material, Table S5).
3.3 Pharmacodynamics
While the majority of subjects showed a daily fluctuation in their peripheral lymphocyte count on day − 1 [16,17,18], there was no remarkable difference in daily fluctuations between day − 1 and day 1 in the placebo group (data not shown).
The percentage change in the lymphocyte count from baseline was increased across all dose groups under both fed and fasted conditions following a single dose of carotegrast methyl (Fig. 3). The fed-to-fasted ratios of geometric LSMs of the AUEC in regard to the change in the lymphocyte count were 0.99, 0.94 and 1.01 at 240 mg, 480 mg and 960 mg doses, respectively, and the ratio of LSMs and the corresponding CIs, for all doses together, were between 0.8 and 1.25 [1.03 (0.88–1.20)], thus indicating the absence of a food effect. The point estimates (standard error) of Emax and half maximal effective concentration (EC50), which were directly obtained from the percentage change in the lymphocyte count from baseline and carotegrast plasma concentrations, were 61.45 (2.97)% and 10.26 (1.93)%, respectively. Although the mean (SD) time, where the change in the lymphocyte count was ≥ 90% of Emax, was prolonged in a dose-dependent manner in both fed and fasted conditions, there was no obvious difference in the mean (SD) time at 960 mg between fed and fasted conditions (Table 3).

Significant increase in lymphocyte count under both fasted (blue) and fed (red) conditions following a single administration of carotegrast methyl. The percentage increases in lymphocyte counts were significant post-dose 1–12 h at the dose of 240 mg, 0.5–15 h at 480 mg and 0.5–12 h at 960 mg under fasted conditions, and were significant post-dose 0.5–8 h at 240 mg, 0.25–8 and 15 h at 480 mg, and 1–8 and 15 h at 960 mg under fed conditions compared with placebo. Data are presented as mean ± SD
3.4 Safety
There were no serious AEs or AEs that led to subject discontinuation and all AEs reported were mild. In fasted conditions, one (16.7%) subject experienced increases in C-reactive protein and WBC count (i.e. more than 10,000 WBC count) at the 240-mg dose, and one (16.7%) subject experienced acute tonsillitis at the 480-mg dose. In fed conditions, one (16.7%) subject experienced an increase in WBC count at the 480-mg dose, and one (20.0%) subject experienced pharyngolaryngeal pain at the 960-mg dose (Table 4). All AEs, other than tonsillitis, were evaluated as adverse drug reactions possibly related to the study drug. No clinically relevant changes in vital signs, ECG or physical examination were noted during the study.
4 Discussion
Carotegrast methyl was designed to provide favourable oral bioavailability. After a single dose of carotegrast methyl, the majority of the drug was metabolised to the active metabolite, carotegrast. The power model did not demonstrate the dose proportionality of Cmax and AUClast of these analytes. This study demonstrated that food reduced the systemic exposure of carotegrast methyl and carotegrast but did not influence the increase in lymphocyte count. The time ≥ 90% of Emax was prolonged dose-dependently regardless of fed or fasted conditions. This may be associated with our finding that the systemic exposure increased with dose increment although the increase was found to be sub-proportional.
The concentration–time profiles of carotegrast methyl and carotegrast, as well as the food-effect analysis, indicated the effect of food on the pharmacokinetics of both carotegrast methyl and carotegrast. Systemic exposure, based on Cmax and AUClast, was reduced by 34–68% and 21–57% for carotegrast methyl and by 19–42% and 5–29% for carotegrast, respectively, following a low-fat meal compared to fasting (Table 2). It is noteworthy that these reductions were more evident on the pharmacokinetics of carotegrast methyl than carotegrast, and only a 5% reduction was observed for carotegrast AUClast at the 960-mg dose under fed conditions. The prolongation of carotegrast methyl t1/2 (\(_{T_{ \rm{max} } - 6 {\text{h}}}\)) under fed conditions was also observed when compared to fasted conditions (Supplemental Material, Table S3) although there were no differences in carotegrast methyl t1/2 (\(_{T_{ \rm{max} } - 6 {\text{h}}}\)) among the three dose groups under either fed or fasted conditions (Table S5). This suggests that food intake may interfere with the absorption of carotegrast methyl. Again, it should be noted that the prolongation of t1/2 (\(_{T_{ \rm{max} } - 6 {\text{h}}}\)) under fed conditions was not markedly observed in carotegrast. Overall, the effect of food on systemic exposure to carotegrast methyl appeared to be more extensive than that of carotegrast. The reason for this is unknown. Considering that the AUClast of carotegrast methyl did not establish dose proportionality, the enzyme CES 1, which is involved in the metabolism of carotegrast methyl, is very unlikely to be inhibited or saturated as a result of carotegrast methyl administration.
