
Abstract
Antibody-drug conjugates (ADCs) are an emerging class of therapeutics for lung cancer, and several are currently in development for this malignancy. The structure of these molecules is based on an antibody that targets a protein on the lung cancer cell surface and a cytotoxic payload attached by a linker. Many protein targets, including TROP2, c-MET, CEACAM5, HER2, and HER3 have been identified. In metastatic non-small cell lung carcinoma (NSCLC) without alterations in oncogenic drivers, platinum-based chemotherapy and immune checkpoint inhibitors (ICIs) targeting the programmed death-1/programmed death-ligand 1 (PD1/PDL1) interaction are the standard first-line treatments. In patients with EGFR-mutated or ALK-rearranged NSCLC, tyrosine kinase inhibitors (TKIs) are recommended. However, although the prognosis of patients with metastatic NSCLC differs between such with and without alterations in oncogenic drivers, most patients eventually experience disease progression. A novel therapeutic class is needed in routine practice to overcome the mechanisms of resistance to ICIs and EGFR/ALK TKIs. Several ADCs have already been approved for other cancers, such as breast cancer and urothelial carcinoma. This review summarizes the knowledge about the efficacy and tolerance profiles of ADCs targeting TROP2, HER2, HER3, CEACAM5 and c-MET in metastatic NSCLC with and without alterations in oncogenic drivers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Antibody–drug conjugates (ADCs) are an emerging class of therapeutics for lung cancer, and several ADCs are currently in development in metastatic NSCLC with or without alterations in oncogenic drivers. |
These ADCs could present future therapeutic options for patients with checkpoint inhibitor-refractory or EGFR TKI-refractory metastatic NSCLC. |
1 Introduction
Lung cancer (LC) is expected to be a major contributor to total global cancer-related deaths in 2023 [1]. Non-small cell lung cancer (NSCLC) accounts for 85% of all LC cases; however, this population is heterogeneous, and molecular analysis of non-squamous NSCLC distinguished patients with oncogenic driver alterations from those without such alterations. In recent years, the therapeutic landscape for LC has changed with the discovery of epidermal growth factor receptor (EGFR)-activating alterations or anaplastic lymphoma kinase (ALK) gene rearrangements that both can be targeted by tyrosine kinase inhibitors (TKIs). The prognosis of the LC population harbouring EGFR or ALK alterations is good, with a 1-year overall survival (OS) rate of between 89 and 84% [2, 3]. Other oncogenic drivers, including BRAF, HER2, KRAS, ROS and MET, have been shown to be associated with a less favourable prognosis than EGFR or the ALK population. Personalized treatments, mainly including dabrafenib (TKI against BRAF) and trametinib (TKI against MEK), have been developed for V600E BRAF-mutated NSCLC, and sotorasib or adagrasib (TKI against KRAS) have been used for KRAS G12C-mutated NSCLC. At diagnosis, the molecular hallmarks of non-squamous NSCLC, namely EGFR, ALK, BRAF, KRAS, HER2, ROS, or MET gene alterations, are analysed to determine therapeutic strategies. However, LC patients harbouring a targetable oncogenic driver alteration constitute a minority of the population. In any case, tumour cells eventually become resistant to targeted therapies, making chemotherapy treatment necessary. In NSCLC without targetable oncogenic driver alterations, platinum-based chemotherapy in combination with programmed death-1/programmed death ligand-1 (PD1/PDL1) blockade is the standard of care in the first-line metastatic setting, with a 12-month OS rate of 69.2% [4]. Immune checkpoint inhibitor (ICI) therapy targeting PD1/PDL1 has led to a revolution in NSCLC research, and with this approach, approximately 20% of patients achieved a long-term durable response [5]. There is no effective therapy for NSCLC patients without a targetable oncogenic driver and who exhibit disease progression under first-line platinum-based chemotherapy and ICI therapy.
Antibody-drug conjugates (ADCs), a novel therapeutic class, could be a therapeutic option in NSCLC patients with and without alterations in oncogenic drivers who do not respond to standard therapies. Within 3 years, seven ADCs have emerged, and research to improve the technologies related to this therapeutic class has proceeded rapidly. This review focuses on the structure, mechanism of action, efficacy, and toxicity of emerging ADCs in NSCLC.
