
Abstract
Background
Tapentadol prolonged release (PR; 100–250 mg twice daily) has been efficacious and well tolerated for managing moderate-to-severe, chronic osteoarthritis hip or knee pain in phase 3 studies with washout of previous analgesic treatment.
Objective
The objective of this study was to evaluate the effectiveness and tolerability of tapentadol PR (50–250 mg twice daily) after direct rotation from World Health Organization (WHO) step III opioids in patients with severe osteoarthritis knee pain who previously responded to WHO step III therapy but showed poor tolerability.
Methods
This open-label, phase 3b study (NCT00982280) was conducted from October 2009 through June 2010 (prematurely terminated due to slow recruitment and study drug shortages) in clinical care settings in Europe and Australia. The study population included patients with severe, chronic osteoarthritis knee pain who had taken WHO step III opioids daily for ≥2 weeks before screening, responded to therapy (average pain intensity [11-point numerical rating scale-3 (NRS-3)] ≤5 at screening), and reported opioid-related adverse effects as their reason for changing analgesics. Patients switched directly from WHO step III therapy to tapentadol. Patients received oral tapentadol PR (50–250 mg twice daily) during 5-week titration and 7-week maintenance periods. Oral tapentadol immediate release (IR) was permitted (≤twice/day, ≥4 h apart) for acute pain episodes due to index pain or withdrawal symptoms following discontinuation of previous opioids (combined dose of tapentadol [PR and IR] ≤500 mg/day). This study was planned to evaluate conversion to tapentadol PR, based on responder rate 1 (percentage of patients with same/less pain [NRS-3] versus Week −1) at Week 6 (primary endpoint), adverse events (AEs), and discontinuation rates. Equianalgesic ratios were calculated for tapentadol prior to WHO step III opioids (PR and PR plus IR formulations).
Results
Of 82 patients enrolled, 63 received study medication. In the per-protocol population, responder rate 1 at Week 6 (last observation carried forward) was 94.3 % (50/53; P < 0.0001 vs. the null hypothesis rate [<60 %]). Mean (standard deviation) pain intensity scores were 4.7 (0.66) at baseline, 2.5 (1.46) at Week 6, and 1.8 (1.41) at Week 12 in the main analysis population (change from baseline at Weeks 6 and 12, P < 0.0001). Tapentadol to transdermal buprenorphine equianalgesic ratios (PR [n = 48], 262.9:1; PR plus IR [n = 48], 281.1:1) and tapentadol to oral oxycodone equianalgesic ratios (PR [n = 4], 4.3:1; PR plus IR [n = 6], 4.6:1) were calculated for the main analysis population. In the safety population, prevalence of AEs reported as associated with prior opioids at Week −1 (reasons for rotation) and related to tapentadol treatment at Week 12 decreased over time; the most common were nausea (46.0 vs. 24.1 %) and constipation (31.7 vs. 7.4 %). Overall, 14.3 % of patients discontinued the study early; reasons included AEs (9.5 %), lack of efficacy (3.2 %), and withdrawal of consent (1.6 %).
Conclusions
Significant improvements in effectiveness were observed for tapentadol PR (50–250 mg twice daily) versus WHO step III opioids in patients with severe, chronic osteoarthritis knee pain who previously responded to WHO step III therapy. Equianalgesic ratios were calculated for tapentadol to transdermal buprenorphine and oral oxycodone and were in line with observations from previous phase 3 studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Opioids are used to manage osteoarthritis pain in patients who have not responded to more conservative pharmacological options [1–3] and are effective for the relief of moderate-to-severe, chronic osteoarthritis pain [4–6]. Evidence suggests that disturbed descending pain inhibition often plays a role in osteoarthritis pain [7–10]. The prevalence of chronic pain following joint replacement surgery ranges from 27 to 44 % [11–13]; central sensitization associated with disturbed descending pain inhibition is thought to contribute to this pain [14]. Central sensitization may lead to variations in patient response to opioid therapy [7, 9]. Constant nociceptive pain resulting from cartilage degradation [15] and disruption of descending inhibitory pain pathways may also contribute to osteoarthritis pain [7, 16, 17]. The multi-mechanistic origin of osteoarthritis pain may complicate pain control; analgesics with multiple mechanisms of analgesic activity, including those that target descending pain pathways (e.g., noradrenaline reuptake inhibition), may be more effective than those with a single mechanism of action (e.g., opioids) [7, 16, 17]. Long-term opioid therapy offers only moderate benefits for patients with osteoarthritis pain and is often associated with poor tolerability that may lead patients to discontinue opioid treatment or switch to a different opioid, resulting in interruption of pain control [4, 18–21].
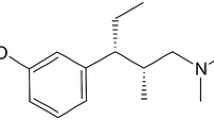
Tapentadol is a centrally acting analgesic with two mechanisms of action, μ-opioid receptor agonism and noradrenaline reuptake inhibition [22, 23]. Previous phase 3 studies have shown that tapentadol prolonged release (PR; 100–250 mg twice daily) is effective and well tolerated for managing moderate-to-severe, chronic pain, such as osteoarthritis pain [24–26], low back pain [25–27], and pain related to diabetic peripheral neuropathy [28, 29]. Most (>75 %) patients in those studies reported severe pain at baseline [24–28]. A separate phase 3b study demonstrated the effectiveness and tolerability of tapentadol PR in patients with severe, chronic osteoarthritis knee pain who had not responded adequately to World Health Organization (WHO) step I or II analgesics or co-analgesics or who were not receiving regular analgesic treatment [30]. The current multicenter, multinational, open-label phase 3b study (NCT00982280) evaluated the effectiveness and tolerability of tapentadol PR in patients with severe, chronic osteoarthritis knee pain who had responded to WHO step III opioid therapy but showed a lack of tolerability; direct rotation of these patients from their previous WHO step III treatment to tapentadol PR was studied and equianalgesic ratios of tapentadol to prior WHO step III opioids were evaluated.
2 Methods
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice guidelines, and applicable local laws. Prior to study enrollment, all patients signed an informed consent document. The study protocol, patient information sheet, and informed consent form were approved by independent ethics committees.
