
Abstract
In recent years, subcutaneous administration of biotherapeutics has made significant progress. The self-administration market for rheumatoid arthritis has witnessed the introduction of additional follow-on biologics, while the first subcutaneous dosing options for monoclonal antibodies have become available for multiple sclerosis. Oncology has also seen advancements with the authorization of high-volume subcutaneous formulations, facilitated by the development of high-concentration formulations and innovative delivery systems. Regulatory and Health Technology Assessment bodies increasingly consider preference data in filing dossiers, particularly in evaluating novel drug delivery methods. The adoption of a pharmacokinetic-based clinical bridging approach has become standard for transitioning from intravenous to subcutaneous administration. Non-inferiority studies with pharmacokinetics as the only primary endpoint have started deviating from traditional randomization schemes, favoring the subcutaneous route and comparing with historical intravenous data. While nonclinical and computational models made progress in predicting safety and immunogenicity for subcutaneously dosed antibodies, clinical trial evidence remains essential due to inter-individual variations and the impact of formulation parameters on anti-drug antibody formation. Ongoing technological advancements and the expanding knowledge base on pharmacokinetic–pharmacodynamic correlation across specialty areas are expected to further accelerate clinical development of subcutaneous biologics.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
In the last 5 years, subcutaneous (SC) administration of biotherapeutics has evolved in the selected disease area archetypes rheumatoid arthritis (RA), multiple sclerosis (MS), and high-volume monoclonal antibodies (mAbs) in oncology. |
Follow-on biologics with similar product presentations to established brands have entered the RA market. Subcutaneously dosed mAbs in prefilled syringe or autoinjector formats have been introduced in the MS market, and additional large-volume SC dosing options are authorized in oncology. |
Preference studies confirm the broad acceptance of high SC dosing volumes over intravenous (IV) infusion. |
The pharmacokinetic-based bridging approach from IV to SC administration, initially developed for SC trastuzumab, is now a standard across specialty areas. |
This article proposes molecule-agnostic measures to accelerate clinical development of SC dosing alternatives and injection device platforms for mAbs, aiming to facilitate decentralized care through reduced dosing complexity. |
1 Introduction
This article serves as a sequel to a previously published review titled “Subcutaneous Administration of Biotherapeutics: An Overview of Current Challenges and Opportunities” in BioDrugs, dated back to 2018 [1]. The update takes into consideration the growing significance of subcutaneous (SC) dosing of biotherapeutics, specifically monoclonal antibodies (mAbs), in various disease domains during the past 5 years. A pivotal driver of this advancement has been the COVID-19 pandemic, which underscored the necessity for administering biotherapeutics via parenteral routes in decentralized healthcare environments [2].
In 2018, despite the existence of prior experience with subcutaneous self-administration of large-volume immunoglobulin products, there was still skepticism regarding the general tolerability and acceptance of high-dose and high-volume mAb administration. Today, this methodology is widely employed across various indications and has become a standard practice [3]. The first large-volume on-body delivery systems and corresponding treatment management strategies have been implemented to enable convenient administration of such biotherapeutics in the comfort of people's homes.
There is a growing recognition that sustainability aspects must play a central role not only in the final presentation of a medical product but also in considering the costs and resources involved in its development [4]. Therefore, it is imperative to capitalize on previous knowledge and experiences in the development and commercialization of SC formulations for high-dose mAbs. The knowledge acquired in one disease area can be effectively applied to other indications, eliminating the need to duplicate assessments for each new molecule that utilizes an established drug delivery technology.
The focus of this review is on mAbs available with both an intravenous (IV) and an SC dosing regimen and their respective follow-on biologics. The article offers recommendations on the rationale, timing, and approach for initiating SC development in comparison with IV development. Furthermore, this article outlines the progression of the molecule-agnostic pharmacokinetic-based clinical bridging pathway for SC dosing alternatives during the past decade and presents potential strategies to expedite further development.
2 Subcutaneous (SC) Administration of Biotherapeutics Across Specialty Areas
In the 2018 review article [1], rheumatoid arthritis (RA), multiple sclerosis (MS), and high-dose mAbs in oncology were chosen as representative disease areas for subcutaneously administered biotherapeutics. The present review gives an overview of approved product presentations in these disease areas as of July 2023.
During that period, RA already served as a field where SC administration at home, including self-administration, was an established norm. This practice was reinforced by a wide range of available devices, including prefilled syringes and pen devices. In late 2017, the first partially reusable autoinjector (AI) for etanercept [5] was introduced to the market, aiming to enhance the injection process in remote healthcare settings.
Since 2018, the market for RA has witnessed the approval of a greater variety of SC biosimilar treatment options (Table 1). To date, the number of follow-on biologics that have received Marketing Authorization (MA) through the European Medicines Agency’s (EMA) centralized procedure has risen from two to ten for adalimumab, and from two to three for etanercept [6]. During the same period, there has been a parallel increase in the number of biosimilar approvals by the United States (US) Food and Drug Administration (FDA) through the abbreviated 351(k) pathway [7]. The approvals for adalimumab have grown from two to nine, while those for etanercept have increased from one to two. It is noteworthy that the dosing regimens and product presentations of these biosimilars are comparable to those of the original versions [8]. Up until 2018, three infliximab biosimilars had been approved both in the European Union (EU) and the US that are administered intravenously, such as the original infliximab product (Remicade) that is exclusively administered through IV dosing [5, 8]. In 2020, the EMA authorized the first SC follow-on biologic for infliximab [9] as an extension to the Marketing Authorization granted for the IV formulation. In contrast, the FDA mandated the manufacturer to submit a new Biologic License Application (BLA) under the 351(a) pathway [10].
While the SC follow-on biologics for adalimumab and etanercept have obtained approvals from both the EMA and FDA, the presence of patents for the original formulations has resulted in delays for initial biosimilar launches in the US. These launches have been pushed back to 2023 and 2029, respectively [11,12,13]. Consequently, the first adalimumab follow-on biologics, known as Amjevita and Hadlima, entered the market in January and July 2023.
A notable regulatory distinction between the EU and the US regarding biosimilars is the classification of ‘interchangeability.’ The term ‘interchangeability’ pertains to the ability of a pharmacist to substitute an equivalent/interchangeable medicine without consulting the prescriber [14]. According to the EMA, once a biosimilar is approved in the EU, it can be considered interchangeable [15]. Under the FDA approval process, there are additional requirements specific to interchangeable biosimilars, such as the conduct of a switching study in which participants alternate between the reference product and the interchangeable biosimilar. Participants are then compared with people exclusively using the reference product [16]. Out of the four follow-on biologics that currently meet the FDA's interchangeability criteria, only one adalimumab biosimilar, Cyltezo, has been classified as interchangeable for the treatment of RA since 2021 [17].
In 2018, the SC injectable market for MS primarily consisted of interferon treatments, available in diverse presentations like prefilled syringes, pens, autoinjectors, and vials for at-home and self-administration. During that period, all authorized mAbs including the anti-α4 integrin mAb natalizumab, the anti-cluster of differentiation 52 (CD52) mAb alemtuzumab, and the anti-cluster of differentiation 20 (CD20) mAb ocrelizumab, were exclusively available with IV dosing regimens [18]. Ofatumumab, an anti-CD20 monoclonal antibody, has received FDA and EMA authorization for SC administration since 2020 and 2021, respectively. This new dosing option utilizes low-volume prefilled syringe and pen devices, allowing for administration outside of centralized settings [5, 8]. A new SC formulation of natalizumab, administered using two prefilled syringes per dose, has been available in the EU since 2021 for healthcare provider-supervised injection [5]. However, in the US, a complete response letter was issued for the supplemental BLA regarding the SC dosing regimen [19]. Additionally, a SC dosing alternative for ocrelizumab with a 6-monthly (q6m) regimen is currently in phase III clinical development [20]. As of now, no follow-on monoclonal antibodies have been approved for MS (Table 2).
Until 2018, in the field of high-volume SC mAb presentations in oncology, trastuzumab was approved by the EMA for HER2-positive breast cancer [5], and rituximab had approvals from both the EMA and FDA for B-cell malignancies [5, 8]. Trastuzumab is now also authorized in the US [8]. The first fixed-dose combination of SC pertuzumab and trastuzumab for HER2-positive breast cancer and daratumumab SC for multiple myeloma were launched on the market [5, 8]. Initially, all these monoclonal antibodies were approved individually with IV dosing regimens (Table 3).
To enhance the dispersion of the injected dosing volume within the interstitial tissue, the mAbs are co-formulated with the recombinant enzyme human hyaluronidase PH20 (rHuPH20). The classification of rHuPH20 differs between regulatory agencies. The EMA considers rHuPH20 as a novel excipient [21], while the FDA defines it as an active ingredient and specifically identifies it as an endoglycosidase [22]. Hence, in the EU, the SC dosing alternatives are authorized as an extension of the existing marketing authorization through a type 2 variation. Both the IV and SC versions of the medication have the same trade name and label in the European Summary of Product Characteristics (SmPC) [5].
In the US, the SC formulations of trastuzumab, rituximab, and daratumumab were filed under a new BLA, and the IV and SC formulations have distinct labels [8]. The SC formulations in the US, which include rHuPH20, are distinguished by specific name affixes. They are known as Hylecta (trastuzumab), Hycela (rituximab), and Faspro (daratumumab), respectively [8]. The pertuzumab-trastuzumab fixed-dose combination obtained a new Market Authorization Application (MAA) and was approved through a new BLA in the US. Previously, the IV dosing regimen for pertuzumab and trastuzumab involved sequential administration of the two monoclonal antibodies using separate formulations [8]. Significant progress is being made in the development of SC dosing alternatives for high-dose mAbs in the field of cancer immunotherapy with a number of molecules currently undergoing phase III clinical investigation [23,24,25].
Unlike the RA market, approved biosimilars for high-dose mAbs such as rituximab and trastuzumab are currently limited to IV dosing regimens [6, 7]. This divergence can be attributed to several factors, including the occasional occurrence of severe infusion-related reactions for mAbs in oncology, the unavailability of the dispersion enhancer rHuPH20, and the continued complexity of high-volume SC administration. Currently, these factors require mAbs in cancer care to be administered in a healthcare institutional setting. This contrasts with the rheumatoid arthritis (RA) space, where at-home dosing of biotherapeutics is a crucial part of the standard of care.
Furthermore, the introduction of branded SC formulations for trastuzumab and rituximab in the EU in 2013 and 2014 [5], respectively, was a significant factor that delayed the potential development of biosimilar SC formulations. In the US, access to the SC versions of these mAbs was not possible until the approval of Rituxan Hycela in 2017 and Herceptin Hylecta in 2019 [8].