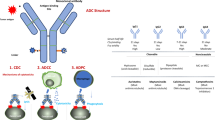
2 Structure of Antibody-Drug Conjugates (ADCs)
Drug conjugate structures are based on four components, each with specific properties: an antigen, an antibody, a cytotoxic payload and a linker [6]. The ideal properties of the antigen include high expression on tumour cells and low/no expression on healthy cells, limiting on/off-target toxicity; additionally, they have high affinity and avidity for antibody recognition and the capacity to internalize into tumour cells after antibody binding to facilitate transmembrane trafficking and enhance ADC action. The antibodies used in the therapeutic landscape are immunoglobulin (Ig) G antibodies with high affinity and avidity for antigens. Humanized antibodies are preferred as they have decreased immunogenicity. The other specific properties of IgG isotopes include a long serum half-life and complement factor C1q binding and activation. The drug payload is a highly potent agent that remains stable in the plasma circulation and exerts its cytotoxic effects only after internalization into tumour cells to limit off-target toxicity. The optimal drug/antibody ratio (DAR) for limiting toxicity and increasing efficacy ranges between 2 and 8 cytotoxic agents per antibody. The linker aims to prevent premature release of cytotoxic agents into non-target tissues. There are two linker types: cleavable and non-cleavable. The first depends on physiological conditions (pH, proteolysis, high intracellular glutathione concentration) for the release of cytotoxic agents, while the second depends on lysosomal degradation.
3 Mechanism of Action of ADCs
The main mechanism of action of ADCs is based on target binding between the antigen and the antibody, which induces internalization of the ADC following the release of the cytotoxic payload. The ADC activity rate is not correlated to the quantity of antigen expressed on the tumour cells. The efficacy of an ADC for the same antigen differs according to tumour histological subtype; for example, the overall response rate (ORR) of sacituzumab govitecan, an ADC against Trop2, ranges between 3 and 35% according to histological subtype [7,8,9]. In the ASCENT trial, the ORR of metastatic breast cancer patients treated with sacituzumab govitecan reached 35%, but no predictive biomarker was identified [7]. Moreover, for the same antigen target, ADCs are efficient regardless of the gene alterations that encode the corresponding antigens; for example, trastuzumab deruxtecan (T-DXd), an ADC against HER2, is efficient in both breast cancer patients with HER2 amplification and LC patients with HER2 mutations [10, 11].
The bystander effect is another mechanism of action. Ogitani et al. [12] suggested a therapeutic effect in HER2-positive and HER2-negative cells treated with an ADC against HER2; both cell types were killed. This effect can be explained by membrane permeability, which leads to payload release into neighbouring cells. Cleavable linkers are less stable in plasma circulation, and this effect accounts for the better efficacy of ADCs with cleavable linker versus those with non-cleavable linkers [13].
Finally, by activating immune cells, ADCs induce tumour cell death. IgG1 is the most common antibody used in ADCs. Through high binding affinity with the Fc gamma receptor, IgG1 induces antibody-dependent cell-mediated cytotoxicity, antibody-dependent phagocytosis, and complement-dependent cytotoxicity [14,15,16]. However, the communication between the Fc gamma receptor and IgG1 activates several IgG-mediated functions in immune cells. This mechanism potentially leads to toxicity and adverse effects, for example thrombopenia induced by trastuzumab emtansine (TDM-1) [17].
4 ADCs in Non-Small Cell Lung Carcinoma
In NSCLC, several ADCs have been developed, and these target TROP2, HER2, HER3, CEACAM5, and c-MET.
4.1 Trophoblast Cell-Surface Antigen 2 (TROP2)
Trophoblast cell-surface antigen 2 (TROP2) is a membrane glycoprotein encoded by the tumour-associated calcium signal transducer 2 gene at chromosome 1p32. This protein is expressed on epithelial cells (respiratory epithelium, breast glands, pancreas, prostate and urothelium) and is involved in cellular proliferation, regeneration, and transformation [18, 19]. In LC, TROP2 is overexpressed in 64% of adenocarcinomas; high expression of this protein is associated with a poor prognosis [20]. Two ADCs, datopotamab deruxtecan (Dato-Dxd) and sacituzumab govitecan, are being developed for LC indications. Both agents are humanized IgG1 molecules with cleavable linkers, and the cytotoxic drugs act as topoisomerase 1 inhibitors.