2.1 Patient Population
This study included men and non-pregnant, non-lactating women diagnosed with knee osteoarthritis based on the following American College of Rheumatology criteria: knee pain and radiographic osteophytes or knee pain, ≥40 years of age, morning stiffness of <30 min duration, and crepitus on motion. Eligible patients must have been experiencing pain requiring a strong (WHO step III) analgesic at the reference joint for ≥3 months. The target population included patients who had been taking WHO step III opioids for ≥2 weeks before screening and had responded to that treatment (average pain intensity during the last 3 days before screening ≤5 on an 11-point numerical rating scale-3 [NRS-3; recalled 3-day average pain intensity (11-point NRS); 0 = “no pain” to 10 = “pain as bad as you can imagine”]). Eligible patients had to report opioid-related adverse effects as their reason for changing analgesics, and subject satisfaction with treatment ratings could not be better than “fair” (5-point verbal rating scale; 0 = “poor” to 4 = “excellent”).
Patients were excluded if they had a history of alcohol or drug abuse; severe renal impairment; moderate or severe hepatic impairment; active hepatitis B or C within 3 months of screening; reported HIV infection (HIV testing was not performed); seizure disorder or epilepsy; mild or moderate traumatic brain injury, stroke, transient ischemic attack, or brain neoplasm within the past year; or severe traumatic brain injury within the past 15 years or residual sequelae suggesting potential transient changes in consciousness. Patients were also excluded if they had concomitant autoimmune inflammatory conditions; osteoarthritis in a flare state; history and clinical signs of crystal-induced, metabolic, infectious, or autoimmune disease at the reference joint; any painful procedure required during the study that could affect efficacy or safety assessments; or the presence of painful conditions other than osteoarthritis of the reference joint that could confound self-assessment of pain. Patients with osteoarthritis at joints other than the reference joint were not excluded from the study if the main source of their pain and disability was the reference joint.
Use of monoamine oxidase inhibitors within 14 days of screening or intra-articular injections of hyaluronic acid at the reference joint within 3 months of screening was prohibited. Patients were permitted to take selective serotonin reuptake inhibitors if they had been taking stable doses for ≥30 days prior to screening and if doses remained stable throughout the study.
2.2 Study Design
This open-label, multicenter, phase 3b study included a 1-week observation period under the previous WHO step III regimen (Week −1, starting with screening and ending with baseline), a 5-week titration and stabilization period, and a 7-week maintenance period. All WHO step III analgesics and potential concomitant WHO step II analgesics were discontinued at the end of the observation period. The average total daily dose (TDD) of WHO step III analgesics (including all formulations of all opioids taken) used over the last 3 days before the baseline visit were converted into a morphine equivalent dose (MED). The starting dose of tapentadol PR (50, 100, or 150 mg twice daily) was determined based on the MED range for the patient’s previous WHO step III analgesic(s) (Table 1).
An interim visit occurred 3–4 days after the baseline visit (i.e., 3–4 days after initiation of study treatment). A first titration corresponding with the interim visit was allowed; doses were then titrated on a weekly basis to the dose providing an optimal balance of pain relief and tolerability within the therapeutic range of tapentadol PR 50–250 mg twice daily. Titration was continued until patients achieved at least similar pain relief (i.e., the same or less pain intensity [11-point NRS-3]) compared with the previous WHO step III analgesic treatment.
Throughout the study, patients continued taking any WHO step I analgesics, co-analgesics, or medications used to control adverse effects related to the prior opioid regimen at the same stable dose, unless they were participating in a tapering substudy as described below. Laxatives could be discontinued if laxative-induced diarrhea occurred after patients switched to tapentadol PR. Patients were permitted to take tapentadol immediate release (IR) 50 mg (≤twice/day, ≥4 h apart) as backup medication throughout the study for acute pain episodes due to index pain or for withdrawal symptoms that occurred during the first days of the titration period following discontinuation of the previous opioid (combined TDD of tapentadol IR and PR, ≤500 mg). During the maintenance period, patients continued on the dose of tapentadol PR determined during titration. For Substudy A, one concomitant WHO step I analgesic or co-analgesic was tapered from Week 9 to Week 11 until the analgesic or co-analgesic was stopped or the patient’s pain intensity score increased; co-analgesics had priority for tapering. For Substudy B, concomitant medications used to control adverse effects related to the previous opioid analgesic were tapered and stopped during Week 7.
2.3 Study Evaluations
Study evaluations were performed using one or more of the following populations: safety (all patients who took ≥1 dose of study drug), main analysis (all patients who took ≥1 dose of study drug and had ≥1 post-baseline pain intensity assessment), and per-protocol (subset of the main analysis population; all patients who received treatment up to and including Week 6 and had no major protocol deviations).
The primary endpoint was responder rate 1 (percentage of patients with the same/less pain compared with Week −1 [on previous WHO step III analgesic]) at Week 6 in the per-protocol population, using the last observation carried forward (LOCF) for imputing missing pain intensity assessments. Responder rate 2 (percentage of patients with the same/less pain and improvement of ≥1 category in subject satisfaction with treatment compared with baseline) at Week 6 (LOCF) was a secondary endpoint in the per-protocol population. Responder rates 1 and 2 were also evaluated at Weeks 6, 8, and 12 in the main analysis population using observed-case analysis. Observed-case analyses for responder rates and all other endpoints included results for all patients who received ≥1 dose of tapentadol PR and had ≥1 post-baseline pain intensity assessment. Additional analyses included pain intensity ratings (NRS-3), subject satisfaction with treatment, Patient Global Impression of Change (PGIC) [31, 32], Clinician Global Impression of Change (CGIC) [33], Western Ontario and McMaster Universities (WOMAC) Osteoarthritis Index [34], EuroQol-5 Dimension (EQ-5D) [35], Short Form-36 (SF-36) [36], Hospital Anxiety and Depression Scale (HADS) [37], and a 4-item sleep questionnaire [38].