The post hoc analyses confirmed an absence of food effect on the change in lymphocyte count following a single dose of carotegrast methyl. This intrigued us as the food effect on the pharmacokinetic parameters (Cmax and AUClast) of neither carotegrast methyl nor carotegrast demonstrated comparable bioavailability and systemic exposure decreased under fed conditions compared to fasting. The absence of a food effect on the pharmacological action could be explained by a reduced food effect on exposure to carotegrast rather than carotegrast methyl, especially at the 960-mg dose. It is important to note that the pharmacodynamic effect was prolonged in a dose-dependent manner; however, there was no obvious difference in the time ≥ 90% of Emax between fed and fasted conditions at the 960-mg dose. This may be due to a minimal food effect on plasma carotegrast concentrations at the 960-mg dose.
Natalizumab, which is widely used for the treatment of relapsing-remitting multiple sclerosis as well as CD, elicited a 30–90% increase in the peripheral lymphocyte count from baseline [19]. Following the administration of natalizumab, α4-integrin saturation levels immediately increased by more than 90% and were sustained at over 70% for 4 weeks [20]. In this study, all doses of carotegrast methyl were estimated to prompt a 60% increase in the peripheral lymphocyte count, with the 960-mg dose providing the longest pharmacodynamic effect (approximately 6 h in fed conditions) with respect to the time achieved above 90% of Emax. This result is in line with our multiple-dose study, which demonstrated that the administration of a 960-mg dose of carotegrast methyl three times daily prompted a 24-h sustained increase in peripheral lymphocytes that was not present following the 240- or 480-mg dose [21]. Our assertion that a 960-mg dosing regimen is required for clinical efficacy is supported by a Japanese phase 2 study in patients with active UC in which 960 mg of carotegrast methyl three times daily for 8 weeks demonstrated clinical efficacy compared to placebo [22]. Similarly, another phase 2 study with a 480-mg dosing regimen failed to show efficacy (unpublished data on file, EA Pharma Co., Ltd., Tokyo, Japan). A large-scale, long-term phase 3 trial investigating the efficacy and safety of 960 mg of carotegrast methyl three times daily is currently ongoing (ClinicalTrials.gov Identifier: NCT03531892).
In this study, two subjects reported AEs in fasted conditions and two subjects reported AEs in fed conditions. Overall, the safety profile of a single dose of carotegrast methyl was favourable and comparable in both fed and fasted conditions. Carotegrast methyl up to a 960-mg dose was well tolerated and had no clinically significant effect on vital signs, ECGs, physical or neurological examinations. All AEs reported were mild and transient.
The limitation of this study is the small sample size (n = 6 per group) used to analyse the effect of food on pharmacokinetic and pharmacodynamic properties. The design of this study was not in line with FDA guidance [23], where a minimum of 12 subjects and high-fat meals are recommended in order to evaluate a food effect. This study was conducted using a low-fat meal as it is representative of a typical breakfast for both a Japanese population and patients with UC. Given that this drug may be administered three times daily after each meal, a further food interaction study with a high-fat meal would be required.