4.1.1 Datopotamab Deruxtecan
The TROPION-PanTumor01 trial is the first human dose-escalation and dose-expansion study evaluating the safety and anti-tumour activity of Dato-DXd [19]. A total of 180 patients with heavily pretreated advanced NSCLC received 4, 6 or 8 mg/kg (D1 = D21) of Dato-DXd. The confirmed ORR ranged between 22 and 26%, with a median time to response of 1.4 months. The median progression-free survival (PFS) ranged between 4.3 and 6.9 months, while the median OS ranged between 10.5 and 12.9 months. The dose was not correlated with tumour activity but rather was associated with a higher rate of toxicity; indeed, a dose of 8 mg/kg/3 weeks was associated with a higher incidence of stomatitis and interstitial lung disease (ILD), leading to treatment interruptions; thus, 6 mg/kg is the selected dose for development [19]. TROPION-Lung05 studied Dato-DXd in previously treated NSCLC patients with actionable genomic alterations. The first results were presented at the European Society of Medical Oncology (ESMO) Congress 2023. The study included patents with actionable genomic alterations who were previously treated with a targeted TKI and platinum-based chemotherapy. Among the 137 patients, 57% and 25% had NSCLC with EGFR mutations and ALK rearrangements, respectively. The ORR was 35.8% (27.8–44.4%). The median duration of response (DOR) and PFS were 7 (4.2–9.8) and 5.4 (4.7–7.0) months, respectively [21]. The phase III TROPION-Lung 01 trial evaluated Dato-Dxd versus docetaxel for previously treated metastatic NSCLC with or without actionable genomic alterations. The first results were presented at the ESMO Congress in 2023. The majority of patients had been previously exposed to platinum-based chemotherapy and checkpoint inhibitors. The ORR and DOR were 26.4% (21.5–31.8) and 7.1 (5.6–10.9) months, respectively, in the Dato-DXd group compared with 12.8% (9.3–17.1) and 5.6 months (5.4–8.1), respectively, in the docetaxel group. PFS was significantly increased for Dato-DXd patients (hazard ratio [HR] 0.75, p = 0.004), with a median of 4.4 months (4.2–5.6). According to subgroup analysis, patients with non-squamous NSCLC (HR 0.63, 95% confidence interval [CI] 0.51–0.78) and patients with actionable genomic alterations (HR 0.38) had a significant improvement in PFS. In the squamous NSCLC population, Dato-DXd seemed to be less effective (HR 1.38, 95% CI 0.94–2.02). The interim OS data suggested that Dato-DXd versus docetaxel was beneficial for patients with a median OS of 12.4 months versus 11 months. Stomatitis and nausea were the most frequent adverse events in the Dato-DXd group. Fewer grade ≥3 adverse events were observed for Dato-Dxd than for docetaxel; however, three patients in the Dato-DXd group presented grade 5 ILD [22]. Dato-DXd is a more tolerable drug and a potential new, meaningful therapy for patients with NSCLC eligible for second-line chemotherapy.
TROPION-Lung02 is a phase Ib study evaluating Dato-DXd plus pembrolizumab (ICI anti-PD1) with or without platinum-based chemotherapy. Patients in the doublet and triplet groups received ≤1 line of treatment and no prior treatment, respectively. The ORR was 38% and 47% for the doublet and triplet groups, respectively. Although immature, the median PFS was 10.8 and 7.8 months for patients receiving doublet and triplet therapies, respectively. Stomatitis (45%) and nausea (45%) were the most common adverse events, while a decreased neutrophil count (8%) and increased amylase level (8%) were the most frequent grade ≥3 events. Three grade 3 ILDs were observed [23]. A phase III study is ongoing and is evaluating Dato-DXd + pembrolizumab + platinum chemotherapy versus pembrolizumab in combination with platinum chemotherapy and pemetrexed in advanced/metastatic NSCLC patients with a PDL1 tumour proportion score (TPS) <50% (ClinicalTrials.gov identifier: NCT05555732). TROPION-Lung 08, a phase III trial, is evaluating Dato-DXd plus pembrolizumab versus pembrolizumab in advanced/metastatic NSCLC patients with a PDL1 TPS ≥50% (ClinicalTrials.gov identifier: NCT05215340).
4.1.2 Sacituzumab Govitecan
Sacituzumab govitecan outcomes in heavily pretreated patients with metastatic NSCLC were reported in the IMMU-132 01 study. For the 47 patients included, the ORR was 19%, and the median PFS and OS were 5.2 and 9.5 months, respectively. Nausea, diarrhoea and fatigue were the most common adverse events [24], and febrile neutropenia was the most serious adverse event. UGT1A1∗28 homozygoty (28/46, 60.9%), compared with heterozygoty (69/180, 38.3%) or UGT1A1∗1 wild-type status (59/177, 33.3%), was associated with a greater rate of febrile neutropenia [8]. EVOKE 01, a phase III trial, is studying sacituzumab govitecan versus docetaxel in patients with NSCLC after progression on platinum chemotherapy and checkpoint inhibitor therapy (ClinicalTrials.gov identifier: NCT05089734). EVOKE 02, a phase II study, studied first-line sacituzumab govitecan plus pembrolizumab with or without platinum chemotherapy in treatment-naïve metastatic NSCLC (ClinicalTrials.gov identifier: NCT05186974). Preliminary results were presented during the World Conference on Lung Cancer, 2023. The ORR was 69% in cohort A (TPS ≥50%) and 44% in cohort B (TPS <50%). The most common adverse effects were diarrhoea, anaemia and asthenia. The limited availability of ADC data in the frontline setting indicate the need for further studies in the metastatic LC population. Moreover, the tolerance of the combination ADC and checkpoint inhibitor needs to be studied in order to assess this therapeutic option in current practice.