Safety and tolerability assessments included adverse event (AE) reporting and laboratory and vital sign evaluations. AEs were categorized as treatment-emergent AEs (TEAEs; AEs that occurred after the first intake of study drug or increased in intensity, frequency, or quality during treatment with study drug), and non-TEAEs (NTEAEs; AEs that occurred or were present prior to the first intake of study drug, including ongoing medical history). AEs were considered at least possibly related to study drug administration if there was evidence suggesting a causal relationship and unlikely or not related if there was no evidence of a causal relationship. AEs were evaluated for at least a possible association with any WHO step III analgesics and co-analgesics. Incidence, intensity, duration, and causality of all AEs were analyzed. The prevalence of AEs reported as associated with treatment at Week −1 (on WHO step III treatment) and related to treatment at Week 12 (on tapentadol PR treatment) were compared.
2.4 Statistical Analyses
A sample size of 178 patients was required to provide 80 % power to perform all three of the following analyses in a stepwise manner (given the rejection of the null hypothesis at the first two steps): comparison of a responder rate 1 of ≥60 % and the null hypothesis responder rate (<60 %; non-inferiority margin, 14.3 %), comparison of a responder rate 2 of ≥60 % and the null hypothesis responder rate (<60 %; non-inferiority margin, 14.3 %), and rejection of the null hypothesis that mean pain intensity score at Week 6 was not equivalent to that at Week −1 (i.e., the difference in means was ≥0.673 away from 0) in favor of the alternate hypothesis that responder rate 1 was ≥60 %.
A one-sample Chi-square test was used to analyze responder rates 1 and 2 in the per-protocol population at Week 6 (LOCF) and in the main analysis population at Weeks 6, 8, and 12 (observed-case). A one-sample paired t-test was used to analyze the changes from baseline to Weeks 6, 8, and 12 in mean pain intensity, WOMAC subscale and global scores, EQ-5D health status index and patient-rated health state (100-mm visual analog scale [VAS]) scores, HADS anxiety and depression subscale scores, and SF-36 subscale and summary scores.
Equianalgesic ratios of tapentadol to previous WHO step III analgesics were calculated. The mean TDD (average TDD during the 3 days prior to the visit) of tapentadol at which a pain intensity score (NRS-3) equivalent to or less than the pain intensity score at baseline (on previous WHO step III regimen) was reached was defined as the equipotent dose. The corresponding mean TDD of WHO step III analgesic was the average of the TDDs taken during the 3 days before baseline. Equianalgesic ratios were calculated for combined tapentadol PR and tapentadol IR to combined WHO step III PR and IR opioid analgesics and for tapentadol PR to WHO step III PR opioid analgesics.
Analyses were performed using two separate datasets (one that included results from Weeks 9–12 for patients who participated in Substudy A and one that excluded those results) because of the possibility that tapering of WHO step I analgesics and co-analgesics during Weeks 9 through 12 might result in pain peaks that could influence effectiveness, function, and quality-of-life analyses. Results presented here are for the dataset that included results from Weeks 9 through 12 for patients who participated in Substudy A in the main analysis population using observed-case analysis (unless otherwise specified); results of analyses for the dataset that excluded results from Weeks 9 through 12 for patients who participated in Substudy A using observed-case analysis and LOCF are summarized in the Electronic Supplementary Material.
3 Results
3.1 Patients
The numbers of patients in the safety (n = 63), main analysis (n = 62), and per-protocol (n = 53) populations were lower than initially planned because this study was terminated prematurely due to slow recruitment and study drug shortages. Baseline and demographic characteristics for the safety population are summarized in Table 2. In the safety population, the mean (standard deviation [SD]) duration of osteoarthritis knee pain was 6.45 (5.92) years. Overall, 14.3 % (9/63) of patients in the safety population discontinued the study early for reasons including AEs (9.5 % [6/63]), lack of efficacy (3.2 % [2/63]), and withdrawal of consent (1.6 % [1/63]).
During Week −1, 14.3 % (9/63) of patients were taking co-analgesics, 52.4 % (33/63) were taking WHO step I analgesics, 11.1 % (7/63) were taking WHO step II opioid analgesics, and 100 % (63/63) were taking WHO step III opioid analgesics (according to the inclusion criteria). The WHO step III analgesics taken during Week −1 were buprenorphine (transdermal system; 77.8 % [49/63]), oxycodone (15.9 % [10/63]), hydromorphone (6.3 % [4/63]), morphine (4.8 % [3/63]), and methadone (1.6 % [1/63]). The mean release rate of transdermal buprenorphine (the most commonly used WHO step III analgesic during Week −1) was 21.46 μg/h, corresponding to an average buprenorphine dose of about 0.52 mg/day. The MEDs of the TDDs of WHO step III opioids that patients were taking at baseline are summarized in Table 3.
Concomitant WHO step I analgesics were taken by 55.6 % (35/63) of patients and concomitant co-analgesics were taken by 14.3 % (9/63) of patients. A total of 55.6 % (35/63) of patients took concomitant medications to treat adverse effects, including adverse effects related to their previous opioid therapy.
3.2 Effectiveness, Function, and Quality-of-Life
In the per-protocol population, responder rate 1 (percentage of patients with the same/less pain compared with Week −1) at Week 6 (LOCF) was 94.3 % (50/53), which was significantly different from the null hypothesis responder rate 1 of <60 % (P < 0.0001). Responder rate 2 (percentage of patients with the same/less pain and an improvement of ≥1 category in subject satisfaction with treatment) at Week 6 (LOCF) was 92.5 % (49/53) in the per-protocol population and was significantly different from the null hypothesis responder rate 2 of <60 % (P < 0.0001). In the main analysis population (n = 62), responder rates 1 and 2 increased from the interim visit to Week 4 (observed-case analysis) and remained at approximately 90 % or above until Visit 12 (Fig. 1).

