
Abstract
Malaria is a mosquito-borne disease caused by protozoan parasites of the genus Plasmodium. Despite significant declines in malaria-attributable morbidity and mortality over the last two decades, it remains a major public health burden in many countries. This underscores the critical need for improved strategies to prevent, treat and control malaria if we are to ultimately progress towards the eradication of this disease. Ideally, this will include the development and deployment of a highly effective malaria vaccine that is able to induce long-lasting protective immunity. There are many malaria vaccine candidates in development, with more than a dozen of these in clinical development. RTS,S/AS01 (also known as Mosquirix) is the most advanced malaria vaccine and was shown to have modest efficacy against clinical malaria in phase III trials in 5- to 17-month-old infants. Following pilot implementation trials, the World Health Organisation has recommended it for use in Africa in young children who are most at risk of infection with P. falciparum, the deadliest of the human malaria parasites. It is well recognised that more effective malaria vaccines are needed. In this review, we discuss malaria vaccine candidates that have progressed into clinical evaluation and highlight the most advanced candidates: Sanaria’s irradiated sporozoite vaccine (PfSPZ Vaccine), the chemoattenuated sporozoite vaccine (PfSPZ-CVac), RTS,S/AS01 and the novel malaria vaccine candidate, R21, which displayed promising, high-level efficacy in a recent small phase IIb trial in Africa.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Malaria is a mosquito-borne disease caused by Plasmodium parasites and it causes significant morbidity and mortality. |
Improved strategies are needed to prevent and control malaria, including the deployment of a highly effective malaria vaccine that is able to induce long-lasting protective immunity. |
RTS,S/AS01 (Mosquirix), a moderately efficacious vaccine targeting the pre-erythrocytic stage of the parasite, has now been recommended for the prevention of P. falciparum malaria in children living in regions with moderate-high malaria transmission. |
More effective malaria vaccines will be required to eradicate malaria; there are many candidates in the malaria vaccine development pipeline, with a number of these currently being evaluated in clinical trials. |
1 Introduction
Plasmodium parasites, the causative agents of malaria, are endemic in 84 countries and resulted in 247 million cases of malaria and 619,000 deaths in 2021, mostly in children < 5 years of age [1]. Malaria disproportionately affects the world’s poorest populations and is transmitted by female mosquitoes of the Anopheles genus. There are eight different Plasmodium spp. that can cause malaria in humans, including the most common species, P. falciparum and P. vivax. P. falciparum is responsible for the majority of morbidity and mortality [1]. It is most commonly found in Africa, which remains the continent with the greatest burden of malaria cases and deaths in the world.
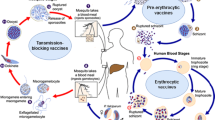
The P. falciparum parasite has a complex life-cycle, requiring both an invertebrate definitive host and a mammalian host. The P. falciparum sporozoite is injected by the mosquito into the human host during a blood meal and travels via the blood to the liver where it invades hepatocytes. Once inside the hepatocyte, the sporozoite undergoes exoerythrocytic schizogony which results in the production of tens of thousands of merozoites. When the hepatocyte ruptures, the merozoites are released into the bloodstream where they invade red blood cells (RBCs). Here, the parasites undergo further development, culminating in erythrocytic schizogony and the production of 16–32 merozoites which are released upon cell rupture and invade new RBCs. Parasites that are sexually committed, develop within the RBCs into male and female gametocytes that are infectious to the mosquito host and can be taken up during a blood meal. The parasite undergoes sexual development in the mosquito, and this concludes with the production of sporozoites that invade the mosquito’s salivary glands and are transmissible to the human host.
The pre-erythrocytic stage of the parasite life-cycle is clinically silent. It is the blood stage that is responsible for the signs and symptoms of malaria, which can include fever, chills, muscle aches, headache, altered consciousness, nausea, vomiting, diarrhea, fatigue, and respiratory distress. Malaria is typically classified as asymptomatic, uncomplicated symptomatic or severe and can rapidly progress to severe disease if it is not diagnosed and treated promptly. Severe malarial disease can manifest as cerebral malaria, severe anaemia, acute respiratory distress syndrome, multi-organ failure and may result in death. It is individuals with limited or no prior exposure to the malaria parasite who are most at risk of severe disease and naturally acquired host immunity is inextricably linked with parasite exposure in malaria endemic regions. In areas of high malaria endemicity, it is young children that are most impacted by symptomatic and severe disease. By the time they are adults, and as a result of repeated exposure to the parasite, they develop partially protective immunity, also known as clinical immunity, which manifests as asymptomatic infection and lower parasite burdens [2].