SKB264, a novel TROP2 ADC that uses 2-methylsulfonyl pyrimidine as a linker to conjugate its payload (KL610023), is a belotecan-derivative topoisomerase I inhibitor (DAR 7.4). This drug was designed to be more effective with a better balance between stability in circulation and release of the cytotoxic drug in tumour cells. A phase I/II study including 43 patients with refractory NSCLC treated with SKB264 showed an ORR of 26%, a 5.3-month median PFS and a 9-month OS of 80.4%. SKB264 was more effective in refractory EGFR-mutant NSCLC patients (ORR 60%, median PFS 11.1 months). Neutropenia (32%), anaemia (30%) and stomatitis (9%) were the most common grade ≥3 adverse events [25].
4.2 Human Epidermal Growth Factor Receptor 2 (HER2)
Human epidermal growth factor receptor 2 (HER2) is a receptor tyrosine kinase of the ERBB family that is involved in the control of cell proliferation, migration, and differentiation. Alterations in HER2 expression through gene amplification (2–3%), gene mutation (1–2%) or protein overexpression (20–40%) lead to uncontrolled cell proliferation (International Association for the Study of Lung Cancer [IASLC] Atlas of Molecular Testing 2023).
4.2.1 Trastuzumab Emtansine (TDM-1)
TDM-1, which targets HER2, is an ADC with a non-cleavable linker, and DM1 is a tubulin polymerization inhibitor (maytansine derivative) [DAR 3.5:1]. No efficacy was observed in a phase II study that included patients with NSCLC with HER2 overexpression. The study was terminated early due to limited efficacy [26]. Li et al. [27] reported tumour activity in HER2-mutant NSCLC patients treated with TDM-1, with an ORR of 44% and a 5-month median PFS.
4.2.2 Trastuzumab Deruxtecan (T-DXd)
T-DXd is designed with a humanized anti-HER2 IgG1 antibody, a cleavable linker and a potent topoisomerase inhibitor payload (exatecan derivative) [DAR 8:1] [12]. Destiny Lung01, a phase II trial, was designed to evaluate the efficacy of T-DXd in refractory HER2-overexpressing (cohort 1) and HER2-mutant (cohort 2) metastatic NSCLC. In cohort 2, the ORR, median PFS and median OS were 54.9%, 8.2 (95% CI 6.0–11.9) and 17.8 (95% CI 13.8–22.1) months, respectively. T-DXd activity was observed across HER2 mutation subtypes, as well as in patients without HER2 protein overexpression [11]. However, in cohort 1, T-DXd was less effective, with an ORR and median PFS of 34.1% and 6.7 months, respectively. Biomarkers of treatment efficacy need to be identified. Nausea (73%) and fatigue (53%) were the most common adverse events and neutropenia (19%) was the most common grade ≥3 adverse event [11]. T-DXd was approved for patients with previously treated HER2-mutant NSCLC in the US in August 2022 and in the EU in October 2023. The 2023 ESMO guidelines recommend T-DXd following first-line therapy for patients with metastatic NSCLC with HER2 mutations (ESMO guidelines 2023).
4.3 HER3
The receptor tyrosine-protein kinase ERBB3 (HER3) is expressed in 83% of NSCLC patients, mainly in patients with EGFR-mutant LC. HER3 expression is associated with metastatic progression and is an acquired mechanism of resistance to EGFR TKIs.
4.3.1 Patritumab Deruxtecan (HER3-DXd)
HER3-DXd is composed of a humanized IgG1 antibody against HER3, a cleavable linker and a topoisomerase I inhibitor (exatecan derivative) with a DAR of 8:1 [27]. A phase I dose-escalation study evaluating HER3-DXd in EGFR TKI-refractory EGFR-mutant NSCLC (n = 97) confirmed 5.6 mg/kg/21 days as the recommended dose [28]. The dose-expansion study included patients with EGFR-mutant NSCLC refractory to EGFR TKIs and platinum chemotherapy (cohort 1, n = 45) and patients with NSCLC without driver oncogenes or EGFR-mutant NSCLC (cohort 2). The most common adverse events were fatigue (64%) and nausea (60%), while the most common grade ≥3 AEs were thrombopenia, neutropenia and fatigue. Treatment-related ILD occurred in 7% of patients. Considering tumoral activity, the ORR was 22% for all pooled patients and 17% for the platinum chemotherapy/osimertinib-refractory NSCLC patients. The median PFS was 8.2 months and OS was not reached [29]. Early clearance (between 3 and 6 weeks after the beginning of HER3-Dxd) of EGFR-activating mutations was associated with longer median PFS (8.3 vs. 4.4 months; HR 0.33, 95% CI 0.13–0.81). Tumour activity in patients with HER3-DXd was observed regardless of the type of EGFR gene alterations [29]. Yu et al. reported the OS data for patients with EGFR-TKI and platinum chemotherapy-refractory EGFR-mutated NSCLC, with a median OS of 15.8 months (95% CI 10.8–21.5) [28]. HERTHENA-lung01 was a phase II trial evaluating HER3-DXd in EGFR-mutant NSCLC after progression on-treatment EGFR TKIs and platinum chemotherapy. The median PFS and OS were 5.5 and 11.9 months, respectively. Similar toxicity profiles were reported [30]. Both the HERTHENA-lung01 and U31402-A-U102 studies reported evidence efficacy of HER3-DXd in refractory EGFR-mutated NSCLC patients. Some studies are ongoing; HERTHENA-lung02 is a phase III trial including EGFR-mutant NSCLC after progression on EGFR TKIs (ClinicalTrials.gov identifier: NCT05338970). A phase I trial evaluated HER3-DXd plus osimertinib in osimertinib-refractory or -naïve EGFR-mutant NSCLC (ClinicalTrials.gov identifier: NCT04676477).