Responder rates 1 and 2 over time (main analysis population; observed-case analysis)a. WHO World Health Organization
aResponder rate 1 was the percentage of patients with the same or less pain compared with Week −1 (on previous WHO step III analgesic); responder rate 2 was the percentage of patients with the same or less pain compared with Week −1 and an improvement of ≥1 category in subject satisfaction with treatment compared with baseline
b P < 0.0001 for testing the hypothesis that the response is 50 %
cThe interim visit occurred 3–4 days after the baseline visit
Significant reductions from baseline were observed in the mean pain intensity score at Weeks 6, 8, and 12 in the main analysis population (P < 0.0001 for the change from baseline for all comparisons; Fig. 2). Mean (SD) pain intensity scores were 4.6 (0.63) at screening, 4.7 (0.66) at baseline, 2.5 (1.46) at Week 6, and 1.8 (1.41) at Week 12. Mean (SD) changes in pain intensity from baseline to Weeks 6 and 12 were −2.2 (1.55) and −2.9 (1.40), respectively (P < 0.0001 for both comparisons). The percentage of patients who rated their satisfaction with treatment as “excellent,” “very good,” or “good” increased from 1.6 % (1/62) at baseline to 92.7 % (51/55) at Week 6 and 94.4 % (51/54) at Week 12 (Fig. 3). Satisfaction with treatment was rated as “poor” by 24.2 % (15/62) of patients at baseline and by no patients at Weeks 6 or 12. On the PGIC and CGIC, respectively, ratings of the patient’s overall condition as “very much improved,” “much improved,” or “minimally improved” were reported by 94.5 % (52/55) of patients and 94.5 % (52/55) of investigators at Week 6 and by 92.6 % (50/54) of patients and 94.4 % (51/54) of investigators at Week 12 (Fig. 4).

Mean pain intensity (NRS-3) over time (main analysis population; observed-case analysis)a. The vertical dotted line indicates start of tapentadol prolonged release. NRS-3 numerical rating scale-3, W week, WHO World Health Organization
aStandard deviations: screening, 0.63; baseline, 0.66; interim, 1.03; W1, 1.33; W2, 1.40; W3, 1.02; W4, 1.14; W5, 1.20; W6, 1.46; W7, 1.47; W8, 1.40; W9, 1.37; W10, 1.60; W11, 1.45; W12, 1.41
bPreplanned statistical analyses of change from baseline were conducted at Weeks 6, 8, and 12; P < 0.0001 for the change from baseline at each timepoint
cOn WHO step III analgesics
dEnrollment to baseline (i.e., point of rotation to tapentadol prolonged release)
eThe interim visit occurred 3–4 days after the baseline visit

Subject satisfaction with treatment at baseline, Week 6, and Week 12 (main analysis population; observed-case analysis)

Ratings on the a PGIC and b CGIC at Weeks 6 and 12 (main analysis population; observed-case analysis). PGIC Patient Global Impression of Change, CGIC Clinician Global Impression of Change
Mean WOMAC, EQ-5D, SF-36, HADS, and sleep questionnaire scores at baseline, Week 6, and Week 12 are reported in Electronic Supplementary Material Tables S5–S9. Significant improvements from baseline in mean WOMAC global score and pain, stiffness, and physical function subscale scores were observed at Weeks 6, 8, and 12 (P < 0.0001 for all comparisons; Fig. 5). On the EQ-5D, mean health status index score improved significantly from baseline to Week 6 (mean [SD] change from baseline, 0.20 [0.288]; P < 0.0001), Week 8 (0.26 [0.270]; P < 0.0001), and Week 12 (0.26 [0.313]; P < 0.0001), as did mean VAS score (Week 6, 29.6 [15.37]; Week 8, 33.0 [17.40]; Week 12, 35.5 [18.33]; P < 0.0001 for all comparisons). Significant improvements from baseline were observed in all individual mean SF-36 domain scores at Weeks 6 and 12 (P ≤ 0.0005 for all comparisons), except for role-emotional (numerical improvements were observed from baseline but they were not statistically significant; Week 6, P = 0.5515; Week 12, P = 0.4976; Fig. 6). The mean SF-36 physical component summary score also improved significantly from baseline to Week 6 (mean [SD] change from baseline, 11.9 [10.52]; P < 0.0001) and Week 12 (15.6 [14.44]; P < 0.0001). No significant changes from baseline were observed in mean SF-36 mental component summary score at Week 6 (mean [SD] change from baseline, 0.5 [7.09]; P = 0.6070) or at Week 12 (0.1 [8.28]; P = 0.9657; Electronic Supplementary Material Table S7).

Mean WOMAC subscale and global scores over time (main analysis population; observed-case analysis). WOMAC Western Ontario and McMaster Universities, SDs standard deviations, W week
aSDs: baseline, 13.19; W6, 16.72; W8, 19.57; W12, 19.00
bSDs: baseline, 9.88; W6, 12.40; W8, 14.26; W12, 14.04
cSDs: baseline, 2.48; W6, 3.45; W8, 3.94; W12, 3.59
dSDs: baseline, 1.76; W6, 1.52; W8, 1.80; W12, 1.78
e P < 0.0001 for the change from baseline

Mean (SD) changes in SF-36 domain scores from baseline to a Week 6a and b Week 12b (main analysis population; observed-case analysis)c,d. SD standard deviation, SF-36 Short Form-36
aSD: physical functioning, 21.99; role-physical, 41.88; bodily pain, 20.67; general health, 13.25; vitality, 13.92; social functioning, 27.39; role-emotional, 29.99; mental health, 11.24
bSD: physical functioning, 27.49; role-physical, 48.84; bodily pain, 29.28; general health, 15.59; vitality, 15.75; social functioning, 27.79; role-emotional, 33.50; mental health, 12.12
cSee Electronic Supplementary Material Table S7 for mean total SF-36 scores at baseline, Week 6, and Week 12
dWeek 6, n = 55; Week 12, n = 53
e P ≤ 0.0005 for the change from baseline
f n = 52
Significant decreases from baseline were observed in mean HADS anxiety and depression scores at Weeks 6, 8, and 12 (P < 0.05 for all comparisons). Mean (SD) HADS anxiety and depression subscale scores at baseline were 5.2 (4.26) and 5.1 (4.64), respectively; these scores were both below the range corresponding to clinically manifested anxiety and depression (scores ≥8 are considered to indicate the likely presence of anxiety or depression [39]). For the HADS anxiety and depression subscale scores, respectively, mean (SD) changes from baseline were −1.1 (2.86) and −0.9 (2.41) at Week 6 and −1.4 (2.76) and −1.1 (3.00) at Week 12.