Existing control strategies for the mosquito vector (insecticides) [3] and the parasite (anti-malarial drugs) [4] are becoming less effective due to the development of resistance. Artemisinin-combination therapies (ACTs) are the frontline treatment for P. falciparum malaria. The emergence and spread of artemisinin resistance as well as partner drug resistance, has contributed to a decline in ACT efficacy [5, 6] in the Greater Mekong Subregion. Resistance to artemisinin has also now been documented in parts of Africa [7]. To progress towards the ambitious goal of malaria eradication, additional control measures will be required. A highly effective malaria vaccine that can induce long-lasting protective immunity will contribute to a reduction in malaria-attributable clinical disease and death and will have a significant positive impact on global public health. In this review we discuss some of the P. falciparum vaccine candidates that have progressed into clinical evaluation, with a focus on the most advanced vaccine candidates.
2 Strategies for Malaria Vaccine Development
Despite a century of research, we still do not have a highly effective malaria vaccine that is able to induce long-lasting protective immunity. This can be attributed to a number of parasite-specific factors including the complexity of the malaria parasite’s life-cycle, the predominantly stage-specific expression of targets of the immune response and the parasite’s ability to evade and modulate the human immune response [8]. The majority of vaccines in development target a single life-cycle stage. The revised Malaria Vaccine Technology Roadmap calls for the development of malaria vaccines by 2030 with at least 75% protective efficacy against P. falciparum clinical malaria, with this level of immunity maintained for at least 2 years [9]. More recently, the WHO’s preferred product characteristics (PPC) document for malaria vaccines outlines three strategic goals: (i) malaria vaccines that prevent human blood-stage infection at the individual level, (ii) malaria vaccines that reduce malaria morbidity and mortality in individuals at risk in malaria-endemic areas and (iii) malaria vaccines that reduce transmission of the parasite and thereby substantially reduce the incidence of human infection in the community [10]. Here, the suggested target levels of efficacy associated with goals (i) and (ii) are more ambitious, that is, a 90% reduction in the incidence of blood-stage infection and clinical malaria over 12 months of follow-up with the acknowledgement that vaccines with lower efficacy against clinical malaria also have the potential for significant public health impact [10]. The realisation of these goals will likely require a multi-pronged approach including further optimisation of current vaccine candidates that have not yet achieved this level of protective efficacy in the field, developing and evaluating other diverse and novel vaccine approaches and combining parasite antigens or sub-optimal vaccine candidates that target different life-cycle stages. Vaccines targeting the pre-erythrocytic stage of the life-cycle do not so far appear to induce protective immune responses that also target blood-stage parasites (for example [11]); thus, if these pre-erythrocytic-stage vaccines are ‘leaky’ and parasites escape the vaccine-induced response, they will exit the liver, develop within the blood and allow transmission. A vaccine approach targeting multiple stages of the life-cycle may enable synergistic activity to induce superior protection.
The majority of vaccine candidates in clinical development can be broadly categorised as whole-parasite or sub-unit vaccines. The rationale for including the whole parasite in the vaccine is to maximise the number of proteins (some of which are conserved between parasite strains) available to the immune system and ensures immunisation with a broad protein repertoire. Such an approach is thought to limit the impact of variation in any single parasite protein [12], which has been seen to negatively affect the efficacy of sub-unit vaccine candidates [13, 14]. The majority of malaria vaccines in development are sub-unit vaccines that contain a single parasite protein to stimulate an antibody response. These individual proteins can vary between different P. falciparum strains [12] and this may impact on vaccine efficacy, manifesting as narrow protection against parasite strains that match the vaccine. Ideally, this sub-unit vaccine approach requires the identification of invariant (conserved), biologically relevant proteins and the induction of persisting, high titre functional antibodies. Sub-unit vaccines generally require inclusion of an adjuvant to ensure adequate immunogenicity; identification of safe and potent human-compatible adjuvants has been a significant hurdle. A number of sub-unit vaccine candidates have progressed from pre-clinical evaluation into clinical trials, but the majority have had limited efficacy when tested in malaria endemic areas. Only a single vaccine candidate, RTS,S/AS01 (Mosquirix), has completed evaluation in phase III trials and although it only has moderate efficacy against clinical malaria, it was recently recommended for use in children who are at high risk of P. falciparum infection in sub-Saharan Africa [15].