3 Preference Studies for High-Volume SC Biotherapeutic Administration
3.1 Evidence Update
The 2018 review [1] examined the advantages of high-volume SC administration (typically >5 mL) compared with IV infusions, focusing on immunoglobulin replacement therapy in primary immunodeficiency and the use of trastuzumab and rituximab in cancer treatment. It was noted that while SC immunoglobulins can be self-administered at home, SC trastuzumab and rituximab require administration by a healthcare provider in a controlled setting. The data indicated that SC administration is preferred due to its reduced invasiveness and dosing complexity, resulting in cost savings for healthcare institutions.
Since 2018, additional insights have emerged regarding preference and healthcare resource utilization for the SC fixed-dose combination of pertuzumab and trastuzumab in HER2-positive breast cancer and daratumumab in multiple myeloma. These newer products have larger injection volumes compared with previously approved monoclonal antibodies, with 15 mL and 10 mL for the loading and maintenance doses of the pertuzumab-trastuzumab combination and 15 mL for daratumumab in all cycles. Clinical trials investigating these higher-volume mAb formulations have confirmed the preference for SC dosing over IV administration and have demonstrated time savings in terms of drug preparation, administration, patient care, and patient chair usage, with reduced involvement of healthcare providers [26,27,28].
In 2022, a systematic literature review on time and resource use costs compared SC and IV administration for people with cancer, analyzing 72 relevant publications [29]. The majority of assessments focused on the high-volume formulations of trastuzumab and rituximab, which were discussed in the previous 2018 review. The extensive evidence from these studies consistently supported the findings of time savings associated with the preparation and administration of SC therapies.
3.2 The Regulatory Perspective
The FDA encourages the submission of patient preference information (PPI) to inform decision-making processes. In the US Prescribing Information (USPI) [8], PPI is utilized to inform labeling claims for SC dosing alternatives compared with IV infusion regimens. While submitting PPI is voluntary, the FDA acknowledges its usefulness, particularly in evaluating the benefit–risk profile of devices when patient decisions are ‘preference sensitive.’ This includes situations where multiple treatment options exist without clear superiority, uncertainty or variability in the evidence supporting one option, or when patient views on benefits and risks vary from those of healthcare professionals [30].
In the EU, the inclusion of patient preference studies (PPS) in regulatory assessments currently occurs on an ad-hoc basis without a formal guideline available [31]. The PREFER initiative, funded by the Innovative Medicines Initiative (IMI), aims to provide systematic methodologies and recommendations for integrating patient perspectives throughout the development, approval, and post-approval stages of new therapies [32]. The initiative has received a positive draft opinion from the EMA, with the agency stating that the inclusion of PPS data in regulatory documents, such as the Clinical Overview or the European Public Assessment Report (EPAR), would be relevant to the regulatory decision-making process and benefit–risk assessment, as well as informing prescribers and users of the medicinal product [33]. The joint qualification of IMI PREFER by EMA and the European Network for Health Technology Assessment (EUnetHTA) concludes that the initiative represents a major advancement in conducting and evaluating PPS in decision-making processes [34].
The patient preference results from the PrefHer, PHranceSCa, and PrefMab studies are included in a dedicated paragraph in section 14, ‘Patient Experience,’ in the USPI for the respective SC formulations of trastuzumab, the pertuzumab-trastuzumab fixed-dose combination, and rituximab. In the corresponding European SmPCs, the data from preference studies are mentioned under Sect. 4.2, ‘Posology and method of administration,’ providing information on switching between the IV and SC formulations. However, for daratumumab, for which no dedicated preference trial had been conducted at the time of approval of the SC formulation, neither the USPI nor the SmPC contain references to ‘patient experience’ or ‘preference’ [5, 8].
4 Developing SC versus IV Formulations for Biotherapeutics—Rational Decision Making and Practical Considerations
Until recently, it was common to initiate a development program directly with a SC dosing regimen only for low-dose/low-volume mAb products with a favorable risk–benefit profile. This was observed in diseases like RA and MS, where the biologicals used are well tolerated, allowing for non-supervised administration after treatment initiation by an experienced healthcare provider. The use of standard prefilled syringe, autoinjector, and pen device platforms, accommodating dosing solutions of approximately 2 mL or less, facilitated self-administration outside of a controlled healthcare setting.
In the past, development programs for higher dose mAbs typically began with an IV regimen, and SC dosing options were later introduced as part of lifecycle management. This approach considered factors such as infusion-related reactions and the absence of suitable drug delivery technologies. However, advancements in high-concentration technologies [35], the availability of dispersion enhancers like hyaluronidase [36], and the development of high-volume on-body delivery systems [37] have made it possible to initiate clinical trials for high-dose mAbs directly with a SC formulation, allowing for a timely launch of the SC product without compromising its development timeline.
This section will discuss considerations specific to high-dose/high-volume mAbs, as many of these mAbs were historically introduced to the market with IV regimens. Given the remaining technical and practical challenges associated with implementing an SC dosing regimen, manufacturers may adopt various approaches, including launching the product with either IV administration only, SC administration only, or both routes of administration.
4.1 Rational Decision Making
Manufacturers have the option to consider different development scenarios for IV versus SC administration (Fig. 1). In scenario 1, only an IV formulation is developed. This is typically the case for molecules where an early and high maximum concentration (Cmax) is necessary for optimal therapeutic effect. While this requirement is not common for mAb-based treatments, other biotherapeutics like alteplase or tenecteplase for acute ischemic stroke or acute myocardial infarction fall into this scenario [5, 8]. Additionally, antibody–drug conjugates that may be cleaved in the interstitial space (data on file, F. Hoffmann-La Roche) cannot be delivered subcutaneously, and drugs that require administration in a controlled healthcare setting do not necessarily require an SC formulation.

Subcutaneous versus intravenous formulation—Development scenarios. Cmax maximum serum concentration, IV intravenous, SC subcutaneous
Scenario 2 involves the development of a mAb with an SC formulation only. This approach is suitable for molecules used in monotherapy or in combination with other subcutaneously or orally administered drugs. Introducing an IV regimen would add complexity and inconvenience to the overall dosing regimen. Additionally, if the molecule is safe and well tolerated, and at-home self-administration is already established in the indication, there is no need to offer an IV treatment alternative.
In scenario 3, manufacturers pursue the development of a mAb with both IV and SC formulations. This strategy allows for greater flexibility in dosing options, taking into consideration both individual preferences and capabilities and country-specific reimbursement models. The approach enables the harmonization of dosing regimens with combination partners and facilitates the development of fixed-dose combinations tailored to specific indications [3].
Considering the preference for SC administration and its potential for decentralized dosing, the authors recommend prioritizing the development of a SC formulation. This is particularly relevant for high-dose mAbs, where the feasibility of a stable high-concentration dosing solution compatible with standard delivery devices should be evaluated early on. For high-dose mAbs, it is advisable to include an IV formulation in phase I clinical trials, even if the intention is to launch a SC formulation exclusively. This allows for a fallback option if SC dosing is not feasible or well tolerated. Moreover, clinical data on the safety of high maximum serum levels (Cmax) with the IV route can be valuable as supporting evidence for future dose adjustments, alternative formulations, or devices that may impact the molecule's absorption profile.
4.2 Clinical Development Pathway of SC Dosing Alternatives for mAbs—Current Status
The clinical development program for subcutaneously dosed mAbs is contingent upon whether this route is the first to enter the market or if a manufacturer is introducing a novel SC dosing alternative for an already established IV regimen. In the case of developing a new molecule with an SC formulation, the regulatory pathway aligns with the same paradigm as an IV formulation. Manufacturers are required to conduct nonclinical assessments encompassing pharmacology, pharmacokinetics, and toxicology, as well as a comprehensive clinical development program to demonstrate efficacy and safety that supports the submission of a new MAA or BLA.
This section provides a high-level overview of the clinical development pathways for SC dosing alternatives for mAbs with established IV regimens in oncology. It discusses their adaptation to other specialty areas and suggests measures to streamline development for future molecules. For more details, refer to earlier review articles on the topic [1, 37].
4.3 Establishing a Molecule-Agnostic Clinical Bridging Approach from IV to SC Dosing for mAbs in Oncology and Hematology
The established bridging approach for transitioning from an IV to an SC regimen for the same mAb relies on utilizing the same antibody in different formulations. It is anticipated that with comparable exposure (measured as area under the serum concentration–time curve [AUC]), the systemic safety profile of the mAb remains unchanged regardless of the administration route. To gather supportive evidence, manufacturers undertake a dedicated preclinical and toxicology bridging program for the SC formulations.
Following SC administration, it is observed that at comparable AUC, Cmax is lower compared with IV administration due to the slower absorption into the systemic circulation. However, to account for the higher AUC at earlier time points with IV administration, minimum or trough concentrations (Cmin/Ctrough) are higher following SC administration (Fig. 2). Based on this, the clinical evidence package is designed to demonstrate several key aspects: (i) pharmacokinetic non-inferiority (Ctrough and/or AUC) between the IV and SC formulations to ensure comparable efficacy, (ii) consistency in safety, tolerability, and immunogenicity profiles, and (iii) non-inferior efficacy. The ultimate goal is to extrapolate the clinical data generated in one indication to other indications for the same mAb.

Impact of SC versus IV delivery on the pharmacokinetic profile of a mAb. Cmax maximum serum concentration, Cmin minimum serum concentration, IV intravenous, mAb monoclonal antibody, SC subcutaneous
4.4 Concept Development and Bridging Programs for Approved Products
Table 4 presents the IV and SC dosing regimens along with the clinical trial designs for high-volume mAb presentations with approved formulations for both routes. All manufacturers employed the pharmacokinetic-based bridging approach as described.
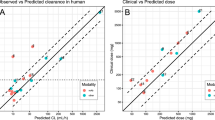
The concept was initially developed for trastuzumab in HER2-positive breast cancer [38]. The IV form of the mAb was marketed with both weekly (q1w) and every 3 weeks (q3w) dosing regimens adjusted according to body weight. The selection of the two IV dose strengths aimed to achieve comparable mean AUC, surpassing the target concentration based on preclinical xenograft studies. Due to the different individual dose levels, the maximum serum levels with the q1w regimen were significantly lower compared with the q3w regimen. However, a comparison of historical data still demonstrated comparable efficacy [39]. Pharmacokinetic modeling, utilizing data from the established IV formulation and data from a phase I/Ib dose-finding and confirmation study with the SC formulation in a mixed population of healthy volunteers and participants with HER2-positive early breast cancer (eBC) in the adjuvant setting [40], predicted that a q3w SC fixed-dose regimen, at comparable AUC, would result in non-inferior Ctrough compared with the body weight-adjusted IV regimens. Furthermore, the predicted Cmax of the SC regimen would fall within the range of the q1w and q3w IV regimens (Fig. 3). The predicted dose was subsequently validated in a phase III non-inferiority study with a 1:1 randomization of participants with HER2-positive eBC [41].