4.4 CEACAM5
CEA-related cell adhesion molecule 5 belongs to the CEACAM family and is overexpressed in NSCLC. CEACAM5 promotes cell proliferation and migration [28].
4.4.1 Tusamitamab Ravtansine (Tus-rav)
Tusamitamab ravtansine (Tus-rav) is composed of a humanized IgG1 antibody against CEACAM5, a cleavable linker, and DM4, a microtubule polymerization inhibitor, as a payload. The dose-expansion study included patients with NSCLC with CEACAM5 expression. In this study, the treatment-related adverse events were keratopathy/keratitis (38% all grade, 10.9% grade ≥3), fatigue (37% all grade, 4% grade ≥3), peripheral neuropathy (27.7% all grade, 1.1% grade ≥3), diarrhoea (22% all grade, 1.1% grade ≥3) and dyspnoea (21.7% all grade, 10.9% grade ≥3). The ORR was correlated with the CEACAM5 expression rate, with ORRs at 7.1% and 20.3% in the moderate and high expression cohorts, respectively [31]. Based on these results, a phase III study comparing Tus-rav (100 mg/m2/4 days) versus docetaxel as second-line therapy after failure under checkpoint inhibitor and platinum-based chemotherapy is ongoing. The enrolled population includes patients with non-squamous NSCLC with CEACAM5 expression (ClinicalTrials.gov identifier: NCT04154956).
4.5 c-MET
The MET proto-oncogene encodes a cell surface receptor tyrosine kinase that is activated by its ligand hepatocyte growth factor. Twenty-five percent of NSCLC cells express the protein MET (according to immunohistochemistry [IHC], a threshold of at least 2+ staining in 50% of cells was considered positive). MET amplification and mutation occurred in 3% and 2%, respectively, of patients with squamous LC.
4.5.1 Telisotuzumab Vedotin (Tel-Ved)
Telisotuzumab vedotin (Tel-Ved) is a first-class ADC composed of an anti-c-MET antibody linked to monomethyl auristatin E, a potent microtubule inhibitor. The phase II Lung-MAP trial is based on the first human study in which the selected dose was 2.7 mg/kg/21 days. The ORR was 18.8% (partial response), which occurred only in the squamous LC population. Therefore, the Lung-MAP trial included a population of patients with squamous LC and positive expression of c-MET (IHC). Among the 28 patients included, 15 had not been treated with ICIs (cohort 1), and 13 had previously received immunotherapy (cohort 2). The ORRs were 18% and 0% in cohorts 1 and 2, respectively. The median PFS and OS were 2.4 and 5.6 months in cohort 1, and 1.6 and 4.8 months in cohort 2, respectively. There were three deaths (12%) possibly attributed to treatment: one patient in cohort 1 died from a bronchopulmonary haemorrhage, and two patients in cohort 2 died from pneumonitis [32]. The Luminosity trial is a phase I/II study that included squamous and non-squamous NSCLC patients with and without EGFR mutations and was based on intermediate (IHC 3+ in 25–50% of the cells) or high (IHC 3+ in 50% of the cells) expression of c-MET (phase I). All patients received Tel-Ved at a dose of 1.9 mg/kg/14 days. The results of the fourth interim analysis were presented at the American Society of Clinical Oncology (ASCO) annual meeting 2023. The ORR was 36.5% in the group with NSCLC without EGFR mutation (52.2% in the high c-MET subgroup and 24.1% in the intermediate c-MET subgroup), whereas it was 11.6% in the group with LC with EGFR mutation and 11.1% in the group with squamous NSCLC. Two deaths possibly attributed to treatment were observed in the squamous NSCLC cohort [33]. Patients with squamous NSCLC and the EGFR-mutated NSCLC population are not eligible to receive Tel-Ved regardless of c-MET expression. A phase III study that included non-squamous NSCLC patients with EGFR wild-type and positive expression of c-MET is in progress to compare Tel-Ved versus docetaxel (ClinicalTrials.gov identifier: NCT04928846).