On the sleep questionnaire, the mean (SD) number of awakenings per night decreased significantly from baseline (2.3 [1.54] awakenings) to Week 6 (1.1 [1.12] awakenings) and Week 12 (1.1 [1.06] awakenings; P < 0.0001 for the change from baseline to Weeks 6 and 12). The mean (SD) number of hours slept per night increased significantly from baseline (6.6 [1.38] h) to Week 6 (7.0 [1.09] h) and Week 12 (7.0 [1.19] h; P < 0.05 for the change from baseline to Weeks 6 and 12). At baseline, Week 6, and Week 12, respectively, overall sleep quality ratings of “excellent” or “good” were reported by 37.1 % (23/62), 63.6 % (35/55), and 68.5 % (37/54) of patients.
Results for effectiveness, function, and quality-of-life measures were comparable for the dataset that included results from Weeks 9 through 12 for patients who participated in Substudy A and the dataset that excluded those results (Electronic Supplementary Material Tables S1–S9). When the LOCF was used for imputing missing data, improvements were generally consistent with those shown when no imputation method was used (observed-case analysis; Electronic Supplementary Material Tables S1–S9).
3.3 Treatment Exposure and Equianalgesia
In the overall population, 83.9 % (52/62) of patients required no adjustment of their tapentadol PR starting dose up to Week 6 and 3.2 % (2/62) of patients required one adjustment to achieve the same or less pain intensity compared with baseline (LOCF; responders, based on responder rate 1 definition). Five (8.1 %) patients were non-responders (based on responder rate 1 definition) and did not adjust their dose of tapentadol PR up to Week 6, one (1.6 %) patient was a non-responder and had one dose adjustment, and two (3.2 %) patients were non-responders and had three dose adjustments. At Week 6, the mean (SD) TDD of tapentadol PR was 232.7 (145.37) mg, and the mean (SD) TDD of tapentadol IR was 7.0 (17.48) mg. The percentages of patients taking tapentadol PR and tapentadol IR in each dose range are summarized in Table 4. The most commonly used dose of tapentadol PR at Week 6 was 50 mg twice daily (taken by 41.8 % [23/55] of patients). Most patients (78.2 % [43/55]) did not take tapentadol IR once they achieved stable dosing with tapentadol PR at Week 6. The equianalgesic ratios for tapentadol to transdermal buprenorphine and oral oxycodone are shown in Table 5. Equianalgesic ratios for tapentadol to other prior WHO step III opioids are not presented due to low patient numbers.
3.4 Safety and Tolerability
Overall, 98.4 % (62/63) of patients in the safety population reported NTEAEs (under the previous analgesic regimen; included ongoing medical conditions reported at screening). Of the 255 NTEAEs reported, 158 (62.0 %) were considered to be non-associated with previous analgesic or co-analgesic treatment and 97 (38.0 %) were considered to be associated with previous analgesic or co-analgesic treatment. According to the study inclusion criteria, all eligible subjects were required to report opioid-related adverse effects as their reason for switching their analgesic treatment.
The prevalence of the AEs reported as associated with previous treatment during Week −1 and related to tapentadol treatment at Week 12 (Fig. 7) generally decreased during the study under treatment with tapentadol. Nausea, constipation, dry mouth, fatigue, and dizziness were among the most commonly reported AEs associated with previous treatment at Week −1 and the most commonly reported reasons for switching to tapentadol PR; the prevalence of these AEs decreased during the study.

Prevalence of AEs (≥2 %) associated with previous treatment at Week −1 and related to tapentadol treatment at Week 12 (safety population; n = 63)a. AE adverse event, WHO World Health Organization
aThe prevalence of these AEs were summarized during Week −1 (the week prior to titration when patients were still on WHO step III treatment) and during Week 12 (the final week of tapentadol prolonged release treatment)
bNausea, constipation, dry mouth, fatigue, and dizziness were reported as the main reasons for switching to tapentadol prolonged release
At least one TEAE was reported from Week 1 to Week 12 by 34.9 % (22/63) of patients. Of the 116 TEAEs reported, 73 (62.9 %) were classified as at least possibly related to study drug. The majority of TEAEs reported were of mild or moderate intensity (85.3 % [99/116]). The most commonly reported (incidence ≥5 %) TEAEs (Fig. 8) included diarrhea, nausea, dizziness, constipation, hyperhidrosis, drug withdrawal syndrome, and fatigue, which are known adverse drug reactions (ADRs) of tapentadol. Although drug withdrawal syndrome is a known ADR of tapentadol, withdrawal occurring at the switch from the previous WHO step III opioid should be regarded as associated with the prior treatment, not with tapentadol PR. Five serious TEAEs were reported in two patients and included abdominal pain, chest pain, renal pain, transient ischemic attack, and dysphagia. Six patients in the safety population (n = 63) had TEAEs that led to premature study discontinuation. The only TEAEs leading to discontinuation reported for >1 patient were nausea (3.2 % [2/63]) and hyperhidrosis (3.2 % [2/63]).

Incidence of TEAEs reported by ≥5 % of patients (safety population; n = 63) (percentages based on the number of individual TEAEs and TEAEs in each SOC). The horizontal dotted lines separate the TEAEs included in each SOC. TEAE treatment-emergent adverse event, SOC system organ class
No clinically relevant changes were observed in vital signs, laboratory values, or physical examination parameters.