Evidence generation with pharmacokinetic-based clinical bridging approach. Hypothesis generation based on available trastuzumab pharmacokinetic data following intravenous administration. The PK profile of the SC formulation is bridged by the q3w and q1w IV regimens. Subcutaneous dose selection concept: Ctrough as least as high as with IV regimen; Cmax bracketed by Cmax of q1w and q3w IV regimens; comparable AUC with IV and SC regimens. *Serum trough concentration (Ctrough) of 20 µg/mL depicts PK target established from preclinical xenograft models. Ctrough serum trough concentration, IV intravenous, mg milligram, kg kilogram, ml milliliter, µg microgram, SC subcutaneous, q1w weekly, q3w every 3 weeks. This figure was published in Bittner B, Schmidt J. Formulation and device lifecycle management: A guidance for researchers and drug developers. 1st ed. William Andrew Publishing (Elsevier); 2022 [37].
Both the population of healthy volunteers and individuals diagnosed with HER2-positive eBC were considered relatively homogeneous and as such ‘sensitive,’ enabling pharmacokinetic comparisons with a reasonably low sample size. This approach of generating comparative pharmacokinetic data in a sensitive population is now recommended in the biosimilar guidelines of both the EMA and the FDA [42, 43]. To underscore the importance of the pharmacokinetic-based clinical bridging approach, the pivotal phase III study selected Ctrough as a co-primary endpoint along with pathological complete response (pCR). Upon initial approval by the EMA in 2013, the data generated in eBC could be extrapolated to metastatic breast cancer (mBC), an indication with the same IV dose and dosing regimen as eBC [44].
The same clinical development concept and adaptive trial design were subsequently implemented for rituximab in B-cell malignancies to bridge from a body surface area-adjusted IV to a fixed SC dosing regimen [45]. In the case of rituximab, conducting clinical trials in healthy volunteers for dose finding was not feasible, necessitating the inclusion of participants diagnosed with follicular lymphoma (FL). Pharmacokinetic-based dose finding and confirmation were performed in participants with FL who responded to IV induction therapy [46], representing a relatively homogeneous and sensitive population with reduced target tissue load. Additional pharmacokinetic and efficacy data were collected in a phase III study in the induction setting, utilizing a 1:1 randomization scheme [47].
Following consultation with European Rapporteurs, supplemental clinical data were required for rituximab indications with different doses and dosing regimens. Consequently, a dedicated pharmacokinetic-based clinical study was conducted in chronic lymphocytic leukemia (CLL) [48], leading to regulatory approval in the EU in 2014. The pivotal studies that supported the filing of the SC formulation in the EU focused solely on Ctrough as the primary endpoint. Similar to trastuzumab, model-based dose selection for rituximab was conducted using available pharmacokinetic data on distribution and elimination from the IV regimen, along with absorption kinetics from the SC studies [46].
Unlike trastuzumab, the initial dose of the rituximab SC regimen is still administered via IV infusion. This deviation is due to the occurrence of sometimes severe infusion-related reactions (IRRs), which can be managed by reducing the infusion rate [49]. This measure was no longer feasible with SC bolus injection.
The SC dosing option for the anti-CD38 monoclonal antibody daratumumab in the treatment of multiple myeloma received authorization from the FDA and EMA in 2020 [5, 8]. In the pivotal phase III non-inferiority study involving individuals with relapsed or refractory multiple myeloma (RRMM), the co-primary endpoints were overall response rate and maximum Ctrough. Participants were randomly assigned in a 1:1 ratio to receive either the SC or IV formulation [50]. Similarly, in 2020, the SC fixed-dose combination of pertuzumab and trastuzumab for HER2-positive breast cancer was approved in both the US and the EU [5, 8]. The clinical development pathway for this combination followed the same approach as described for trastuzumab. Leveraging the established pharmacokinetic-based bridging approach, the Ctrough was the primary endpoint in the pivotal phase III non-inferiority study, while total pathological complete response (tpCR) served as a secondary endpoint [51].
The SC formulation of ravulizumab, an anti-complement component 5 (C5) monoclonal antibody, was approved by the US in 2022 for the treatment of adult patients with paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS) to inhibit complement-mediated thrombotic microangiopathy (TMA) [5, 8]. The pivotal phase III non-inferiority study for the SC formulation was conducted in individuals with PNH [52], a rare blood disorder with a global prevalence of 16 cases per million people [53]. Participants were enrolled with a 2:1 randomization scheme, receiving either the SC or IV formulation, and had prior treatment with the anti-C5 monoclonal antibody eculizumab. The first dose of ravulizumab was administered intravenously in both groups [52]. The second dose was given 15 days later following the predefined administration route. Subsequently, individuals in the SC arm received weekly doses for the remainder of the trial, while those in the IV arm did not receive additional doses until the primary analysis at day 71, following the approved every 8 weeks (q8w) regimen. After the primary analysis, all participants were offered the option to continue treatment with the SC formulation during an extension period of up to 172 days.
It is worth mentioning that unlike the approved high-volume mAb formulations in oncology, which are available in vial presentations for manual or semi-manual injection using a handheld syringe or infusion pump, SC ravulizumab was developed specifically with an on-body delivery system. This platform had previously received approval for the lipid-lowering anti-proprotein convertase subtilisin kexin type 9 (PCSK9) mAb, evolocumab [5, 8]. The 3.5-mL device was already integrated into the phase III trial described for SC ravulizumab [52].
4.5 Applying the Established Molecule-Agnostic Clinical Development Approach Across Specialty Areas—Ongoing Developments for High-Dose/High-Volume mAbs
Several other high-dose/high-volume mAbs are currently undergoing clinical development across different specialty areas, following the described pharmacokinetic-based bridging approach. For detailed trial designs, refer to Xu et al. [54], who systematically evaluated prior experiences in IV to SC development programs for therapeutic proteins.
In summary, pembrolizumab, nivolumab, atezolizumab in cancer immunotherapy, and ocrelizumab in multiple sclerosis have either completed or are in late-stage clinical development. The manufacturers are utilizing the pharmacokinetic-based clinical bridging approach, incorporating dose-finding studies and non-inferiority assessments with AUC or Ctrough as primary endpoints [20, 55,56,57].
4.6 Accelerating the Molecule-Agnostic Clinical Development Approach—The Target Product Profile
As the focus on healthcare cost and resource optimization grows, along with the demand for at-home dosing options, accelerating the development of SC dosing alternatives for mAbs has become a prominent concern for researchers and drug developers. To progress in a scientifically robust manner, three key questions need to be addressed:
-
1.
How can the clinical bridging program be streamlined by incorporating predictive nonclinical models, in vitro experiments, and computational tools?
-
2.
Can evidence from nonclinical and clinical bridging programs be applied to different mAbs and indications?
-
3.
What optimizations can be made to clinical trial designs and study conduct to achieve earlier key results while maintaining scientific validity?
Manufacturers start with establishing the target product profile for an SC dosing alternative by outlining the desired efficacy, safety, and tolerability profile, dosing regimen, treatment setting, extrapolation to other indications. This approach serves as a basis to define strategies for generating evidence for filing and commercialization (Table 5).
The next section examines the different components of the target product profile and provides an overview of available insights to help expedite the clinical bridging program. It explores the necessity of clinical trial data for bridging from IV to SC dosing, as well as strategies to minimize the size and duration of the development program. Focus is on scenarios where a mAb is currently approved or in advanced stages of clinical development using an IV dosing regimen. The objective for the manufacturer is to develop an SC dosing alternative that demonstrates non-inferior efficacy, safety, and tolerability.
4.6.1 Safety and Tolerability Profile
4.6.1.1 Infusion-Related Reactions
When evaluating the advantages and disadvantages of conducting larger clinical trials to bridge between IV and SC formulations of the same mAb, it is crucial to consider the clinical manifestation of potentially severe or fatal infusion-related reactions (IRRs) [58]. These reactions can be associated with cytokine release syndrome, characterized by an increase in inflammatory cytokines occurring approximately 90 min after the first infusion [59]. The incidence and severity of these systemic reactions are carefully assessed as an essential component of all clinical bridging studies [37], and early detection and management of IRRs are mandated [60].
It has been observed that SC dosing of certain mAbs, such as alemtuzumab in multiple sclerosis and daratumumab in multiple myeloma, may reduce the incidence and severity of IRRs. due to slower absorption into the systemic circulation from the interstitial tissue [61, 62]. However, this phenomenon is not consistently observed for all mAbs, and to make marketing claims regarding this potential advantage, manufacturers need to support them with data from clinical trial investigations. Additionally, although there have been advancements in developing predictive nonclinical models for T cell-associated toxicities, this research field is still in its early stages [63]. Therefore, ongoing and future clinical development programs will provide valuable clinical data to further refine and improve these predictive models.
4.6.1.2 Immunogenicity
SC mAb administration is perceived to have a higher immunogenicity risk compared with IV infusion, potentially due to lymphatic absorption [64]. Anti-drug antibodies (ADAs) can neutralize the mAb, affecting efficacy and causing adverse immune reactions [65].
Significant advancements have been made in the development and validation of nonclinical models and computational technologies to predict the immunogenicity risk of mAbs [66, 67]. While these models provide valuable insights and aid in mAb selection, they cannot fully replace clinical immunogenicity assessments. Nonclinical data cannot directly translate to human responses, and ADA formation varies between individuals. Additionally, factors like product ingredients, impurities, aggregation, and subvisible particle concentration can also trigger ADA formation [68]. Therefore, it is essential to evaluate the potential impact of SC dosing on the immunogenicity profile of mAbs in clinical trials. These trials assess the presence of ADAs that may interfere with the biological and clinical activity of the mAb, both at the population and individual level. By obtaining early data, clinicians can develop appropriate strategies to manage immunogenicity risks [69].
For detailed information on the conduct of clinical immunogenicity assessment, it is recommended to refer to the guidelines provided by the FDA and EMA [70, 71].
4.6.1.3 Local Tolerability
Local injection-site reactions are a common side effect with the SC administration of mAbs (refer to USPI and SmPC for trastuzumab, rituximab, daratumumab, pertuzumab-trastuzumab fixed-dose combination [5, 8]). These reactions are typically mild and temporary, characterized by symptoms such as erythema, pruritus, pain, inflammation, rash, induration, itching, and edema [72]. Various factors can contribute to the occurrence of local manifestations, including the mAb formulation (such as pH, volume, excipients), administration technique, and individual characteristics such as body weight, gender, and age [73, 74]. Consequently, local tolerability data for each specific SC mAb presentation need to be collected early in the development process. This data can be obtained through preclinical models and further complemented by clinical data, ideally collected during early dose-finding studies. Ongoing efforts are focused on developing models that simulate large-volume mAb injections using anisotropic porohyperelastic models and data-driven tissue layer geometries, aiming to enhance the understanding of the underlying mechanics and transport processes [75].