3 Clinical Evaluation of Malaria Vaccines
A malaria vaccine’s efficacy can be evaluated in both controlled human malaria infection (CHMI) challenge studies and field trials. Following the demonstration of safety and immunogenicity in a phase I trial, the use of CHMI enables a controlled assessment of the vaccine’s protective efficacy in a relatively small number of individuals prior to embarking on larger, more expensive field trials. In CHMI studies, following vaccination, individuals are deliberately exposed to the malaria parasite either by mosquito bite or by direct injection of sporozoites or parasitised red blood cells (pRBCs) (reviewed in [16]) to assess vaccine efficacy. For pre-erythrocytic vaccine candidates, the study endpoint is generally blood-stage infection, detected by microscopy or qPCR. For blood-stage vaccine candidates, detection of a sub-patent blood-stage infection or the parasite multiplication rate are used. Although CHMI studies have historically involved malaria-naïve adults, more recently they have also been undertaken in malaria-exposed adults in malaria endemic areas. The latter enables a preliminary assessment of how the vaccine will perform in the presence of pre-existing anti-malarial immunity.
In field trials, protection against natural infection is assessed in residents of malaria endemic areas. For vaccines targeting pre-erythrocytic or asexual blood-stage parasites, efficacy can be measured against different endpoints, including blood-stage infection (e.g., time to first infection; incidence of infections of a defined parasite density), clinical disease (e.g., time to first or only episode of clinical malaria), severe malaria and death. In field trials, for an accurate assessment of efficacy against blood-stage infection, drug treatment should be administered prior to the start of the follow-up period.
A recent CHMI study suggests that it may also be necessary to administer drug treatment during the vaccination period as blood-stage parasitemia was shown to negatively impact on the efficacy of a pre-erythrocytic malaria vaccine [17]. While the precise mechanism for this is unknown, asymptomatic P. falciparum infections are known to be associated with immunosuppression [18]. Based on these observations, it is possible that vaccine efficacy has been underestimated in studies where drug treatment has not been used to clear blood-stage parasitemias prior to vaccination.
4 Asexual Blood-Stage Vaccines
Vaccines targeting the asexual blood-stage of the malaria parasite aim to reduce parasite burden and prevent clinical disease. The majority of vaccine candidates endeavour to achieve this by inducing antibodies that prevent invasion of the merozoite into new RBCs, prevent adhesion of the pRBCs to the vasculature in critical organs and promote phagocytosis of pRBCs. Many of the challenges that researchers have faced developing blood-stage vaccines are related to the selection of the vaccine antigen, for example, difficulties with antigen production, polymorphic vaccine antigens that are immunologically distinct, and redundancy in merozoite invasion ligands which can result in the parasite switching invasion pathways to evade an immune response. It should be noted that some of these challenges are also relevant to the development of pre-erythrocytic-stage and transmission-blocking vaccines. Additionally, from a biological perspective, antibodies targeting merozoite antigens have a very limited timeframe in which to neutralise the parasite prior to invasion of the RBC.
4.1 Whole-Parasite Blood-Stage Vaccines
Few whole-parasite blood-stage vaccine candidates have progressed beyond pre-clinical evaluation (reviewed in [19]). A single dose of a chemically attenuated P. falciparum whole-parasite blood-stage vaccine candidate was shown to induce species and strain-transcending parasite-specific cellular responses in malaria-naïve adults [20]; further clinical studies are required to assess its efficacy. Unlike the majority of vaccine candidates targeting blood-stage parasites, pre-clinical studies demonstrated that this chemically attenuated vaccine approach may induce antibody-independent protection [21]. A genetically attenuated P. falciparum whole-parasite blood-stage vaccine (with the knob-associated histidine-rich protein (KAHRP) gene deleted) was also evaluated in malaria-naïve adults [22]. Parasite-specific immune responses were detected in individuals who developed parasitemia following vaccination, however, drug treatment was required to terminate these infections. For both approaches, as the parasites were administered within intact RBCs from a universal blood donor, the risk of alloimmunisation was considered minimal. However, the induction of antibodies against RBC antigens was observed in a small proportion of volunteers [20, 22]. Further refinement of whole-parasite vaccine candidates containing RBC material will be required to address this risk of alloimmunisation.