The efficacy and toxicity outcomes for each ADC are summarized in Table 1. Table 2 summarizes the ongoing clinical trials for each ADC.
5 Discussion
TDM-1 was the first approved ADC in 2013 and was approved for metastatic breast cancer with HER2 overexpression [34]. In 2022, seven ADCs against HER2 (TDM-1, T-DXd), TROP2 (sacituzumab govitecan), Nectin-4 (enfortumab vedotin), tissue factor (tisotumab vedotin), and folate receptor alpha (mirvetuximab soravtansine-gynx) were approved for the treatment of solid tumours (HER2-positive breast cancer, triple-negative breast cancer, urothelial carcinoma, cervical cancer, and ovarian cancer) [35]. Among the several ADCs currently in development for LC treatment, only T-DXd is US FDA approved for metastatic HER2-mutant NSCLC [11]. In the future, the therapeutic landscape of LC could change with the approval of other drug conjugates. Many clinical trials evaluating the survival outcomes of patients with NSCLC with or without alterations in oncogenic drivers treated with ADC compared with patients treated with standard therapies are ongoing. This therapeutic class could become an alternative for EGFR TKI-resistant NSCLC and chemoimmunotherapy-refractory wild-type NSCLC. Moreover, the combination of ADCs with other drugs, such as checkpoint inhibitors, could be an interesting therapeutic option in NSCLC. Studies are ongoing to determine the toxicity profiles and anti-tumour effects of these combinations.
To optimize the efficacy/toxicity ratio of ADCs and expand the knowledge on their mechanisms of action, further preclinical studies are needed. The target should be a protein present on tumour cells, tumour-associated cells or in the tumoral microenvironment [6]. To limit toxicity, the target must be highly expressed in tumour cells and expressed at a low level in normal tissue. However, anti-tumour activity is not correlated with the expression of the target. For example, in the IMMU-132 study evaluating sacituzumab govitecan, the expression of TROP2 samples was not sufficient for patient inclusion in the study [8]. Exploratory analyses of pivotal studies will be interesting to determine the subgroup of patients who had the most favourable benefit from these drugs.
In terms of the tolerance profile, severe adverse events were observed in the first clinical studies. Close monitoring is recommended in current practice for the early identification of toxicity. The literature suggests that toxicity is not correlated with the expression of the target in healthy tissue but rather depends on the payload. Similar toxicity was reported in three studies evaluating three different ADCs but with the same payload (enfortumab vedotin, polatuzumab vedotin and tisotumab vedotin) [36,37,38]. The target was different, but the same class of payloads was used. A meta-analysis of studies evaluating the clinical outcomes of ADCs in oncology between 2000 and 2023 revealed that off-target delivery of the payload is the major factor influencing toxicity [39]. In the future, new-generation ADCs with the most favourable toxicity profile are needed to limit off-target effects.
6 Conclusions
Several ADCs for LC are currently under development. T-DXd is the only approved ADC for patients with metastatic HER2-mutated NSCLC. ADCs targeting TROP2, HER2, HER3, CEACAM5, and c-MET showed anti-tumour activity with manageable adverse effects. Interestingly, this novel therapeutic class is active in NSCLC with and without alterations in oncogenic drivers. In the future, the therapeutic landscape could be changed to propose new drug options for patients with checkpoint inhibitor-refractory or EGFR TKI-refractory NSCLC. Studies evaluating the combination of ICIs or EGFR TKIs with an ADC are needed. New-generation ADCs are under development to improve the toxicity/anti-tumour activity ratio.
References
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clinicians. 2023;73:17–48.
Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382:41–50.
Mok T, Camidge DR, Gadgeel SM, Rosell R, Dziadziuszko R, Kim D-W, et al. Updated overall survival and final progression-free survival data for patients with treatment-naive advanced ALK-positive non-small-cell lung cancer in the ALEX study. Ann Oncol. 2020;31:1056–64.
Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N Engl J Med. 2018;378:2078–92.
De Castro G, Kudaba I, Wu Y-L, Lopes G, Kowalski DM, Turna HZ, et al. Five-year outcomes with pembrolizumab versus chemotherapy as first-line therapy in patients with non–small-cell lung cancer and programmed death ligand-1 tumor proportion score ≥1% in the KEYNOTE-042 study. J Clin Oncol. 2023;41:1986–91.
Chau CH, Steeg PS, Figg WD. Antibody–drug conjugates for cancer. Lancet. 2019;394:793–804.
Bardia A, Hurvitz SA, Tolaney SM, Loirat D, Punie K, Oliveira M, et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N Engl J Med. 2021;384:1529–41.
Bardia A, Messersmith WA, Kio EA, Berlin JD, Vahdat L, Masters GA, et al. Sacituzumab govitecan, a Trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann Oncol. 2021;32:746–56.