3.5 Substudies A and B
In Substudy A (n = 21), 81.0 % (17/21) of patients reduced their doses of WHO step I analgesics and 19.0 % (4/21) reduced their doses of co-analgesics from Week 9 to Week 11. The WHO step I analgesics that were tapered included paracetamol (acetaminophen), metamizole (dipyrone), diclofenac, ketoprofen, and meloxicam; the co-analgesics that were tapered included pregabalin, gabapentin, and flupirtine. Overall, 94.1 % (16/17) of patients in Substudy A tapered and completely stopped taking their doses of WHO step I analgesics, and 75 % (3/4) of patients tapered and completely stopped taking their co-analgesics. In Substudy A, responder rate 1 was 95.2 % (20/21) and responder rate 2 was 90.5 % (19/21) at Week 6 (LOCF); mean (SD) pain intensity score (11-point NRS-3) decreased significantly from baseline (4.5 [0.60]) to Week 6 (mean [SD] change from baseline, −2.2 [1.63]; P < 0.0001), Week 8 (−3.0 [1.60]; P < 0.0001), and Week 12 (−3.2 [1.57]; P < 0.0001).
All patients who participated in Substudy B (n = 21) tapered and completely stopped taking their side-effect medication during Week 7.
4 Discussion
The selected population of this trial was defined as patients with severe pain related to knee osteoarthritis who had responded to previous strong (WHO step III) opioid therapy. A pain intensity score (NRS-3) ≤5 at baseline was believed to be adequate to define the respective population because pain intensity was measured under treatment with strong opioids (and not after a washout period, as is typical in phase 3 trials). The need for strong opioid therapy was assumed as a prerequisite for patients who entered the trial. The objective of the trial was generally not to obtain better analgesia in this strong-opioid responder population but to reach comparable effectiveness outcomes with better tolerability after rotation. Results were more positive than anticipated and showed that treatment with tapentadol PR (50–250 mg twice daily) resulted, on average, in better pain relief compared with previous WHO step III opioids in patients with severe, chronic osteoarthritis knee pain who had responded to WHO step III opioids but reported a lack of tolerability.
The percentage of patients reporting the same or less pain compared with the 1-week observation period on their previous WHO step III analgesic regimen increased from the interim visit to Week 4, then remained above 90 % for the remainder of the study. A similar high percentage of patients (~90 %) reported the same or less pain compared with Week −1 and an improvement in their satisfaction with treatment from Week 4 of tapentadol treatment throughout the remainder of the study. Although the patients included in this study had responded to their previous WHO step III opioid regimen, tapentadol PR treatment resulted in significant improvements in mean pain intensity score compared with Week −1 at all weeks of major comparison (Weeks 6, 8, and 12). This unexpected and pronounced improvement in pain intensity upon switching from prior opioid therapy for strong-opioid responders was the main underlying reason for the trial being positive and reaching the primary endpoint despite the small sample size. The improvements observed in mean pain intensity score with tapentadol PR treatment were accompanied by improvements in health status, quality-of-life, function, and anxiety and depression.
Overall, tapentadol treatment was effective for the management of severe osteoarthritis pain. Osteoarthritis pain has historically been described as nociceptive pain and is frequently used as a model of nociceptive pain in regulatory guidelines [7–9, 19, 40]. However, the diversity of underlying conditions associated with osteoarthritis, the effects of sensitization and chronicity, and the frequently missing correlation between cartilage damage (radiological findings) and pain need to be considered when determining pain origins [7]. The efficacy of compounds such as duloxetine, which act on the descending inhibitory pathway, show the relevance of targeting this modulatory pain pathway in osteoarthritis [41]. Further, the efficacy of opioids may result from effects on the ascending pathway, targeting particularly pain associated with continuing mechanical damage and tissue degradation [15, 42]. Thus, combining an opioid and a noradrenergic mechanism of action may be of particular relevance in the management of severe osteoarthritis pain.
Tapentadol PR was associated with improvements in tolerability relative to previous WHO step III treatments (based on the prevalence of AEs reported as associated with previous treatment during Week −1 and related to tapentadol treatment at Week 12). In particular, the AEs of nausea, constipation, dry mouth, and fatigue, which were among the most commonly reported AEs associated with previous treatment during Week −1, were reduced by 50–75 % by Week 12 of tapentadol PR treatment. In combination with the results indicating maintenance or improvement of pain relief for patients rotating from WHO step III opioids to tapentadol PR, the low incidence of drug withdrawal symptoms and low rate of treatment discontinuation indicate that rotation directly from WHO step III opioids to tapentadol PR went smoothly. A high percentage of patients achieved appropriate pain relief with just the starting dose of tapentadol PR (most frequently 50 mg twice daily). Overall, the tolerability profile of tapentadol PR in this study is consistent with that observed in previous placebo-controlled trials [24, 27]. Improvements in tolerability versus earlier trials can be explained by a lower dose range (achieved by allowing for the optional use of concomitant analgesics [e.g., NSAIDs], as is common in practice conditions) and opioid pretreatment.
The equianalgesic ratio calculated in the current study versus oxycodone was in line with observations from previous phase 3 studies [24, 27]; however, the equianalgesic ratios calculated in the current study were limited by small sample sizes. The most commonly used analgesic during Week −1 was transdermal buprenorphine, with a mean release rate of 20 μg/h (corresponds to MED = 51.5 mg/day). Given the low dose that patients taking buprenorphine needed to attain sufficient pain relief during Week −1, many of these patients were able to achieve adequate pain relief with low doses of tapentadol PR, resulting in favorable AE and discontinuation profiles and further improvements in pain intensity.
For practical aspects when rotating to tapentadol, it might be recommended that patients be treated with doses no more than 30 % below the calculated equianalgesic dose of the previous strong opioid or that an equianalgesic conversion be targeted (in patients judged to be at higher risk for developing withdrawal symptoms). These strategies might help to avoid the risk of withdrawal, considering that tapentadol has less of an opioid component to overcome withdrawal symptoms related to the previous opioid treatment.