4.6.2 Efficacy Profile
The pharmacokinetic-based bridging approach has become the standard method for developing SC dosing alternatives for mAbs with IV infusion regimens. Initially, both pharmacokinetic and efficacy measures were used as co-primary endpoints, but recent development programs have focused on pharmacokinetic parameters as the only primary endpoint. This shift is supported by the available clinical evidence showing that despite lower Cmax levels, SC versions of a given mAb exhibit non-inferior efficacy to the IV formulation when overall mAb exposure (AUC) and Ctrough are comparable. Nonclinical xenograft models for trastuzumab and rituximab have further validated the acceptability of this approach in regulatory evaluations, demonstrating that the described differences in Cmax do not significantly impact tumor growth inhibition [38, 45].
The authors believe that the pharmacokinetic-based bridging approach is widely validated with a robust database of various mAbs. Originally designed for high-dose mAbs in oncology, this approach is now gaining acceptance and being applied in other therapeutic areas. While clinical trial data are still essential to evaluate and manage hypersensitivity and immunogenicity reactions, efficacy data can serve as supplementary evidence in the filing process.
4.6.3 Dosing Regimen
4.6.3.1 Fixed-Dose Versus Body-Size-Adjusted Dosing
In the past, mAb dosing regimens were often adjusted based on body size to ensure appropriate dosing for each individual. However, with improved understanding of the distribution and elimination kinetics of biotherapeutics, this approach is now considered partially outdated [76], especially for mAbs with limited distribution into adipose tissue. Studies have shown that body weight-adjusted dosing regimens for certain mAbs, such as trastuzumab or pembrolizumab, may result in lower exposures in lower-weight individuals and higher exposure in people with higher body weight, while a fixed dose can lead to the opposite trend [77, 78].
In current practice, IV mAbs are typically developed with a fixed dose or transitioned to a fixed dose as part of their lifecycle management. The initial IV to SC bridging programs for trastuzumab and rituximab included a shift from a body size-adjusted dosing regimen to a fixed dose [37]. Later IV mAbs such as pertuzumab and atezolizumab were initially launched with a fixed dose [79, 80], while for nivolumab and pembrolizumab fixed dosing was implemented as a lifecycle management [78, 81].
4.6.3.2 Administration Route—First Dose IV Versus SC
For some SC dosing regimens, the first dose is still administered as an IV infusion [47, 52], a less convenient and more resources-intense clinical practice. Especially during the early days of developing SC dosing alternatives for mAbs, arguable concerns were raised about the safety when administering a mAb that exhibits IRRs as a fast SC bolus injection. Such perceptions did arise from the procedure to reduce the severity of manifesting IRRs, that is, stopping or slowing down the injection rate [82, 83]. It was discussed that when injecting the full dose within only a few minutes, this measure to handle possible IRRs is precluded.
Based on the available database, SC administration of high-volume mAbs has shown a favorable hypersensitivity profile, particularly when accompanied by suitable pre-medication protocols [84, 85]. The slow appearance rate of mAbs in the plasma (Tmax of approximately 2–14 days) [86, 87] supports the feasibility of initiating treatment directly with the SC formulation.
4.6.3.3 Dosing Frequency
To ensure accurate extrapolation of data from the pivotal non-inferiority trial to other indications, the initial SC projects utilizing a pharmacokinetic-based clinical bridging approach required consistent application of the same dose and dosing frequency for the mAb. Therefore, a separate trial was conducted for SC rituximab in CLL, utilizing a distinct IV dose and dosing regimen [37].
In most of the non-inferiority studies mentioned, the dosing frequency remained consistent between the IV and SC cohorts. However, recent developments have seen manufacturers deviate from this approach while still employing the pharmacokinetic-based bridging strategy. Notably, phase III trials for SC ravulizumab and ocrelizumab have emerged as the first examples of administering the SC and IV formulations with different administration regimens.
In the pivotal ravulizumab clinical study, the established IV formulation with a q8w regimen was compared with a SC version administered with a q1w regimen [52]. In the ocrelizumab phase III trial, a pharmacokinetic comparison was made for the first dose in anti-CD20 treatment-naive patients. Here the IV dose is split, given 2 weeks apart, while the SC dose is administered as a single injection [20].
With the advancing knowledge of the pharmacokinetic–pharmacodynamic correlations for mAbs, it is expected that future approvals will enable extrapolation of pivotal study data to indications with different dosing regimens. Here, modeling, simulation, and meta-analyses of data from other mAbs are facilitating the design of rational trials [88,89,90].
4.6.3.4 Combination Therapy
Developing a SC dosing option for mAbs used in combination therapy holds significant value for end users. The first step involves assessing the feasibility of aligning the dosing frequency of the combination partners. This assessment can be performed through pharmacokinetic modeling using the available data from the IV regimen, which determines the dose levels required to achieve target saturation based on the dosing schedule. Given the relatively long elimination half-lives of mAbs [91], fixed-dose combinations of two or more mAbs can be feasibly administered subcutaneously at intervals ranging from approximately once weekly to once every month.
If the combination partners in question have been previously studied or authorized in IV combination therapy, the clinical development program and extrapolation to other indications can follow a similar path as described for mAbs used in monotherapy or concomitant chemotherapy, as exemplified by the pertuzumab-trastuzumab fixed-dose combination [51]. However, when considering a fixed-dose combination with at least one mAb that has not been investigated in clinical trials, such as the IV combination of the LAG-3-blocking antibody relatlimab and the PD1-blocking antibody nivolumab [92], the development pathway follows that of a new molecule, regardless of the administration route.
4.6.4 Treatment Setting
4.6.4.1 Decentralized Treatment Setting
SC dosing regimens provide the opportunity for decentralized administration, allowing individuals and healthcare providers to select the most suitable location for drug administration based on personal preferences and capabilities. These options include hospitals, infusion or community centers, physician's offices, or even at-home dosing. For manufacturers, gaining insights into preferences, health economic considerations in comparison to IV infusions, and the overall feasibility of this approach is crucial to enable implementation of a flexible care setting right from the initial launch of the SC version.
From a regulatory standpoint, biotherapeutics intended for dosing in a decentralized setting must exhibit safety, tolerability, and ease of administration without direct supervision from a healthcare professional [37]. Currently, the high-volume SC formulations described are not yet approved for non-supervised at-home or self-administration.
For SC dosing alternatives to established IV regimens, evidence on systemic safety is derived from pivotal clinical trials conducted for the IV dosing regimen. Non-clinical toxicology and tolerability data, and the clinical data package for the SC version complement this evidence. Special attention is given to local tolerability and hypersensitivity reactions. Based on this evidence package, treatment management plans, including safety measures, observation time, pre-medication schemes, and educational materials, are generated to support drug administration in a flexible care setting [93,94,95].
Due to its less invasive dosing procedure, the SC formulation is inherently more suitable for decentralized dosing compared with IV infusion regimens. However, for products with dosing volumes exceeding the capacity of current prefilled syringe or autoinjector platforms, user-friendly and intuitive device technologies are required. Today, the first on-body delivery system platform for administering biologics with volumes larger than 3 mL, such as eculizumab and ravulizumab [5, 8], has been approved, while other large-volume device options are currently undergoing clinical trial investigation [96].
To ensure safe and effective use of the device within the intended environment, comprehensive human factors (HF) engineering programs are necessary to assess the general usability of the device. These programs aim to demonstrate the on-body delivery system's usability by the target users, including individuals diagnosed with a chronic condition and/or lay care partners, particularly in the case of at-home dosing [97].
On-body delivery systems designed for dosing volumes of 10 mL or more, which are required for most of the SC mAb presentations described, are relatively new in the field of SC administration technology. Consequently, these device types were not yet available for testing during pivotal clinical studies. As a result, manufacturers will be expected to establish the equivalence of drug delivery between manual or semi-manual injection from a vial presentation used in the pivotal bridging trial for the SC formulation and the automated device [98]. For a proposed molecule-independent device bridging approach please refer to section 4.8.
4.7 Accelerating the Molecule-Agnostic Clinical Development—Clinical Trial Design and Conduct
From the authors’ perspective, manufacturers can expedite the clinical bridging approach from IV to SC administration of mAbs by taking into account the aspects of the target product profile discussed earlier. This assessment draws from existing precedents in various specialty areas and considers the growing body of evidence and regulatory acceptance of predictive modeling and simulation tools as a complement to data obtained from controlled clinical trials.
4.7.1 Study Population
Selection of the appropriate clinical trial population is crucial when considering the target indications for the SC dosing alternative of a mAb. Three fundamental scenarios can be contemplated. In the simplest scenario, where the IV version of the mAb is authorized or developed for a single indication, the clinical bridging study to the SC regimen would involve individuals diagnosed with that specific disease.
In the second scenario, where the IV regimen is approved or in development for multiple indications, manufacturers should conduct the pivotal bridging study in the most sensitive population. This population should have an underlying medical condition suitable for detecting potential impacts of the SC administration route on efficacy, safety, immunogenicity, and pharmacokinetics. Ideally, the population is relatively homogeneous with limited variability in endpoints and minimal confounding factors. Initial dose finding can be done with healthy volunteers if supported by safety data. Alternatively, a mixed population trial can be conducted to gather data for different medical conditions, although this may require a larger sample size due to increased variability.
In a third scenario, manufacturers may opt to develop the SC dosing alternative for a new indication where the IV formulation is not yet approved or in clinical development. In this case, the clinical trial would be designed to demonstrate the efficacy of the SC formulation in the target population. The trial would aim to establish either non-inferiority or superiority compared with the standard of care. This approach would require robust evidence of efficacy to support the approval of the SC dosing alternative in the new indication.
4.7.2 Endpoints in the Pivotal Clinical Study
In the clinical development pathways for approved SC dosing alternatives, the inclusion of a co-primary pharmacokinetic endpoint has been a crucial aspect since the initial project of SC trastuzumab. The emphasis was on the hypothesis that achieving non-inferior Ctrough and/or AUC would result in non-inferior efficacy compared with the IV dosing regimen. As more evidence supported the validity of this bridging approach, pharmacokinetics became the only primary endpoint in phase III trials, with efficacy and safety serving as secondary endpoints.
Similarly, in the initial clinical studies, the pharmacokinetic assessment was typically conducted at steady-state of the mAb or after several treatment cycles [41, 47]. However, more recent SC developments have implemented a different approach, where the pharmacokinetic comparison is performed during the first treatment cycle [20, 55, 57]. By obtaining earlier pharmacokinetic data, this approach can potentially expedite the filing process. Subject to regulatory approval, manufacturers may be able to submit the dossier with a statistical evaluation of the primary endpoint, accompanied by supporting data on efficacy, safety, and immunogenicity in accordance with regulatory guidelines.
Manufacturers gain insight into the preference for SC dosing by incorporating preference assessments in the clinical studies early on. This can be achieved through dedicated preference questionnaires in cross-over studies [99] or by randomizing participants with prior IV experience to the SC version [100]. Another approach is to allow participants to continue treatment on their preferred administration route once the final SC dose has been determined within a trial [52].