Tagawa ST, Balar AV, Petrylak DP, Kalebasty AR, Loriot Y, Fléchon A, et al. TROPHY-U-01: a phase II open-label study of sacituzumab govitecan in patients with metastatic urothelial carcinoma progressing after platinum-based chemotherapy and checkpoint inhibitors. J Clin Oncol. 2021;39:2474–85.
Modi S, Saura C, Yamashita T, Park YH, Kim S-B, Tamura K, et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N Engl J Med. 2020;382:610–21.
Li BT, Smit EF, Goto Y, Nakagawa K, Udagawa H, Mazières J, et al. Trastuzumab deruxtecan in HER2-mutant non–small-cell lung cancer. N Engl J Med. 2022;386:241–51.
Ogitani Y, Hagihara K, Oitate M, Naito H, Agatsuma T. Bystander killing effect of ds -8201a, a novel anti-human epidermal growth factor receptor 2 antibody–drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016;107:1039–46.
Nguyen TD, Bordeau BM, Balthasar JP. Mechanisms of ADC toxicity and strategies to increase ADC tolerability. Cancers. 2023;15:713.
Fu Z, Li S, Han S, Shi C, Zhang Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Sig Transduct Target Ther. 2022;7:93.
Radocha J, Van De Donk NWCJ, Weisel K. Monoclonal antibodies and antibody drug conjugates in multiple myeloma. Cancers. 2021;13:1571.
Tai Y-T, Mayes PA, Acharya C, Zhong MY, Cea M, Cagnetta A, et al. Novel anti–B cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123:3128–38.
Uppal H, Doudement E, Mahapatra K, Darbonne WC, Bumbaca D, Shen B-Q, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res. 2015;21:123–33.
Dum D, Taherpour N, Menz A, Höflmayer D, Völkel C, Hinsch A, et al. Trophoblast cell surface antigen 2 expression in human tumors: a tissue microarray study on 18,563 tumors. Pathobiology. 2022;89:245–58.
Shimizu T, Sands J, Yoh K, Spira A, Garon EB, Kitazono S, et al. First-in-human, phase I dose-escalation and dose-expansion study of trophoblast cell-surface antigen 2–directed antibody-drug conjugate datopotamab deruxtecan in non–small-cell lung cancer: TROPION-PanTumor01. J Clin Oncol. 2023;41(29):4678–87.
Inamura K, Yokouchi Y, Kobayashi M, Ninomiya H, Sakakibara R, Subat S, et al. Association of tumor TROP2 expression with prognosis varies among lung cancer subtypes. Oncotarget. 2017;8:28725–35.
Paz-Ares L, Ahn M-J, Lisberg AE, Kitazono S, Cho BC, Blumenschein G, et al. 1314MO TROPION-Lung05: datopotamab deruxtecan (Dato-DXd) in previously treated non-small cell lung cancer (NSCLC) with actionable genomic alterations (AGAs). Ann Oncol. 2023;34:S755–6.
Ahn M-J, Lisberg A, Paz-Ares L, Cornelissen R, Girard N, Pons-Tostivint E, et al. LBA12 Datopotamab deruxtecan (Dato-DXd) vs docetaxel in previously treated advanced/metastatic (adv/met) non-small cell lung cancer (NSCLC): results of the randomized phase III study TROPION-Lung01. Ann Oncol. 2023;34:S1305–6.
Goto Y, Su W-C, Levy BP, Rixe O, Yang T-Y, Tolcher AW, et al. TROPION-Lung02: Datopotamab deruxtecan (Dato-DXd) plus pembrolizumab (pembro) with or without platinum chemotherapy (Pt-CT) in advanced non-small cell lung cancer (aNSCLC). J Clin Oncol. 2023;41(16 Suppl):9004.
Heist RS, Guarino MJ, Masters G, Purcell WT, Starodub AN, Horn L, et al. Therapy of advanced non–small-cell lung cancer with an SN-38-anti-trop-2 drug conjugate Sacituzumab Govitecan. J Clin Oncol. 2017;35:2790–7.
Fang W, Cheng Y, Chen Z, Wang W, Yin Y, Li Y, et al. SKB264 (TROP2-ADC) for the treatment of patients with advanced NSCLC: efficacy and safety data from a phase 2 study. J Clin Oncol. 2023;41(16 Suppl):9114.
Hotta K, Aoe K, Kozuki T, Ohashi K, Ninomiya K, Ichihara E, et al. A phase II study of trastuzumab emtansine in HER2-positive non-small cell lung cancer. J Thorac Oncol. 2018;13:273–9.
Li BT, Shen R, Buonocore D, Olah ZT, Ni A, Ginsberg MS, et al. Ado-trastuzumab emtansine for patients with HER2 -mutant lung cancers: results from a phase II basket trial. J Clin Oncol. 2018;36:2532–7.