4.2 Sub-Unit Blood-Stage Vaccines
CHMI challenge studies and field trials in malaria endemic areas have generally yielded disappointing results for sub-unit blood-stage vaccine candidates. No blood-stage vaccine candidate has progressed into phase III trials. Falciparum malaria protein 1 (FMP1), the 42kDa C-terminal fragment of merozoite surface protein-1 (MSP-1) formulated with the adjuvant AS02, was shown to have limited efficacy against clinical malaria in Kenyan children [23]. P falciparum apical membrane antigen-1 (AMA-1) was tested in different vaccine formulations in malaria endemic areas. When tested in Malian children, the AMA-C1/Alhydrogel [24] and the FMP2.1/AS02A vaccines [25] did not provide significant protection against clinical malaria. In the latter study there was, however, evidence of allele-specific vaccine efficacy (64%) against clinical malaria in a secondary analysis [25]. More recently, it was shown that FMP2.1/AS01 was not protective against CHMI in malaria-naïve adults [26]. Although the trial was not designed to evaluate vaccine efficacy, there was some evidence of short-lived protection against clinical malaria in Burkinabe children following vaccination with a merozoite surface protein-3 (MSP3) long-synthetic peptide adjuvanted with aluminium hydroxide [27]. P. falciparum serine repeat antigen-5 formulated with aluminium hydroxide (BK-SE36) was evaluated in Ugandan children and adults; there was some evidence of protection against high density parasitemia infections and symptomatic episodes [28]. Vaccination of malaria-naïve adults with the more conserved P. falciparum antigen, reticulocyte-binding protein homolog 5 (Rh5) formulated with AS01B (Rh5.1/AS01B), resulted in a significant reduction in parasite growth rates in vaccinees following challenge with CHMI [29].
A number of vaccine candidates containing multiple blood-stage antigens have also been evaluated. Vaccination with the ‘Combination B’ vaccine comprising P. falciparum ring-infected erythrocyte surface antigen (RESA) and two merozoite surface proteins (MSP1 and MSP2) formulated in the adjuvant, montanide ISA720, resulted in a 62% reduction in parasite densities in a phase I/IIb trial in Papua New Guinean children [14]. There was also evidence of vaccine-induced selection of infecting parasites in this study, attributable to the MSP2 component of the vaccine. A vaccine containing GMZ (a fusion protein of fragments of P. falciparum MSP3 and glutamate-rich protein) formulated with aluminium hydroxide was 14 and 27% effective against episodes of malaria and severe malaria, respectively, in African children [30]. Vaccines containing GMZ that were formulated with the adjuvants CAF01 or alhydrogel were not protective however, when assessed in malaria-exposed adults following challenge with sporozoite CHMI [31].
Strategies including (i) further identification of invariant, biologically relevant proteins with non-redundant functions, (ii) using a multi-allelic/multi-protein vaccine approach and (iii) identifying and utilising new adjuvants to maximise the induction and persistence of high titre antibodies with relevant functionality, will be needed to maximise chances of developing an effective sub-unit malaria vaccine targeting the asexual blood-stage of the malaria parasite.
4.3 Placental Malaria Vaccines
Despite pre-existing naturally acquired anti-malarial immunity, pregnant women living in malaria endemic areas have an increased susceptibility to malaria infection. This can result in severe outcomes for both mother and foetus including maternal anaemia, hypertension, low birthweight, stillbirth, and spontaneous abortion. This susceptibility decreases over successive pregnancies, indicating that women do eventually develop immunity against this pregnancy-associated malaria [32, 33]. Malaria during pregnancy is associated with the accumulation of P. falciparum pRBCs in the placenta; this is mediated by the P. falciparum variant surface antigen, VAR2CSA, binding to chondroitin sulfate A in the intervillous spaces of the placenta [34]. Placental malaria vaccines are focused on preventing the sequestration of the pRBCs in the placenta. Two sub-unit vaccine candidates (PRIMVAC and PAMVAC) containing different recombinant N-terminal fragments of VAR2CSA adjuvanted with alhydrogel or glucopyranosyl lipid adjuvant in stable emulsion (GLA-SE) (PRIMVAC and PAMVAC) or in a liposomal formulation with QS21 (GLA-LSQ) (PAMVAC) were recently evaluated in phase I trials to assess safety and immunogenicity [35, 36]. Anti-adhesion antibodies were induced against homologous parasites, but further optimisation may be needed to improve the functional activity of antibodies against heterologous parasites.
5 Transmission-Blocking Vaccines
Transmission-blocking vaccines typically contain surface antigens from the parasite’s sexual/mosquito stages and aim to induce antibodies that prevent the parasite from developing within the mosquito, thereby disrupting its transmission. These vaccines do not provide a direct benefit to the vaccinee, instead with sufficient vaccine coverage they aim to induce community-based immunity. The main challenges with this vaccine approach have been in relation to antigen production, sub-optimal immunogenicity of vaccine candidates and the rapid waning of antibodies following vaccination. Only results from clinical studies involving the sub-unit vaccine candidates Pfs230 (gametocyte/gamete antigen) and Pfs25 (a post-fertilisation antigen expressed in the mosquito) have been reported.