4.7.3 Pharmacokinetic Modeling to Complement Clinical Trial Data
Pharmacokinetic modeling represents an established approach that plays a crucial role in informing clinical dosing regimens and enhancing trial design [101]. These methods were instrumental in expediting the phase 1/1b dose-finding studies for trastuzumab and rituximab when transitioning from IV to SC administration [40, 102]. The assumption underlying this approach was that the switch in administration route would primarily affect the absorption rate into the systemic circulation, while distribution and elimination patterns of the mAb remained unchanged. Blood sampling was scheduled based on the absorption rate observed with other mAbs [103]. Interim analyses were conducted using samples from participants who had completed the expected absorption period, while enrollment and sampling continued. The obtained pharmacokinetic data were incorporated into the existing pharmacokinetic model previously derived from IV data [104]. This allowed real-time prediction of the complete SC profile and facilitated selection of the final SC dose using an adaptive trial design.
Model-informed drug development approaches have the potential to further expedite IV to SC bridging strategies in the future by leveraging comprehensive data collected throughout the molecule's development program [105]. The FDA defines model-informed drug development as the development and application of exposure-based biological and statistical models derived from preclinical and clinical data sources to inform drug development and regulatory decision making [106]. The agency has initiated a pilot program to discuss model-informed drug development approaches in medical product development, specifically in the Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER), from fiscal years 2023 to 2027 [107]. These efforts aim to enhance collaboration between manufacturers and regulatory authorities to optimize the use of modeling and simulation in drug development.
As a result, there is a growing tendency to utilize model-informed drug development approaches in the development of novel formulations and the bridging of dosing frequencies, thereby supporting lifecycle management in the post-approval phase [108]. The COVID-19 pandemic has further accelerated the adoption of model-informed drug development as a valuable tool to supplement the regulatory review process, particularly in situations where clinical studies may not have encompassed all proposed doses, indications, and populations [109].
4.7.4 Randomization Scheme
Traditionally, pivotal phase III non-inferiority trials for SC versus IV administration used a 1:1 randomization scheme (refer to Sect. 4.4). However, recent developments have seen manufacturers adopt unequal allocation in these trials. For example, the bridging study of ravulizumab employed a 2:1 allocation in favor of the SC route. While asymmetrical randomization schemes may be subject to scrutiny, there are situations where this approach is scientifically and ethically justified [110]. In the case of ravulizumab, such justification is evident. PNH is a rare disease with a low prevalence, making patient recruitment challenging for clinical trials [111]. Additionally, if an IV formulation of the mAb is already in late-stage development or on the market, a substantial safety database exists, supporting a larger sample size for the SC cohort. Since SC administration is not expected to impact the systemic safety profile, the primary focus shifts to pharmacokinetics as the main endpoint, with local tolerability and immunogenicity as key secondary measures. A well-powered pharmacokinetic comparability study typically requires a smaller sample size compared with an efficacy trial [112].
In the phase III clinical bridging study for SC ocrelizumab, participants are initially randomized in a 1:1 ratio to receive either SC or IV administration. After completing the first treatment cycle, all participants are switched to the SC arm. This study design allows for the collection of both IV and SC data within the same trial, reducing variability compared with historical data comparisons. In the future, single-arm trials focusing solely on the SC formulation, supported by model-informed drug development approaches, may be considered to generate additional evidence for the SC regimen.
During the COVID-19 pandemic, recruiting participants for trials involving IV administration posed challenges and incurred higher costs, as it required on-site administration or the assistance of nursing home care services. In contrast, SC trials were more straightforward to operate, thanks to the simplified dosing process that facilitated decentralized administration. Given the increased convenience, feasibility, and cost effectiveness of SC dosing in this context, an unequal allocation could have been justified.
4.7.5 Dosing Frequency Comparison
When dealing with mAb formulations requiring a dosing volume of 3 mL or more, it can be challenging to maintain the less frequent dosing regimen with the IV version, especially in decentralized care settings [113]. This challenge has led to the adoption of a more frequent dosing schedule for the SC route, allowing for smaller individual dosing volumes and enabling the use of autoinjector devices for SC self-injections. In such instances, as demonstrated in studies of SC tocilizumab and abatacept [5, 8, 115,116,117], the lower dosing frequency of the SC regimen did necessitate the demonstration of efficacy as the primary endpoint in pivotal clinical bridging studies (refer to Sect. 4.7.4 for supporting evidence).
Notably, in most pivotal clinical studies for high-dose mAbs, the dosing frequency was consistent between the IV and SC routes [37]. This is a result of the introduction of novel formulation and device technologies that reduce the overall SC dosing volume or facilitate injection of volumes exceeding 5 mL or more [113, 114].
With the use of model-informed drug development approaches, it is anticipated that future bridging studies for different administration routes will increasingly compare unequal dosing frequencies. This approach allows for leveraging the available pharmacokinetic-pharmacodynamic correlation from the IV regimen and supports the design of IV to SC bridging trials that are powered for a pharmacokinetic endpoint.
4.7.6 Clinical Trial Conduct
From an operational standpoint, pharmacokinetic-based dose finding and confirmation can be conducted either within the same study or in independent sequential studies. When the safety profile of the mAb allows for administration to healthy participants, conducting dose finding separately may be preferred. The population of healthy participants typically exhibits reduced pharmacokinetic variability and limited confounding factors, such as concomitant medications or comorbidities. As a result, a smaller sample size is required compared with studies involving participants with a medical condition. Additionally, trials involving healthy participants are generally faster to recruit compared with those involving individuals requiring a medical intervention.
If evaluation of a product in healthy volunteers is not suitable from a pharmacokinetic perspective because of a lack of or reduced expression of the cell-bound or soluble targets, an alternative and still cost-efficient trial design would integrate dose finding and confirmation within a single phase Ib/III trial [46] involving the target population. This study would employ an adaptive design with interim pharmacokinetic analyses and model-based dose selection, enabling the initiation of dosing in the non-inferiority, dose confirmation phase as soon as the appropriate SC dose has been identified. By combining these aspects, time and resources can be optimized, streamlining the overall development process.
In support of a decentralized care setting, enabling clinical trial participants to receive the dose at home provides valuable insights into potential challenges at an early stage. Clinical trial evidence in this context supports the development of training and educational materials for self-administration.
4.8 Accelerating the Molecule-Agnostic Clinical Development—Molecule-Independent Device Bridging Approach
Ensuring the timely availability of automated injection devices, such as autoinjectors or on-body delivery systems, is crucial for facilitating at-home and self-administration of biotherapeutics. However, developing an autoinjector or on-body delivery system for use in the phase III program may not be practical due to the molecule-specific technical development requirements. Subsequently, pharmacokinetic comparability studies may be considered to demonstrate comparable performance between the prefilled syringe or manually filled handheld syringe used in phase III and the automated device [98, 118].
In view of the more consistent injection with an automated device and in line with previous communication [119], in the opinion of the authors, for mAbs utilizing device platforms that were previously validated with another mAb, referencing to these data rather than conducting an additional dedicated pharmacokinetic comparability study should be justified. The underlying prerequisite is that the autoinjector contains the same formulation (i.e., including the same excipients at the same quantities) and dosing volume as that used in the pivotal phase III study for the respective mAb. Technical design verification and validation, including a summative human factors study, would still be conducted for each mAb individually (Fig. 4). Deviations from this approach would be assessed on a case-by-case basis.

Molecule-independent device bridging approach – concept, supporting evidence, and underlying assumptions. AI autoinjector, mAb monoclonal antibody, OBDS on-body delivery system, PK pharmacokinetic
The authors refer to this concept as a molecule-independent device bridging approach (Fig. 4). Eligible mAbs would be characterized by slow absorption from the SC tissue into the systemic circulation [120]. The rationale is that the pharmacokinetic profiles of SC administration using different devices, such as prefilled syringe, handheld syringe, or automated devices, are expected to be similar. This is because the rate-limiting factor for absorption into the systemic circulation is the release from the interstitial space via lymph flow, rather than the specific injection method. For more information on this concept, please refer to previously published material [37].
5 Summary and Outlook
In the opinion of the authors, over the past 5 years, SC administration of high-dose/high-volume mAbs has transitioned from an emerging field to a commonplace practice. This shift is supported by the increasing number of globally authorized high-volume mAbs since the publication of the initial review article in 2018 [1].
The pharmacokinetic-based clinical bridging approach from IV to SC administration has now become a well-established standard in different specialty areas. The concept did allow for extrapolation of data from the pivotal phase III trial in one indication to other indications with the same IV dose and dosing frequency.
Despite advancements in nonclinical and computational models, clinical trial data are still necessary to assess hypersensitivity reactions, immunogenicity, and local tolerability with the SC route. These events depend on factors such as the mAb itself, formulation composition, as well as on individual traits. While pharmacokinetic modeling has already been used in early programs, the structured application of model-based drug development concepts is now being employed to streamline development approaches. This allows for the possibility of omitting 1:1 randomization in phase III clinical studies and instead favoring unequal allocation in support of the novel SC route. Pharmacokinetic modeling enables the assessment of the primary endpoint as early as cycle 1 and facilitates non-inferiority studies comparing IV and SC formulations with different dosing frequencies.
In the coming years, high-volume SC mAbs are expected to be widely available and commonly used in decentralized care settings. This shift will be facilitated by the introduction of large-volume on-body delivery systems. The clinical bridging program for these mAbs will primarily focus on pharmacokinetic endpoints, although the size and design of the trials may vary. This can range from traditional non-inferiority studies to superiority studies compared with standard of care, or even single-arm trials utilizing historical data for comparison with the IV version. As high-dose SC mAbs become easier to administer, it is expected that more of these medications will be developed exclusively in SC formulations.
References
Bittner B, Richter W, Schmidt J. Subcutaneous administration of biotherapeutics: an overview of current challenges and opportunities. BioDrugs. 2018;32(5):425–40. https://doi.org/10.1007/s40259-018-0295-0.
McCreary EK, Bariola JR, Wadas RJ, Shovel JA, Wisniewski MK, Adam M, et al. Association of subcutaneous or intravenous administration of casirivimab and imdevimab monoclonal antibodies with clinical outcomes in adults with COVID-19. JAMA Netw Open. 2022;5(4): e226920. https://doi.org/10.1001/jamanetworkopen.2022.6920.
Bittner B. Drug delivery improvements to enable a flexible care setting for monoclonal antibody medications in oncology—analogue-based decision framework. Expert Opin Drug Deliv. 2023;20(4):457–70. https://doi.org/10.1080/17425247.2023.2184343.
Wynendaele E, Furman C, Wielgomas B, Larsson P, Hak E, Block T, et al. Sustainability in drug discovery. Med Drug Discov. 2021;12: 100107. https://doi.org/10.1016/j.medidd.2021.100107.
The Electronic Medicines Compendium. 2023. Available at: https://www.medicines.org.uk/emc/about-the-emc#gref. (Accessed 15 Jun 2023).