Possible limitations of this study are the high number of patients taking buprenorphine (which may not be representative of opioid consumption patterns in a general patient population), the open-label study design, and the lack of a placebo or active control to allow comparison of results obtained with tapentadol. The design of this effectiveness trial was intended to better approximate clinical practice than randomized, placebo-controlled trials. Effectiveness studies evaluate outcomes that reflect the overall effects of a study drug (e.g., quality of life, patient-reported outcomes, changes in pain intensity over time), providing results that may be more broadly applicable to the general population observed in clinical practice than the more narrow efficacy outcomes obtained in a randomized, controlled trial [43]. Further, the use of a placebo control in this study would have been unethical because this study was designed to assess direct rotation from prior strong opioid therapy to study treatment (tapentadol PR) in a preselected population of patients with severe pain who had responded to opioid therapy but showed a lack of tolerability; rotating these patients from their prior opioid treatment to placebo would have resulted in withdrawal reactions and unnecessary pain peaks. Overall results were unexpectedly positive in this strong-opioid responder population with severe pain and in line with those of previous randomized, double-blind, placebo- and active-controlled phase 3 studies of tapentadol PR for the management of moderate-to-severe, chronic pain [24, 25, 27, 28]; significant improvements in pain intensity were observed with tapentadol PR treatment over the course of those studies. In addition, results (as described previously) were consistent regardless of whether an imputation method (LOCF) was used or not. In this study, direct rotation from strong opioids to tapentadol was studied for the first time in a phase 3b clinical trial setting and provided valuable data for clinical practice.
Although the populations of the tapering substudies (Substudies A and B) were relatively small (n = 21 for each substudy), which limited the statistical evaluation of these data, results support those observed for the overall population. Pain relief was maintained with tapentadol PR treatment after tapering of WHO step I analgesics or co-analgesics, and there was no increase in adverse effects after tapering of medications used to treat adverse effects associated with previous WHO step III opioid therapy.
5 Conclusions
In this phase 3b study, patients with severe osteoarthritis pain who had responded to WHO step III opioids switched directly from their previous WHO step III therapy to tapentadol PR without disruption of pain relief and often experienced improvements in tolerability, as well as further improvements in pain intensity, function, and quality of life. Equianalgesic ratios were calculated for tapentadol to transdermal buprenorphine and oral oxycodone and were in line with observations from previous phase 3 studies [24, 27].
References
Altman RD, Smith HS. Opioid therapy for osteoarthritis and chronic low back pain. Postgrad Med. 2010;122(6):87–97.
Seed SM, Dunican KC, Lynch AM. Osteoarthritis: a review of treatment options. Geriatrics. 2009;64(10):20–9.
Zhang W, Moskowitz RW, Nuki G, et al. OARSI recommendations for the management of hip and knee osteoarthritis, part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis Cartilage. 2008;16(2):137–62.
Roth SH, Fleischmann RM, Burch FX, et al. Around-the-clock, controlled-release oxycodone therapy for osteoarthritis-related pain: placebo-controlled trial and long-term evaluation. Arch Intern Med. 2000;160(6):853–60.
Caldwell JR, Rapoport RJ, Davis JC, et al. Efficacy and safety of a once-daily morphine formulation in chronic, moderate-to-severe osteoarthritis pain: results from a randomized, placebo-controlled, double-blind trial and an open-label extension trial. J Pain Symptom Manage. 2002;23(4):278–91.
Hale M, Tudor IC, Khanna S, et al. Efficacy and tolerability of once-daily OROS hydromorphone and twice-daily extended-release oxycodone in patients with chronic, moderate to severe osteoarthritis pain: results of a 6-week, randomized, open-label, noninferiority analysis. Clin Ther. 2007;29(5):874–88.
Arendt-Nielsen L, Nie H, Laursen MB, et al. Sensitization in patients with painful knee osteoarthritis. Pain. 2010;149(3):573–81.
Gwilym SE, Keltner JR, Warnaby CE, et al. Psychophysical and functional imaging evidence supporting the presence of central sensitization in a cohort of osteoarthritis patients. Arthritis Rheum. 2009;61(9):1226–34.
Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152(3 Suppl):S2–15.
Hochman JR, French MR, Bermingham SL, et al. The nerve of osteoarthritis pain. Arthritis Care Res (Hoboken). 2010;62(7):1019–23.
Nikolajsen L, Brandsborg B, Lucht U, et al. Chronic pain following total hip arthroplasty: a nationwide questionnaire study. Acta Anaesthesiol Scand. 2006;50(4):495–500.
Puolakka PA, Rorarius MG, Roviola M, et al. Persistent pain following knee arthroplasty. Eur J Anaesthesiol. 2010;27(5):455–60.
Wylde V, Hewlett S, Learmonth ID, et al. Persistent pain after joint replacement: prevalence, sensory qualities, and postoperative determinants. Pain. 2011;152(3):566–72.
Lundblad H, Kreicbergs A, Jansson KA. Prediction of persistent pain after total knee replacement for osteoarthritis. J Bone Joint Surg Br. 2008;90(2):166–71.
Loeser RF. Molecular mechanisms of cartilage destruction in osteoarthritis. J Musculoskelet Neuronal Interact. 2008;8(4):303–6.
Management of chronic pain syndromes: issues and interventions. Pain Med. 2005;6(Suppl 1):S1–20.
Curatolo M, Arendt-Nielsen L, Petersen-Felix S. Central hypersensitivity in chronic pain: mechanisms and clinical implications. Phys Med Rehabil Clin N Am. 2006;17(2):287–302.
Benyamin R, Trescot AM, Datta S, et al. Opioid complications and side effects. Pain Physician. 2008;11(2 Suppl):S105–20.
Furlan AD, Sandoval JA, Mailis-Gagnon A, et al. Opioids for chronic noncancer pain: a meta-analysis of effectiveness and side effects. CMAJ. 2006;174(11):1589–94.
Porreca F, Ossipov MH. Nausea and vomiting side effects with opioid analgesics during treatment of chronic pain: mechanisms, implications, and management options. Pain Med. 2009;10(4):654–62.
Nuesch E, Rutjes AW, Husni E, et al. Oral or transdermal opioids for osteoarthritis of the knee or hip. Cochrane Database Syst Rev. 2009;(4):CD003115.
Tzschentke TM, Christoph T, Kögel B, et al. (-)-(1R,2R)-3-(3-Dimethylamino-1-ethyl-2-methyl-propyl)-phenol hydrochloride (tapentadol HCl): a novel μ-opioid receptor agonist/norepinephrine reuptake inhibitor with broad-spectrum analgesic properties. J Pharmacol Exp Ther. 2007;323(1):265–76.