Various Pfs25 vaccine formulations have been evaluated in clinical studies. A Pfs25 virus-like particle (VLP) vaccine was evaluated in malaria-naïve humans and although immunogenic, the induced antibodies had only weak functional activity [37]. To increase immunogenicity, Pfs25 was conjugated to the Pseudomonas aeruginosa ExoProtein A (EPA) and formulated with alhydrogel. In malaria-naïve adults, the functional antibodies induced by the vaccine blocked P. falciparum transmission to mosquitoes in vitro [38]. When this formulation was tested in adults in a malaria endemic area it also induced antibodies with significant functional activity in vitro; however, four doses were required, and the antibodies waned rapidly [39]. In a further study, Pfs25-EPA or Pfs230D1-EPA were formulated with alhydrogel and administered as two doses to malaria-naïve adults. Only serum from Pfs230D1/alhydrogel vaccinees had substantial levels of functional activity, which was complement-dependent [40].
6 Pre-erythrocytic-Stage Vaccines
Vaccines targeting the pre-erythrocytic stage of the malaria parasite aim to prevent the sporozoite from invading and completing development within the hepatocyte. Once the human host is infected, sporozoites are thought to invade hepatocytes within 30 minutes; this is a narrow window in which antibodies need to neutralise the sporozoite to prevent invasion of hepatocytes in the liver. Following successful sporozoite invasion, CD8+ T cells can recognise parasite antigens on the surface of hepatocytes and kill these parasitised cells. If a pre-erythrocytic vaccine is able to completely block infection, it induces sterile protective immunity.
6.1 Whole-Parasite Pre-erythrocytic-Stage Vaccines
Substantial work has been undertaken to develop a whole-parasite pre-erythrocytic-stage vaccine. Early studies in mice and humans confirmed that immunising humans via the bites of irradiated mosquitoes infected with P. berghei and P. falciparum, respectively, could protect against challenge with infectious sporozoites [41,42,43,44]. This strategy was further refined [45] and has progressed as Sanaria’s live attenuated PfSPZ Vaccine which consists of aseptic, purified, radiation-attenuated sporozoites which have been harvested from mosquitoes and administered predominantly via direct venous injection (DVI). Vaccine efficacy against infection has been evaluated in numerous CHMI trials in both malaria-naïve and malaria-exposed adults and in field trials (Tables 1, 2). For this vaccine, the route of immunisation was shown to be critical, with intradermal and subcutaneous vaccination being sub-optimally protective [46]. In malaria-naïve adults, a dose-dependent immunological threshold for high-level protection was proposed, with five doses of 1.35 × 105 sporozoites providing complete protection using homologous CHMI [47]. Recent studies in malaria endemic areas demonstrated 52 and 51% efficacy against infection in adults following five doses of 2.7 × 105 PfSPZ Vaccine [48] or three doses of 1.8 × 106 PfSPZ Vaccine [49], respectively, with drug treatment administered during the vaccination period to eliminate pre-existing parasites. No significant efficacy against infection was observed in Kenyan infants at 6 months, the primary statistical endpoint of the study [50]; here, drug treatment was not administered during the vaccination period. An age-dependent lack of PfSPZ vaccine-specific T-cell responses, including the infrequent detection of Vδ2+Vγ9+ T cells at the time of immunisation, was also proposed as a possible explanation for the poor vaccine efficacy in these infants. Further optimisation of the vaccine regimen was undertaken in malaria-naïve adults, culminating in an accelerated 4-week, three-dose vaccine regimen with 9 × 105 PfSPZ that provided similar protection against homologous and heterologous CHMI (overall vaccine efficacy of 77 and 79%, respectively) for 9–10 weeks [51]. When this regimen was tested in malaria-exposed adults using challenge with homologous CHMI, a vaccine efficacy of 51% was observed [52], with drug treatment administered prior to vaccination to clear pre-existing parasites.