VFA (Verband der forschenden Arzneimittelhersteller). 2023. Übersicht über zentralisiert in der EU zugelassene Biosimilars. Available at: https://www.vfa.de/download/biosimilars-uebersicht-originalpraeparate.pdf. (Accessed 15 Jun 2023).
US FDA. Biosimilar product information. 2023. Available at: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information. (Accessed 15 Jun 2023).
US FDA. 2023. Drugs@FDA: FDA-approved drugs. Available at: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm. (Accessed 15 Jun 2023).
European Pharmaceutical Review. 2020. EC approves subcutaneous formulation of Remsima. Available at: https://www.europeanpharmaceuticalreview.com/news/124819/ec-approves-subcutaneous-formulation-of-remsima/. (Accessed 15 Jun 2023).
Drug Discovery World. 2023. First subcutaneous formulation of infliximab submitted to FDA. Drug Discovery World. 2023. Available at: https://www.ddw-online.com/first-subcutaneous-formulation-of-infliximab-submitted-to-fda-21205-202301/. (Accessed 15 Jun 2023).
The American Journal of Managed Care. 2023. First Humira biosimilar, Amjevita, launches in the United States. AJMC. 2023. Available at: https://www.ajmc.com/view/first-humira-biosimilar-amjevita-launches-in-the-united-states. (Accessed 15 June 2023).
BioPharma-Reporter. 2021. Sandoz sees route to market with Enbrel biosimilar blocked until 2029. Available at https://www.biopharma-reporter.com/Article/2021/05/18/Sandoz-sees-route-to-market-with-Enbrel-biosim-blocked-until-2029.
Cardinal Health. 2023. 2023 Biosimilars Report: Tracking market expansion and sustainability amidst a shifting industry. Available at: https://www.cardinalhealth.com/content/dam/corp/web/documents/Report/cardinal-health-biosimilars-report-2023.pdf. (Accessed 15 Jun 2023).
Ramzan I. Interchangeability of biosimilars: a global perspective for pharmacists. Pharm J. 2020. https://doi.org/10.1211/PJ.2020.20208123.
EMA. 2023. Statement on the scientific rationale supporting the interchangeability of biosimilar medicines. Available at: https://www.ema.europa.eu/en/documents/public-statement/statement-scientific-rationale-supporting-interchangeability-biosimilar-medicines-eu_en.pdf. (Accessed 15 Jun 2023).
US FDA. 2022. Review and approval of biosimilar and interchangeable biosimilar products. Available at: https://www.fda.gov/drugs/biosimilars/review-and-approval#interchangeable%20biosimilar%20products. (Accessed 15 Jun 2023).
US FDA. 2021. FDA approves Cyltezo as the first interchangeable biosimilar to Humira. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-cyltezo-first-interchangeable-biosimilar-humira. (Accessed 15 Jun 2023).
Bittner B. Customer-centric product presentations for monoclonal antibodies. AAPS Open. 2023;9(1):3. https://doi.org/10.1186/s41120-022-00069-y.
Biogen. 2021. Biogen provides regulatory update on the supplemental biologic license application (sBLA) for subcutaneous administration of TYSABRI® (natalizumab) Available at: https://investors.biogen.com/news-releases/news-release-details/biogen-provides-regulatory-update-supplemental-biologic-license. (Accessed 15 Jun 2023).
ClinicalTrials.gov. 2023. A phase III, non-inferiority, randomized, open-label, parallel group, multicenter study to investigate the pharmacokinetics, pharmacodynamics, safety and radiological and clinical effects of subcutaneous ocrelizumab versus intravenous ocrelizumab in patients with multiple sclerosis (Ocarina II). Identifier NCT05232825. Available at: https://clinicaltrials.gov/ct2/show/NCT05232825. (Accessed 15 Jun 2023).
EMA. 2014. MabThera (H-C-165-X-83): EPAR - Assessment Report - Extension. Available at: https://www.ema.europa.eu/en/documents/variation-report/mabthera-h-c-165-x-83-epar-assessment-report-extension_en.pdf. (Accessed 15 Jun 2023).
US FDA. 2017. FDA summary basis for regulatory action: Rituxan Hycela. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761064Orig1s000CrossR.pdf. (Accessed 15 Jun 2023).
Burotto M, Zvirbule Z, Mochalova A, Runglodvatana Y, Herraez Baranda LA, Liu S, et al. 61MO IMscin001 (part 2: randomized phase III): Pharmacokinetics (PK), efficacy and safety of atezolizumab (atezo) subcutaneous (SC) vs intravenous (IV) in previously treated locally advanced or metastatic non-small cell lung cancer (NSCLC). ESMO IOTECH. 2020;16(1): 100166. https://doi.org/10.1016/j.iotech.2022.100166.
Rodrigo Chacon M, Cutuli HJ, Bracarda S, Maruzzo M, Bourlon MT, Jacobs C, et al. Subcutaneous nivolumab versus intravenous nivolumab in patients with previously treated, advanced, or metastatic clear cell renal cell carcinoma. J Clin Oncol. 2022;40(16_suppl):TPS4621. https://doi.org/10.1200/JCO.2022.40.16_suppl.TPS4621.
ClinicalTrials.gov. 2023. A Study of Subcutaneous (SC) Pembrolizumab Coformulated With Hyaluronidase (MK-3475A) vs Intravenous Pembrolizumab in Adult Participants With Metastatic Non-small Cell Lung Cancer (NSCLC) (MK-3475A-D77). Identifier: NCT05722015. Available at: https://clinicaltrials.gov/ct2/show/NCT05722015. (Accessed 15 Jun 2023).
O’Shaughnessy J, Sousa S, Cruz J, Fallowfield L, Auvinen P, Pulido C, et al. Preference for the fixed-dose combination of pertuzumab and trastuzumab for subcutaneous injection in patients with HER2-positive early breast cancer (PHranceSCa): a randomised, open-label phase II study. Eur J Cancer. 2021;152:223–32. https://doi.org/10.1016/j.ejca.2021.03.047.
Slavcev M, Spinelli A, Absalon E, Masterson T, Heuck C, Lam A, et al. Results of a time and motion survey regarding subcutaneous versus intravenous administration of daratumumab in patients with relapsed or refractory multiple myeloma. Clinicoecon Outcomes Res. 2021;13:465–73. https://doi.org/10.2147/CEOR.S302682.
Hamadeh IS, Moore DC, Martin A, Karabinos A, Hill H, Ndiaye A, et al. Transition from intravenous to subcutaneous daratumumab formulation in clinical practice. Clin Lymphoma Myeloma Leuk. 2021;21(7):470–5. https://doi.org/10.1016/j.clml.2021.02.014.
McCloskey C, Ortega MT, Nair S, Garcia MJ, Manevy F. A Systematic review of time and resource use costs of subcutaneous versus intravenous administration of oncology biologics in a hospital setting. PharmacoEconomics - Open. 2023;7(1):3–36. https://doi.org/10.1007/s41669-022-00361-3.
US FDA. Patient preference information – voluntary submission, review in premarket approval applications, humanitarian device exemption applications, and de novo requests, and inclusion in decision summaries and device labeling. Guidance for Industry, Food and Drug Administration Staff, and Other Stakeholders. 2016. Available at: https://www.fda.gov/media/92593/download
Whichello C, Schölin Bywall K, Mauer J, Watt S, Cleemput I, Pinto CA, et al. An overview of critical decision-points in the medical product lifecycle: Where to include patient preference information in the decision-making process? Health Policy. 2020;124(12):1325–32. https://doi.org/10.1016/j.healthpol.2020.07.007.
Smith MY, Janssens R, Jimenez-Moreno AC, Cleemput I, Muller M, Oliveri S, et al. Patients as research partners in preference studies: learnings from IMI-PREFER. Res Involv Engagem. 2023;9(1):21. https://doi.org/10.1186/s40900-023-00430-9.
EMA.Qualification Opinion on the IMI-PREFER project. 2022. Available at: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/qualification-opinion-imi-prefer_en.pdf.
Barbier L, Muller M, Cleemput I, Strammiello V, Dickinson S, Kübler J, et al. EMA/EUnetHTA Qualification of the IMI PREFER patient preference framework and points to consider for method selection: key experiences, outcomes, values and implications. Value in Health. 2022;25(12):S2. https://doi.org/10.1016/j.jval.2022.09.018.
Zarzar J, Khan T, Bhagawati M, Weiche B, Sydow-Andersen J, Alavattam S. High concentration formulation developability approaches and considerations. MAbs. 2023;15(1):2211185. https://doi.org/10.1080/19420862.2023.2211185.
Frost GI. Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration. Expert Opin Drug Deliv. 2007;4(4):427–40. https://doi.org/10.1517/17425247.4.4.427.
Bittner B, Schmidt J. Formulation and device lifecycle management: a guidance for researchers and drug developers. 1st ed. William Andrew Publishing (Elsevier); 2022.
Bittner B, Richter WF, Hourcade-Potelleret F, McIntyre C, Herting F, Zepeda ML, et al. Development of a subcutaneous formulation for trastuzumab—nonclinical and clinical bridging approach to the approved intravenous dosing regimen. Drug Res. 2012;62(9):401–9. https://doi.org/10.1055/s-0032-1321831.
Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20(3):719–26. https://doi.org/10.1200/JCO.2002.20.3.719.
Wynne C, Harvey V, Schwabe C, Waaka D, McIntyre C, Bittner B. Comparison of subcutaneous and intravenous administration of trastuzumab: a phase I/Ib trial in healthy male volunteers and patients with HER2-positive breast cancer. J Clin Pharmacol. 2013;53(2):192–201. https://doi.org/10.1177/0091270012436560.
Ismael G, Hegg R, Muehlbauer S, Heinzmann D, Lum B, Kim SB, et al. Subcutaneous versus intravenous administration of (neo)adjuvant trastuzumab in patients with HER2-positive, clinical stage I-III breast cancer (HannaH study): a phase 3, open-label, multicentre, randomised trial. Lancet Oncol. 2012;13(9):869–78. https://doi.org/10.1016/S1470-2045(12)70329-7.
US FDA. 2016. Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product. Guidance for Industry. Available at: https://www.fda.gov/media/88622/download. (Accessed 15 Jun 2023).
EMA. Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues. 2012. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical_en.pdf. (Accessed 15 Jun 2023).
EMA. Herceptin (H-C-278-X-0060): EPAR - Assessment Report – Extension. 2013. Available at: https://www.ema.europa.eu/en/documents/variation-report/herceptin-h-c-278-x-0060-epar-assessment-report-extension_en.pdf. (Accessed 15 Jun 2023).
Bittner B, Richter WF, Hourcade-Potelleret F, Herting F, Schmidt J. Non-clinical pharmacokinetic/pharmacodynamic and early clinical studies supporting development of a novel subcutaneous formulation for the monoclonal antibody rituximab. Drug Res. 2014;64(11):569–75. https://doi.org/10.1055/s-0033-1363993.