5 Conclusion
In conclusion, a single dose of carotegrast methyl temporarily increased the peripheral lymphocyte count. This increase was not affected by food, particularly at the 960-mg dose in which the effect of food was lowest on the plasma concentration of carotegrast. The 960-mg dose was estimated to provide a prolonged pharmacodynamic effect, especially in terms of a sustained increase in the lymphocyte count. In this phase 1 study, a single dose of carotegrast methyl up to 960 mg was well tolerated in healthy Japanese male subjects.
References
Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2018;390(10114):2769–78. https://doi.org/10.1016/s0140-6736(17)32448-0.
Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46–54. https://doi.org/10.1053/j.gastro.2011.10.001(e42).
de Lange KM, Barrett JC. Understanding inflammatory bowel disease via immunogenetics. J Autoimmun. 2015;64:91–100. https://doi.org/10.1016/j.jaut.2015.07.013.
Zhang YZ, Li YY. Inflammatory bowel disease: pathogenesis. World J Gastroenterol. 2014;20(1):91–9. https://doi.org/10.3748/wjg.v20.i1.91.
Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369(9573):1627–40. https://doi.org/10.1016/s0140-6736(07)60750-8.
Eksteen B, Liaskou E, Adams DH. Lymphocyte homing and its role in the pathogenesis of IBD. Inflamm Bowel Dis. 2008;14(9):1298–312. https://doi.org/10.1002/ibd.20453.
Park SC, Jeen YT. Anti-integrin therapy for inflammatory bowel disease. World J Gastroenterol. 2018;24(17):1868–80. https://doi.org/10.3748/wjg.v24.i17.1868.
Sugiura T, Kageyama S, Andou A, Miyazawa T, Ejima C, Nakayama A, et al. Oral treatment with a novel small molecule alpha 4 integrin antagonist, AJM300, prevents the development of experimental colitis in mice. J Crohns Colitis. 2013;7(11):e533–42. https://doi.org/10.1016/j.crohns.2013.03.014.
Stuve O, Bennett JL. Pharmacological properties, toxicology and scientific rationale for the use of natalizumab (Tysabri) in inflammatory diseases. CNS Drug Rev. 2007;13(1):79–95. https://doi.org/10.1111/j.1527-3458.2007.00003.x.
Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366(20):1870–80. https://doi.org/10.1056/NEJMoa1107829.
Ho PR, Koendgen H, Campbell N, Haddock B, Richman S, Chang I. Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: a retrospective analysis of data from four clinical studies. Lancet Neurol. 2017;16(11):925–33. https://doi.org/10.1016/s1474-4422(17)30282-x.
Japan Clinical Trials Information [Internet]: Japan Pharmaceutical Information Center (Japic). Indentifier JapicCTI-195009 an ascending single dose, randomised, double-blind, placebo-controlled phase 1 study of the tolerability, pharmacokinetics and pharmacodynamics of AJM300 in healthy adult males in the fasting and fed states; 16 Oct 2019; [1 page]. 2005. https://www.clinicaltrials.jp/cti-user/trial/Search.jsp. Accessed 12 Nov 2019.
Japan Clinical Trials Information [Internet]: Japan Pharmaceutical Information Center (Japic). Indentifier JapicCTI-195018 Phase 1 study to investigate safety, tolerability, pharmacokinetics and pharmacodynamics of single oral doses of AJM300 in healthy male subjects―randomised, double-blind, controlled trial. 28 Oct 2019; [1 page]. 2005. https://www.clinicaltrials.jp/cti-user/trial/Search.jsp. Accessed 12 Nov 2019.
U.S. Department of Health and Human Services Food and Drug Administration. Bioanalytical method validation Guidance for industry. 2001. https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf. Accessed 1 Mar 2019.
Igarashi T, Yabe T, Noda K. Study design and statistical analysis of toxicokinetics: a report of JPMA investigation of case studies. J Toxicol Sci. 1996;21(5):497–504.
Miyawaki T, Taga K, Nagaoki T, Seki H, Suzuki Y, Taniguchi N. Circadian changes of T lymphocyte subsets in human peripheral blood. Clin Exp Immunol. 1984;55(3):618–22.