A chemoattenuated, whole-parasite pre-erythrocytic-stage vaccine has also been pursued. This encompasses sporozoite immunisation combined with an anti-malarial drug, which controls the infection while still enabling sufficient parasite exposure to induce a protective immune response. For this vaccine approach, sporozoites have been administered via either the bites of laboratory-reared P. falciparum infected mosquitoes (chemoprophylaxis with P. falciparum sporozoites [CPS]) or by DVI (P. falciparum sporozoite chemoprophylaxis vaccine [PfSPZ-CVac]). Vaccine regimens incorporating different anti-malarial drugs have been examined and this has been reviewed comprehensively elsewhere [53]; chloroquine (CQ) has been most frequently used. An early study evaluating three immunisations with P. falciparum sporozoites administered via 12–15 mosquito bites + CQ reported 100% sterile protection against sporozoite CHMI [54]. More recently the focus has been on immunisation via DVI of purified sporozoites, PfSPZ-CVac, which enables the administration of a precise dose of sporozoites as well as being more feasible for a mass vaccination programme. A number of studies in malaria-naïve adults (Table 1) have focused on optimising the vaccination regimen to maximise protective efficacy, eventually culminating in 100% protection against heterologous CHMI following three doses of 2 × 105 sporozoites + CQ, with the sporozoites administered at monthly intervals [55]. In malaria-exposed adults, vaccine efficacy was 55% following three doses of 1 × 105 sporozoites + CQ and challenge with homologous CHMI [56]. However, when evaluated in adults in a high transmission setting, no significant protection against infection or clinical malaria was observed following three doses of 2.048 × 105 sporozoites administered with CQ [57] (Table 2). This lack of efficacy was attributed to vaccine hyporesponsiveness. Three factors were proposed to contribute to this: pre-existing immune responses reducing the number of sporozoites that were able to effectively invade and develop within the liver and induce protective immune responses, immune dysregulation as a result of lifelong exposure to P. falciparum and the lack of drug treatment prior to vaccination to clear pre-existing parasites. Further studies are focused on evaluating a more condensed vaccination regimen (Table 1), with a Day 0, 5, 28 schedule yielding 77% efficacy in malaria-naïve adults following challenge with heterologous CHMI [58].
Different genetically attenuated P. falciparum sporozoite vaccine candidates have also been developed and evaluated in malaria-naïve human volunteers (e.g., [59, 60]). Here, the aim is to arrest the development of the genetically attenuated parasites (GAPs) in the liver prior to the parasites entering the bloodstream. The PfGAP3KO vaccine (P. falciparum with deletions in the P52, P36 and SAP1 genes) is the most recent candidate to undergo clinical evaluation. Following three or five immunisations with 200 PfGAP3KO-infected mosquito bites/immunisation, 50% of vaccinees developed sterile protective immunity against homologous CHMI [59]. Genetically attenuated parasites that arrest growth late in the liver have been shown in animal models to induce stronger protective immune responses than those that arrest earlier and this may be explained by exposure to a broader repertoire of parasite antigens [61]. Late liver stage-arresting replication-competent (LARC) P. falciparum GAPs have been developed by two different research groups through the targeted deletion of the Mei2 gene [62, 63]. In both instances, the parasite does not produce exoerythrocytic merozoites, and this prevents initiation of the blood-stage infection. These LARC GAPs are being further developed as next-generation, live-attenuated sporozoite vaccines.
Historically, all whole-sporozoite vaccines have relied on mosquitoes as a source of infectious sporozoites. For the PfSPZ Vaccine and PfSPZ-CVac, which contain purified sporozoites, the manufacturing process has been a challenge for large-scale vaccine production as it is labour-intensive, involving the manual dissection of salivary glands of infected mosquitoes to obtain the sporozoites. Recently, Sanaria have developed an in vitro method for the production of infectious sporozoites without the need for mosquitoes [64]. While further optimisation is required, this development has the potential to transform the manufacture of PfSPZ-based vaccines and facilitate their production for mass vaccination programmes.
6.2 Non-circumsporozoite Protein Sub-Unit Pre-erythrocytic-Stage Vaccines
Vaccines based on thrombospondin-related anonymous protein (TRAP) have utilised viral vectors to deliver ME-TRAP, a fusion protein consisting of a multi-epitope string (ME) and TRAP, in heterologous prime-boost regimens. The ME string consists of 20 epitopes, predominantly CD8+ T-cell epitopes from P. falciparum pre-erythrocytic antigens. These vaccines aim to induce a protective CD8+ T-cell response that can target parasitised hepatocytes. The most recent iteration of the ME-TRAP vectored vaccine regimen used non-replicating chimpanzee adenovirus 63 (ChAd63) and Modified Vaccinia Virus Ankara (MVA) expressing ME-TRAP. In a phase IIb trial, vaccine efficacy against infection was 67% in Kenyan adults over a 2-week follow-up period [65]. Significant vaccine efficacy was not observed in Senegalese adults [66] or in Burkinabe infants [67] against infection or clinical malaria, respectively.