Salar A, Avivi I, Bittner B, Bouabdallah R, Brewster M, Catalani O, et al. Comparison of subcutaneous versus intravenous administration of rituximab as maintenance treatment for follicular lymphoma: results from a two-stage, phase IB study. J Clin Oncol. 2014;32:1782–91. https://doi.org/10.1200/JCO.2013.52.2631.
Davies A, Merli F, Mihaljevic B, Siritanaratkul N, Solal-Céligny P, Barrett M, et al. Pharmacokinetics and safety of subcutaneous rituximab in follicular lymphoma (SABRINA): stage 1 analysis of a randomised phase 3 study. Lancet Oncol. 2014;15(3):343–52. https://doi.org/10.1016/S1470-2045(14)70005-1.
Assouline S, Buccheri V, Delmer A, Gaidano G, McIntyre C, Brewster M, et al. Pharmacokinetics and safety of subcutaneous rituximab plus fludarabine and cyclophosphamide for patients with chronic lymphocytic leukaemia. Br J Clin Pharmacol. 2015;80(5):1001–9. https://doi.org/10.1111/bcp.12662.
Paul F, Cartron G. Infusion-related reactions to rituximab: frequency, mechanisms and predictors. Expert Rev Clin Immunol. 2019;15(4):383–9. https://doi.org/10.1080/1744666X.2019.1562905.
Mateos MV, Nahi H, Legiec W, Grosicki S, Vorobyev V, Spicka I, et al. Subcutaneous versus intravenous daratumumab in patients with relapsed or refractory multiple myeloma (COLUMBA): a multicentre, open-label, non-inferiority, randomised, phase 3 trial. Lancet Haematol. 2020;7(5):370–8. https://doi.org/10.1016/S2352-3026(20)30070-3.
Tan AR, Im SA, Mattar A, Colomer R, Stroyakovskii D, Nowecki Z, et al. Fixed-dose combination of pertuzumab and trastuzumab for subcutaneous injection plus chemotherapy in HER2-positive early breast cancer (FeDeriCa): a randomised, open-label, multicentre, non-inferiority, phase 3 study. Lancet Oncol. 2021;22(1):85–97. https://doi.org/10.1016/S1470-2045(20)30536-2.
Yenerel MN, Sicre de Fontbrune F, Piatek C, Sahin F, Füreder W, Ortiz S, et al. Phase 3 Study of subcutaneous versus intravenous ravulizumab in eculizumab-experienced adult patients with PNH: primary analysis and 1-year follow-up. Adv Ther. 2023;40(1):211–32. https://doi.org/10.1007/s12325-022-02339-3.
Piggin M. Nothing about us should be without us. Lancet Haematol. 2023;10(4):E248. https://doi.org/10.1016/S2352-3026(23)00054-6.
Xu Z, Leu JH, Xu Y, Nnane I, Liva SG, Wang-Lin SX, et al. Development of therapeutic proteins for a new subcutaneous route of administration after the establishment of intravenous dosages: a systematic review. Clin Pharmacol Ther. 2023;113:1011–29. https://doi.org/10.1002/cpt.2823.
ClinicalTrials.gov. Study of pembrolizumab (MK-3475) subcutaneous (SC) versus pembrolizumab intravenous (IV) administered with platinum doublet chemotherapy in participants with metastatic squamous or nonsquamous non-small cell lung cancer (NSCLC) (MK-3475-A86). Identifier NCT04956692. 2023. Available at: https://www.clinicaltrials.gov/ct2/show/NCT04956692. (Accessed 15 Jun 2023).
ClinicalTrials.gov. A study of subcutaneous nivolumab versus intravenous nivolumab in participants with previously treated clear cell renal cell carcinoma that is advanced or has spread (CheckMate-67T). Identifier NCT04810078. 2023. Available at: https://clinicaltrials.gov/ct2/show/NCT04810078. (Accessed 15 Jun 2023).
ClinicalTrials.gov. A study to investigate atezolizumab subcutaneous in patients with previously treated locally advanced or metastatic non-small cell lung cancer. Identifier NCT03735121. 2023. Available at: https://clinicaltrials.gov/ct2/show/NCT03735121. (Accessed 15 Jun 2023).
Doessegger L, Banholzer ML. Clinical development methodology for infusion-related reactions with monoclonal antibodies. Clin Transl Immunol. 2015;4(7): e39. https://doi.org/10.1038/cti.2015.14.
Kulkarni HS, Kasi PM. Rituximab and cytokine release syndrome. Case Rep Oncol. 2012;5(1):134–41. https://doi.org/10.1159/000337577.
Cáceres MC, Guerrero-Martín J, Pérez-Civantos D, Palomo-López P, Delgado-Mingorance JI, Durán-Gómez N. The importance of early identification of infusion-related reactions to monoclonal antibodies. Ther Clin Risk Manag. 2019;15:965–77. https://doi.org/10.2147/TCRM.S204909.
Patel K, Parmar S, Shah S, Shore T, Gergis U, Mayer S, et al. Comparison of subcutaneous versus intravenous alemtuzumab for graft-versus-host disease prophylaxis with fludarabine/melphalan–based conditioning in matched unrelated donor allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2016;22(3):456–61. https://doi.org/10.1016/j.bbmt.2015.10.022.
Usmani SZ, Nahi H, Mateos MV, van de Donk NWCJ, Chari A, Kaufman JL, et al. Subcutaneous delivery of daratumumab in relapsed or refractory multiple myeloma. Blood. 2019;134(8):668–77. https://doi.org/10.1182/blood.2019000667.
Donnadieu E, Luu M, Alb M, Barbao P, Bonini C, Bousso P, et al. Time to evolve: predicting engineered T cell-associated toxicity with next-generation models. J Immunother Cancer. 2022;10: e003486. https://doi.org/10.1136/jitc-2021-003486.
Hamuro L, Kijanka G, Kinderman F, Kropshofer H, Bu D, Zepeda M, et al. Perspectives on subcutaneous route of administration as an immunogenicity risk factor for therapeutic proteins. J Pharm Sci. 2017;106(10):2946–54. https://doi.org/10.1016/j.xphs.2017.05.030.
Mosch R, Guchelaar HJ. Immunogenicity of monoclonal antibodies and the potential use of HLA haplotypes to predict vulnerable patients. Front Immunol. 2022;13: 885672. https://doi.org/10.3389/fimmu.2022.885672.
Vaisman-Mentesh A, Gutierrez-Gonzalez M, DeKosky BJ, Wine Y. The molecular mechanisms that underlie the immune biology of anti-drug antibody formation following treatment with monoclonal antibodies. Front Immunol. 2020;11:1951. https://doi.org/10.3389/fimmu.2020.01951.
Kim J, McFee M, Fang Q, Abdin O, Kim PM. Computational and artificial intelligence-based methods for antibody development. Trends Pharmacol Sci. 2023;44(3):175–89. https://doi.org/10.1016/j.tips.2022.12.005.
Jarvi NL, Balu-Iyer SV. Immunogenicity challenges associated with subcutaneous delivery of therapeutic proteins. BioDrugs. 2021;35(2):125–46. https://doi.org/10.1007/s40259-020-00465-4.
Vandivort TC, Horton DB, Johnson SB. Regulatory and strategic considerations for addressing immunogenicity and related responses in biopharmaceutical development programs. J Clin Transl Sci. 2020;4(6):547–55. https://doi.org/10.1017/cts.2020.493.
US FDA. Immunogenicity assessment for therapeutic protein products. Guidance for Industry. 2014. Available at: https://www.fda.gov/media/85017/download. (Accessed 15 Jun 2023).
EMA. Guideline on immunogenicity assessment of therapeutic proteins. 2017. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf. (Accessed 15 Jun 2023).
Alten R, Kellner H, Boyce M, Yonemura T, Ito T, Genovese MC. Systematic analysis of injection-site pain and reactions caused by subcutaneous administration of the adalimumab biosimilar FKB327 versus the adalimumab reference product via different delivery methods. GaBI J. 2020;9(3):108–15. https://doi.org/10.1136/annrheumdis-2019-eular.1292.
Singh SK, Mahler HC, Hartman C, Stark CA. Are injection site reactions in monoclonal antibody therapies caused by polysorbate excipient degradants? J Pharm Sci. 2018;107(11):2735–41. https://doi.org/10.1016/j.xphs.2018.07.016.
St Clair-Jones A, Prignano F, Goncalves J, Paul M, Sewerin P. Understanding and minimising injection-site pain following subcutaneous administration of biologics: a narrative review. Rheumatol Ther. 2020;7(4):741–57. https://doi.org/10.1007/s40744-020-00245-0.
De Lucio M, Leng Y, Hans A, Bilionis I, Brindise M, Ardekani AM, et al. Modeling large-volume subcutaneous injection of monoclonal antibodies with anisotropic porohyperelastic models and data-driven tissue layer geometries. J Mech Behav Biomed Mater. 2023;138: 105602. https://doi.org/10.1016/j.jmbbm.2022.105602.
Egorin MJ. Horseshoes, hand grenades, and body-surface area-based dosing: aiming for a target. J Clin Oncol. 2003;21(2):182–3. https://doi.org/10.1200/JCO.2003.10.084.
Quartino AL, Hillenbach C, Li J, Li H, Wada RD, Visich J, et al. Population pharmacokinetic and exposure–response analysis for trastuzumab administered using a subcutaneous “manual syringe” injection or intravenously in women with HER2-positive early breast cancer. Cancer Chemother Pharmacol. 2016;77:77–88. https://doi.org/10.1007/s00280-015-2922-5.
Freshwater T, Kondic A, Ahamadi M, Li CH, de Greef R, de Alwis D, et al. Evaluation of dosing strategy for pembrolizumab for oncology indications. J Immunother Cancer. 2017;5:43. https://doi.org/10.1186/s40425-017-0242-5.
Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389(10066):255–65. https://doi.org/10.1016/S0140-6736(16)32517-X.
Swain SM, Kim SB, Cortés J, Ro J, Semiglazov V, Campone M, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013;14(6):461–71. https://doi.org/10.1016/S1470-2045(13)70130-X.
Bei D, Osawa M, Uemura S, Ohno T, Gobburu J, Roy A, et al. Benefit-risk assessment of nivolumab 240 mg flat dose relative to 3 mg/kg Q2W regimen in Japanese patients with advanced cancers. Cancer Sci. 2020;111(2):528–35. https://doi.org/10.1111/cas.14252.
Roselló S, Blasco I, García Fabregat L, Cervantes A, Jordan K, ESMO Guidelines Committee. Management of infusion reactions to systemic anticancer therapy: ESMO Clinical Practice Guidelines. Ann Oncol. 2017;28(4):100–18. https://doi.org/10.1093/annonc/mdx216.
Szebeni J, Simberg D, González-Fernández Á, Barenholz Y, Dobrovolskaia MA. Roadmap and strategy for overcoming infusion reactions to nanomedicines. Nat Nanotechnol. 2018;13(12):1100–8. https://doi.org/10.1038/s41565-018-0273-1.