Yu HA, Baik C, Kim D-W, Johnson ML, Hayashi H, Nishio M, et al. Translational insights and overall survival in the U31402-A-U102 study of patritumab deruxtecan (HER3-DXd) in EGFR-mutated NSCLC. Ann Oncol. 2024. https://doi.org/10.1016/j.annonc.2024.02.003.
Jänne PA, Baik C, Su W-C, Johnson ML, Hayashi H, Nishio M, et al. Efficacy and safety of patritumab deruxtecan (HER3-DXd) in EGFR inhibitor-resistant, EGFR-mutated non-small cell lung cancer. Cancer Discov. 2022;12:74–89.
Yu HA, Goto Y, Hayashi H, Felip E, Chih-Hsin Yang J, Reck M, et al. HERTHENA-Lung01, a phase II trial of patritumab deruxtecan (HER3-DXd) in epidermal growth factor receptor-mutated non–small-cell lung cancer after epidermal growth factor receptor tyrosine kinase inhibitor therapy and platinum-based chemotherapy. J Clin Oncol. 2023;41:5363–75.
Gazzah A, Ricordel C, Cousin S, Cho BC, Calvo E, Kim TM, et al. Efficacy and safety of the antibody-drug conjugate (ADC) SAR408701 in patients (pts) with non-squamous non-small cell lung cancer (NSQ NSCLC) expressing carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5). J Clin Oncol. 2020;38(15 Suppl):9505.
Waqar SN, Redman MW, Arnold SM, Hirsch FR, Mack PC, Schwartz LH, et al. A phase II study of telisotuzumab vedotin in patients with c–MET-positive stage IV or recurrent squamous cell lung cancer (LUNG-MAP sub-study S1400K, NCT03574753). Clin Lung Cancer. 2021;22:170–7.
Camidge DR, Bar J, Horinouchi H, Goldman JW, Moiseenko FV, Filippova E, et al. Telisotuzumab vedotin (Teliso-V) monotherapy in patients (pts) with previously treated c-Met–overexpressing (OE) advanced non-small cell lung cancer (NSCLC). J Clin Oncol. 2022;40(16 Suppl):9016.
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–91.
Tarantino P, Carmagnani Pestana R, Corti C, Modi S, Bardia A, Tolaney SM, et al. Antibody–drug conjugates: smart chemotherapy delivery across tumor histologies. CA Cancer J Clinicians. 2022;72:165–82.
De Bono JS, Concin N, Hong DS, Thistlethwaite FC, Machiels J-P, Arkenau H-T, et al. Tisotumab vedotin in patients with advanced or metastatic solid tumours (InnovaTV 201): a first-in-human, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20:383–93.
Rosenberg J, Sridhar SS, Zhang J, Smith D, Ruether D, Flaig TW, et al. EV-101: a phase I study of single-agent enfortumab vedotin in patients with nectin-4–positive solid tumors, including metastatic urothelial carcinoma. J Clin Oncol. 2020;38:1041–9.
Palanca-Wessels MCA, Czuczman M, Salles G, Assouline S, Sehn LH, Flinn I, et al. Safety and activity of the anti-CD79B antibody–drug conjugate polatuzumab vedotin in relapsed or refractory B cell non-Hodgkin lymphoma and chronic lymphocytic leukaemia: a phase 1 study. Lancet Oncol. 2015;16:704–15.
Zhu Y, Liu K, Wang K, Zhu H. Treatment-related adverse events of antibody–drug conjugates in clinical trials: a systematic review and meta-analysis. Cancer. 2023;129:283–95.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflict of interest
Laura Bender Somme: MSD, Novartis, Daiichi Sankyo, Takeda. Christos Chouaid: AstraZeneca, BI, GSK, Roche, Sanofi Aventis, BMS, MSD, Lilly, Novartis, Pfizer, Takeda, Bayer and Amgen. Roland Schott: Bristol-Myers Squibb/Celgene, Roche, AstraZeneca. Laurent Greillier: BMS, MSD, Takeda, Pfizer, Roche, Novartis, Amgen, Lilly, Jansen. Fabien Moinard-Butot and Jean-Baptiste Barbe-Richaud declare they have no conflicts of interest that might be relevant to the contents of this manuscript.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Data availability
Not applicable.
Code availability
Not applicable.
Author contributions
LBS, RS and CC performed the material preparation, data collection and analysis. LBS wrote the first draft of the manuscript. All authors commented on previous versions of the manuscript, and read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Somme, L.B., Chouaid, C., Moinard-Butot, F. et al. Antibody-Drug Conjugates as Novel Therapeutic Agents for Non-Small Cell Lung Carcinoma with or without Alterations in Oncogenic Drivers. BioDrugs 38, 487–497 (2024). https://doi.org/10.1007/s40259-024-00660-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-024-00660-7