6.3 Circumsporozoite Protein-Based Sub-Unit Pre-erythrocytic-Stage Vaccines
The cloning of the first malaria genes in the 1980s [68,69,70] led to the development of circumsporozoite protein (CSP)-based vaccine candidates. Most of the sub-unit vaccines targeting the pre-erythrocytic stage of the parasite are based on the CSP, which is expressed on both the sporozoite and liver-stage parasites. The RTS,S/AS01 and R21 vaccines have been evaluated in field trials (discussed further below). These vaccines both contain truncated CSP without the amino terminus. The amino terminal region is critical for sporozoite attachment and invasion; antibodies targeting this region are associated with protection. Thus, full-length (FL) CSP vaccine candidates are also in early clinical development, with a recent study showing that a FL CSP vaccine adjuvanted with GLA-LSQ was safe and immunogenic in malaria-naïve humans [71]. Efficacy of this vaccine candidate has not yet been reported. A gene-based vaccine approach has also been evaluated using a heterologous prime-boost regimen. Volunteers were primed with both PfCSP and PfAMA-1 DNA and boosted with adenoviral vectors encoding the genes expressing PfCSP and PfAMA-1 [72]. PfAMA-1 was chosen as a second antigen due to its expression on sporozoites and liver-stage parasites and its’ potential to induce blood-stage immunity. This regimen induced the highest level of sterile immunity in humans that has been achieved with a gene-based anti-parasite vaccine; 4/15 volunteers (27%) were protected against homologous CHMI. Protection was associated with AMA-1-specific CD8+ T-cell responses. Further vaccine optimisation is required to improve efficacy.
6.3.1 RTS,S—The Clinical Trials
RTS,S, the most advanced pre-erythrocytic vaccine, utilises the hepatitis B surface antigen (HBsAg) VLP vaccine platform. RTS,S contains 189 amino acids from a single allele of the P. falciparum CSP including NANP conserved repeats (the ‘R’) from the central region and the C-terminus of the non-repeat region which contains T-cell epitopes (the ‘T’). The NANP repeats contain the immunodominant B-cell epitopes while the C-terminal region contains numerous polymorphisms and three known T-cell epitopes consisting of highly variable CD4+ and CD8+ T-cell epitopes and a conserved ‘universal’ CD4+ T-cell epitope. This truncated CSP fragment is fused to approximately 25% of the HBsAg (the ‘S’) that is in the vaccine. When expressed in yeast cells, this chimeric fusion protein (RTS) and the remaining un-fused HBsAg (the ‘, S’), self-assemble to form VLPs with the CSP and S sequences displayed on their surface. In the 1980s, GlaxoSmithKline (GSK) and the Walter Reed Army Institute of Research (WRAIR) entered into a collaborative research partnership to develop the RTS,S vaccine. There have been numerous refinements to the RTS,S vaccine over the years, with a major focus on evaluating different adjuvants, including GSK’s proprietary adjuvant systems (Table 3), to maximise the vaccine’s efficacy.
Numerous studies have been undertaken in malaria-naïve individuals using CHMI to evaluate vaccine efficacy against infection (Table 4). Initially, two different RTS,S formulations (RTS,S/Alum and RTS,S/Alum/Monophosphoryl lipid A [MPL] [AS04]) were evaluated in malaria-naïve adults. Only the RTS,S/AS04 vaccine demonstrated any efficacy, completely protecting 2/8 vaccinees from challenge with CHMI [73]. A further study in malaria-naïve adults evaluated the efficacy of RTS,S vaccines formulated with the adjuvants AS04, AS03 (an oil-in-water emulsion) or AS02 (an oil-in-water emulsion with MPL and the saponin QS21). Poor efficacy was observed in the AS04 (1/8) and AS03 (2/7) adjuvanted vaccine groups, however 6/7 volunteers (85.7%) who received the AS02 adjuvanted vaccine were completely protected [74]. Both the AS03 and AS02 groups received fractional third doses. Subsequently, a number of phase Ia/II challenge studies were undertaken for the RTS,S/AS02 vaccine to optimise antigen dose and vaccine regimen [75,76,77]. Across all studies, when two to three doses of RTS,S 50 µg were administered, protective efficacy against challenge with CHMI was approximately 40–50%.
Heterologous prime-boost vaccine regimens have been examined for RTS,S/AS02. RTS,S/AS02 was administered to naïve volunteers and volunteers previously immunised with PfCSP DNA to assess safety and immunogenicity [78]. Although priming with PfCSP DNA did not result in improved CD4+ T-cell or antibody responses [78, 79], there was an expansion of the PfCSP-specific CD8+ T-cell responses induced by the DNA vaccination [79]. These CD8+ T-cell responses were not observed in individuals who received RTS,S/AS02 alone, indicating that this heterologous prime-boost vaccine regimen resulted in an enhanced, broader immune response. An additional approach employed the viral vector MVA, which expressed the entire CSP as well as two additional T-cell epitopes (CS). RTS,S/AS02 vaccination with MVA-CS as the primary or final booster vaccination resulted in equivalent vaccine efficacy of 33% against challenge with CHMI [80].