Suzuki S, Toyabe S, Moroda T, Tada T, Tsukahara A, Iiai T, et al. Circadian rhythm of leucocytes and lymphocytes subsets and its possible correlation with the function of the autonomic nervous system. Clin Exp Immunol. 1997;110(3):500–8.
Scheiermann C, Kunisaki Y, Frenette PS. Circadian control of the immune system. Nat Rev Immunol. 2013;13(3):190–8. https://doi.org/10.1038/nri3386.
Ghosh S, Goldin E, Gordon FH, Malchow HA, Rask-Madsen J, Rutgeerts P, et al. Natalizumab for active Crohn’s disease. N Engl J Med. 2003;348(1):24–32. https://doi.org/10.1056/NEJMoa020732.
European Medical Agency (EMA). Tysabri: EPAR—scientific discussion. 2007. https://www.ema.europa.eu/documents/scientific-discussion/tysabri-epar-scientific-discussion_en.pdf. Accessed 12 Nov 2019.
Fukase H, Kajioka T, Oikawa I, Ikeda N, Furuie H. AJM300, a novel oral antagonist of alpha4-integrin, sustains an increase in circulating lymphocytes: a randomised controlled trial in healthy male subjects. Br J Clin Pharmacol. 2019. https://doi.org/10.1111/bcp.14151.
Yoshimura N, Watanabe M, Motoya S, Tominaga K, Matsuoka K, Iwakiri R, et al. Safety and efficacy of AJM300, an oral antagonist of alpha4 integrin, in induction therapy for patients with active ulcerative colitis. Gastroenterology. 2015;149(7):1775–83. https://doi.org/10.1053/j.gastro.2015.08.044(e2).
U.S. Food and drug administration. Guidance for industry: food-effect bioavailability and fed bioequivalence studies. 2002. https://www.fda.gov/media/70945/download. Accessed 1 Apr 2019.
Acknowledgements
The authors thank the subjects for their participation, the staff at the CPC clinic and EA Pharma Co., Ltd. for their kind assistance in the conduct of the study as well as for data management and processing. The authors acknowledge the Toray Research Center, Inc. for the pharmacokinetic analysis and BELSYSTEM24 Holdings, Inc. (currently A2 Healthcare Corporation) for the statistical analysis. We would also like to thank Mie Yamamoto, PhD (SunFlare Co., Ltd.), who wrote the first draft of the manuscript based on input from the authors and revised subsequent drafts, and Jo Whitcombe (SunFlare Co., Ltd.), for proofreading of the manuscript. This study was funded by EA Pharma Co., Ltd. and the medical writing assistance and the proofreading were funded by EA Pharma Co., Ltd. and Kissei Pharmaceutical Co., Ltd.
Author information
Authors and Affiliations
Contributions
All authors were involved in study design and carried out the study. HF participated in data acquisition and analysis, IO carried out the statistical analysis and, together with the rest of the authors, interpreted the data. All authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript and critically reviewed and approved the final version of the manuscript for submission.
Corresponding author
Ethics declarations
Conflict of interest
T. Kajioka, I. Oikawa and N. Ikeda are employees of EA Pharma Co., Ltd. H Furuie received consultancy fees from EA Pharma Co., Ltd. H. Fukase received funds for conducting clinical investigations in connection with this trial.
Ethical approval
The study was approved by the CPC Clinic ethics committee, Kagoshima, Japan (Reference number: CPC2008-05). All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional review board and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Written informed consent was obtained from all individual participants prior to study enrolment.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Fukase, H., Kajioka, T., Oikawa, I. et al. Food Effect on a Single High Dose of Carotegrast Methyl, an Oral Antagonist of α4-Integrin, in Healthy Male Subjects: A Randomised, Placebo-Controlled, Double-Blind Study. Clin Drug Investig 40, 237–247 (2020). https://doi.org/10.1007/s40261-019-00879-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-019-00879-1