Perumal JS, Foo F, Cook P, Khan O. Subcutaneous administration of alemtuzumab in patients with highly active multiple sclerosis. Mult Scler. 2012;18(8):1197–9. https://doi.org/10.1177/1352458511435716.
Kim EB, O’Donnell E, Branagan AR, Burke JN, Harrington CC, Agyemang E, et al. Real-world observations and practical considerations of subcutaneous daratumumab administration in multiple myeloma. Blood. 2021;138(1):5018. https://doi.org/10.1182/blood-2021-153751.
Varkhede N, Forrest ML. Understanding the monoclonal antibody disposition after subcutaneous administration using a minimal physiologically based pharmacokinetic model. J Pharm Pharm Sci. 2018;21(1s):130s-s148. https://doi.org/10.18433/jpps30028.
Zhao L, Ji P, Li Z, Roy P, Sahajwalla CG. The antibody drug absorption following subcutaneous or intramuscular administration and its mathematical description by coupling physiologically based absorption process with the conventional compartment pharmacokinetic model. J Clin Pharmacol. 2013;53(3):314–25. https://doi.org/10.1002/jcph.4.
Brekkan A, Jönsson S, Karlsson MO, Hooker AC. Reduced and optimized trial designs for drugs described by a target mediated drug disposition model. J Pharmacokinet Pharmacodyn. 2018;45:637–47. https://doi.org/10.1007/s10928-018-9594-9.
Wang Z, Verstockt B, Sabino J, Vermeire S, Ferrante M, Declerck P, et al. Population pharmacokinetic-pharmacodynamic model-based exploration of alternative ustekinumab dosage regimens for patients with Crohn’s disease. Br J Clin Pharmacol. 2022;88(1):323–35. https://doi.org/10.1111/bcp.14971.
Davda JP, Dodds MG, Gibbs MA, Wisdom W, Gibbs J. A model-based meta-analysis of monoclonal antibody pharmacokinetics to guide optimal first-in-human study design. MAbs. 2014;6(4):1094–102. https://doi.org/10.4161/mabs.29095.
Keizer RJ, Huitema ADR, Schellens JHM, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:493–507. https://doi.org/10.2165/11531280-000000000-00000.
Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutiérrez E, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386(1):24–34. https://doi.org/10.1056/NEJMoa2109970.
Dineen-Griffin S, Garcia-Cardenas V, Williams K, Benrimoj SI. Helping patients help themselves: a systematic review of self-management support strategies in primary health care practice. PLoS One. 2019;14(8): e0220116. https://doi.org/10.1371/journal.pone.0220116.
Kalyango JN, Hall M, Karamagi C. Home medication management practices and associated factors among patients with selected chronic diseases in a community pharmacy in Uganda. BMC Health Serv Res. 2012;12:323. https://doi.org/10.1186/1472-6963-12-323.
Schildmeijer KGI, Unbeck M, Ekstedt M, Lindblad M, Nilsson L. Adverse events in patients in home healthcare: a retrospective record review using trigger tool methodology. BMJ Open. 2018;8(1): e019267. https://doi.org/10.1136/bmjopen-2017-019267.
Woodley WD, Yue W, Morel DR, Lainesse A, Pettis RJ, Bolick NG. Clinical evaluation of an investigational 5 mL wearable injector in healthy human subjects. Clin Transl Sci. 2021;14(3):859–69. https://doi.org/10.1111/cts.12946.
DeGrazio F, Paskiet D. Injectable combination product development: facilitating risk-based assessments for efficiency and patient centric outcomes. J Pharm Sci. 2020;109(7):2101–15. https://doi.org/10.1016/j.xphs.2020.03.020.
Hu P, Wang J, Florian J, Shatzer K, Stevens AM, Gertz J, Ji P, et al. Systematic review of device parameters and design of studies bridging biologic-device combination products using prefilled syringes and autoinjectors. AAPS J. 2020;22(2):52. https://doi.org/10.1208/s12248-020-0433-8.
Rummel M, Kim TM, Aversa F, Brugger W, Capochiani E, Plenteda C, et al. Preference for subcutaneous or intravenous administration of rituximab among patients with untreated CD20+ diffuse large B-cell lymphoma or follicular lymphoma: results from a prospective, randomized, open-label, crossover study (PrefMab). Ann Oncol. 2017;28(4):836–42. https://doi.org/10.1093/annonc/mdw685.
Salar A, Avivi I, Bittner B, Bouabdallah R, Brewster M, Catalani O, et al. Comparison of subcutaneous versus intravenous administration of rituximab as maintenance treatment for follicular lymphoma: results from a two-stage, phase Ib study. J Clin Oncl. 2014;32(17):1782–91. https://doi.org/10.1200/JCO.2013.52.2631.
Tang Y, Cao Y. Modeling pharmacokinetics and pharmacodynamics of therapeutic antibodies: progress, challenges, and future directions. Pharmaceutics. 2021;13:422. https://doi.org/10.3390/pharmaceutics13030422.
Hourcade-Potelleret F, Lemenuel-Diot A, McIntyre C, Brewster M, Lum B, Bittner B. Use of a population pharmacokinetic approach for the clinical development of a fixed-dose subcutaneous formulation of trastuzumab. CPT Pharmacometrics Syst Pharmacol. 2014;3(1): e87. https://doi.org/10.1038/psp.2013.63.
Richter WF, Jacobsen B. Subcutaneous absorption of biotherapeutics: knowns and unknowns. Drug Metab Dispos. 2014;42(11):1881–9. https://doi.org/10.1124/dmd.114.059238.
Fukushima Y, Charoin JE, Brewster M, Jonsson NE. Population pharmacokinetic analysis of trastuzumab (Herceptin®) based on data from three different dosing regimens. PAGE. 2007;16:1121.
Kimko H, Pinheiro J. Model-based clinical drug development in the past, present and future: a commentary. Br J Clin Pharmacol. 2015;79(1):108–16. https://doi.org/10.1111/bcp.12341.
US FDA. Focus area: model-informed product development. 2022. Available at: https://www.fda.gov/science-research/focus-areas-regulatory-science-report/focus-area-model-informed-product-development#:~:text=MIPD%20encompasses%20model%2Dinformed%20drug,development%20or%20regulatory%20decision%2Dmakin. (Accessed 15 Jun 2023).
US FDA. Model-informed drug development paired meeting program. 2023. Available from: https://www.fda.gov/drugs/development-resources/model-informed-drug-development-paired-meeting-program. (Accessed 15 Jun 2023).
Madabushi R, Seo P, Zhao L, Zhu H. Review: role of model-informed drug development approaches in the lifecycle of drug development and regulatory decision-making. Pharm Res. 2022;39:1669–80. https://doi.org/10.1007/s11095-022-03288-w.
Xiong Y, Fan J, Kitabi E, Zhang X, Bi Y, Grimstein M, et al. Model-informed drug development approaches to assist new drug development in the COVID-19 Pandemic. Clin Pharmacol Ther. 2022;111(3):572–8. https://doi.org/10.1002/cpt.2491.
Hey SP, Kimmelman J. The questionable use of unequal allocation in confirmatory trials. Neurology. 2014;82(1):77–9. https://doi.org/10.1212/01.wnl.0000438226.10353.1c.
Richards SJ, Painter D, Dickinson AJ, Griffin M, Munir T, Arnold L, et al. The incidence and prevalence of patients with paroxysmal nocturnal haemoglobinuria and aplastic anaemia PNH syndrome: a retrospective analysis of the UK’s population-based haematological malignancy research network 2004–2018. Eur J Haematol. 2021;107(2):211–8. https://doi.org/10.1111/ejh.13640.
Dunvald AD, Iversen DB, Svendsen ALO, Agergaard K, Kuhlmann IB, Mortensen C, et al. Tutorial: statistical analysis and reporting of clinical pharmacokinetic studies. Clin Transl Sci. 2022;15(8):1856–66. https://doi.org/10.1111/cts.13305.
Bittner B, Schmidt J. Subcutaneous drug delivery devices: enablers of a flexible care setting. In: Chappel E, editor. Drug delivery devices and therapeutic systems. London: Elsevier Academic Press; 2021. p. 159–79.
Woodley WD, Morel DR, Sutter DE, Pettis RJ, Bolick NG. Clinical evaluation of large volume subcutaneous injection tissue effects, pain, and acceptability in healthy adults. Clin Transl Sci. 2022;15(1):92–104. https://doi.org/10.1111/cts.13109.
Burmester GR, Rubbert-Roth A, Cantagrel A, Hall S, Leszczynski P, Feldman D, et al. Efficacy and safety of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional DMARDs in patients with RA at week 97 (SUMMACTA). Ann Rheum Dis. 2016;75(1):68–74. https://doi.org/10.1136/annrheumdis-2015-207281.
Kivitz A, Olech E, Borofsky M, Zazueta BM, Navarro-Sarabia F, Radominski SC, et al. Subcutaneous tocilizumab versus placebo in combination with disease-modifying antirheumatic drugs in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2014;66(11):1653–61. https://doi.org/10.1002/acr.22384.
Genovese MC, Covarrubias A, Leon G, Mysler E, Keiserman M, Valente R, et al. Subcutaneous abatacept versus intravenous abatacept: a phase IIIb noninferiority study in patients with an inadequate response to methotrexate. Arthritis Rheum. 2011;63(10):2854–64. https://doi.org/10.1002/art.30463.
US FDA. Bridging for drug-device and biologic-device combination products. Guidance for Industry. 2019. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bridging-drug-device-and-biologic-device-combination-products. (Accessed 15 Jun 2023).
Lambert WJ. Why Do the majority of submissions for bridging from a prefilled syringe to an autoinjector include bioequivalence studies in order to demonstrate comparability? AAPS J. 2020;22(3):72. https://doi.org/10.1208/s12248-020-00453-0.
Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):576–88. https://doi.org/10.1002/psp4.12224.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Not applicable.
Conflicts of Interest
Beate Bittner and Johannes Schmidt are employees of F. Hoffmann La Roche and own stock in Roche. The authors declare they have no financial interests. Beate Bittner and Johannes Schmidt serve on the board of the Subcutaneous Drug Delivery and Development Consortium.
Ethical Approval
Not applicable.
Patient Consent to Participate/Publish
Not applicable.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Author Contributions
Beate Bittner was the lead author of the manuscript, defined the concept and scope of the work, conducted the literature search, and wrote the document. Johannes Schmidt conducted the literature search, contributed to the preparation of the tables and figures, and provided input into the scope and concept. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bittner, B., Schmidt, J. Advancing Subcutaneous Dosing Regimens for Biotherapeutics: Clinical Strategies for Expedited Market Access. BioDrugs 38, 23–46 (2024). https://doi.org/10.1007/s40259-023-00626-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-023-00626-1