Tzschentke TM, De Vry J, Terlinden R, et al. Tapentadol HCl. Drugs Future. 2006;31(12):1053–61.
Afilalo M, Etropolski MS, Kuperwasser B, et al. Efficacy and safety of tapentadol extended release compared with oxycodone controlled release for the management of moderate to severe chronic pain related to osteoarthritis of the knee: a randomized, double-blind, placebo- and active-controlled phase III study. Clin Drug Investig. 2010;30(8):489–505.
Lange B, Kuperwasser B, Okamoto A, et al. Efficacy and safety of tapentadol prolonged release for chronic osteoarthritis pain and low back pain. Adv Ther. 2010;27(6):381–99.
Wild JE, Grond S, Kuperwasser B, et al. Long-term safety and tolerability of tapentadol extended release for the management of chronic low back pain or osteoarthritis pain. Pain Pract. 2010;10(5):416–27.
Buynak R, Shapiro DY, Okamoto A, et al. Efficacy and safety of tapentadol extended release for the management of chronic low back pain: results of a prospective, randomized, double-blind, placebo- and active-controlled phase III study. Expert Opin Pharmacother. 2010;11(11):1787–804.
Schwartz S, Etropolski M, Shapiro DY, et al. Safety and efficacy of tapentadol ER in patients with painful diabetic peripheral neuropathy: results of a randomized-withdrawal, placebo-controlled trial. Curr Med Res Opin. 2011;27(1):151–62.
Vinik A, Shapiro DY, Rauschkolb C, et al. Efficacy and tolerability of tapentadol extended release (ER) in patients with chronic, painful diabetic peripheral neuropathy (DPN): results of a phase 3, randomized-withdrawal, placebo-controlled study [abstract]. J Pain. 2012;13(Suppl 4):S72.
Steigerwald I, Müller M, Kujawa J, et al. Effectiveness and safety of tapentadol prolonged release with tapentadol immediate release on-demand for the management of severe, chronic osteoarthritis-related knee pain: results of an open-label, phase 3b study. J Pain Res. 2012;5:121–38.
Dworkin RH, Turk DC, Farrar JT, et al. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain. 2005;113(1–2):9–19.
Farrar JT, Young JP Jr, LaMoreaux L, et al. Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. Pain. 2001;94(2):149–58.
Schneider LS, Olin JT, Doody RS, et al. Validity and reliability of the Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis Assoc Disord. 1997;11(Suppl 2):S22–32.
Bellamy N. WOMAC Osteoarthritis Index: user guide IV. Brisbane: WOMAC; 2000.
Selai CE, Trimble MR, Price MJ, et al. Evaluation of health status in epilepsy using the EQ-5D questionnaire: a prospective, observational, 6-month study of adjunctive therapy with anti-epileptic drugs. Curr Med Res Opin. 2005;21(5):733–9.
Ware JE Jr, Donald Sherbourne C. The MOS 36-item Short-Form Health Survey (SF-36). I. Conceptual framework and item selection. Med Care. 1992;30(6):473–83.
Bjelland I, Dahl AA, Haug TT, et al. The validity of the Hospital Anxiety and Depression Scale. An updated literature review. J Psychosom Res. 2002;52(2):69–77.
Haythornthwaite JA, Hegel MT, Kerns RD. Development of a sleep diary for chronic pain patients. J Pain Symptom Manage. 1991;6(2):65–72.
Snaith RP. The Hospital Anxiety And Depression Scale. Health Qual Life Outcomes. 2003;1:29.
Combe R, Bramwell S, Field MJ. The monosodium iodoacetate model of osteoarthritis: a model of chronic nociceptive pain in rats? Neurosci Lett. 2004;370(2–3):236–40.
Ho KY, Tay W, Yeo MC, et al. Duloxetine reduces morphine requirements after knee replacement surgery. Br J Anaesth. 2010;105(3):371–6.
Schaible HG. Peripheral and central mechanisms of pain generation. Handb Exp Pharmacol. 2007;177:3–28.
Agency for Healthcare Research and Quality. http://www.ahrq.gov. Accessed 2 Oct 2006.
Acknowledgments
This study was sponsored by Medical Affairs, Grünenthal Europe and Australia, Grünenthal GmbH, Aachen, Germany. Tapentadol is a compound developed by Grünenthal GmbH. Grünenthal is the legal sponsor of this trial of tapentadol, the results of which have been submitted for publication. Ilona Steigerwald is an employee of Grünenthal GmbH, Aachen, Germany and has, as such, been involved in the planning and conduct of the study and development of the final integrated trial report and this manuscript. Ilona Steigerwald is a physician and senior member of management in Medical Affairs and certifies that the data, results, and interpretation have not been altered by her position. Michael Schenk has participated in two investigator meetings in Germany and received support from Grünenthal for travel to those meetings. Michael Schenk is an employee of a pain center that included patients in the study; the pain center received compensation from Grünenthal that was per patient and reflected the efforts of the pain center on behalf of the study. All compensation was paid directly to the pain center, not to the individual employees (including Michael Schenk). Uwe Lahne and his clinic received compensation for his role as an investigator for this study. Peter Gebuhr has no conflicts of interest to disclose. Dietmar Falke is an employee of Grünenthal GmbH, Aachen, Germany. Barbara Hoggart has participated in an advisory board and has given lectures to other healthcare professionals; she received support from Grünenthal for these activities. Editorial support for the writing of this manuscript was provided by Megan Knagge, PhD, of MedErgy, and was funded by Medical Affairs, Grünenthal GmbH, Aachen, Germany. The authors retained full editorial control over the content of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Steigerwald, I., Schenk, M., Lahne, U. et al. Effectiveness and Tolerability of Tapentadol Prolonged Release Compared With Prior Opioid Therapy for the Management of Severe, Chronic Osteoarthritis Pain. Clin Drug Investig 33, 607–619 (2013). https://doi.org/10.1007/s40261-013-0102-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-013-0102-0