In parallel with the studies in malaria-naïve adults, evaluation of RTS,S/AS02 commenced in malaria-exposed individuals (Table 5). In adults, following three doses, vaccine efficacy against infection and clinical malaria was 34 and 31%, respectively [81]. Efficacy waned substantially from 9 weeks following the final vaccine dose. A fourth dose the following year did not improve efficacy over this first 9 weeks of follow-up. Evaluation of the RTS,S/AS02A vaccine candidate in paediatric populations commenced with initial studies focused on dose optimisation, immunogenicity, and safety [82]. Half of the standard dose and volume of RTS,S/AS02A was used for proof-of-concept efficacy studies in children aged 1–4 years in Mozambique [83,84,85,86]. Vaccine efficacy against first clinical episode over 6 months of follow-up was 29.9–35.4%, 45% against infection and 57.7% against severe malaria. This vaccine efficacy was maintained at only one of the two study sites over 18 months of follow-up. This disparity may be due to differences in study design, follow-up and treatment, resulting in differential exposure to low-density parasitemias at the different sites [86]. Subsequently, a specific paediatric formulation (RTS,S/AS02D) was developed, containing half the amount of each active ingredient present in the adult formulation (RTS,S/AS02A) in the same volume [87]. In infants, vaccine efficacy against first clinical episode was 65.8% over a 3-month follow-up [88] and this decreased to 33% (non-significant) after 14 months [89]. At a different study site, this efficacy against clinical malaria was not confirmed [90]. At both sites, vaccine efficacy against infection was 65% over 6 months of follow-up [88, 90].
Based on promising studies in the rhesus non-human primate model, a RTS,S vaccine formulated with the adjuvant AS01 (a liposome-based adjuvant system with MPL and QS21) was compared with the RTS/AS02 vaccine in malaria-naïve adults. Vaccine efficacy, reflecting the percentage of vaccinees who were completely protected (i.e. had sterile immunity), was higher in individuals who received the RTS,S/AS01B vaccine (50%) compared with the RTS,S/AS02A vaccine (32%) [91]. Following this, comparative field trials were undertaken. Unadjusted vaccine efficacy against infection was not significant and was similar for the two vaccine formulations (29.5 vs 31.7%), however, following adjustment for confounders, the AS02A adjuvanted vaccine was superior [92].
A paediatric RTS,S/AS01 formulation, RTS,S/AS01E, was also developed, containing half of the active ingredients of the adult RTS,S/AS01B formulation. Vaccine efficacy against clinical malaria was evaluated in infants aged 5–17 months over 8 months of follow-up at two different African sites and it ranged from 53 to 56% [93]. This efficacy was sustained over 15 months of the 7-year follow-up [94,95,96]. The final analysis showed that vaccine efficacy in year 1 was lower in the cohorts with high malaria parasite exposure (39.5%) compared with low exposure (57.5%) and waned more rapidly in participants with high exposure [96]. A separate study showed that delaying the third dose did not improve vaccine efficacy against clinical malaria over a year of follow-up [97]. A pooled analysis of phase II data for RTS,S vaccines concluded that vaccine efficacy against clinical disease was of limited duration and that it declined with increasing transmission intensity, highlighting the importance of evaluating malaria vaccines across areas of varying malaria transmission and the need to account for these factors when reporting pooled vaccine efficacy data from different sites [98].
RTS,S/AS01E was the first malaria vaccine candidate to progress into a phase III clinical trial. In 2011, the first analysis from children (aged 5–17 months) in this trial showed a vaccine efficacy of 55.8% against clinical malaria and 47.3% against severe malaria over 12 months [99]. The vaccine did not perform as well in 6- to 12-week-old infants; efficacy against clinical and severe malaria was 31.5 and 36.6%, respectively [100]. This trend was observed in a follow-up study which also reported that there was evidence of significant waning of protection over an 18-month follow-up period [101]. The final analyses from this trial were reported in 2015 and included evaluating the impact of a booster dose at 20 months [102]. Vaccine efficacy against clinical malaria was shown to wane over the extended follow-up period. During the full follow-up period of 3–4 years, this efficacy in children and infants who did not receive a booster dose was 28.3 and 18.3%, respectively, while it was slightly improved in children (36.3%) and infants (25.9%) who received the booster dose. Significant efficacy against severe malaria over this period was only observed in older children who received the booster dose. A meningitis safety signal was also reported in the final analysis, reflecting the greater incidence of meningitis in children who received the RTS,S/AS01E vaccine compared with the control group. This finding was further investigated in the expanded pilot implementation studies (mentioned further below). A further analysis demonstrated that in children, the RTS,S/AS01 vaccine had greater efficacy over 12 months of follow-up against clinical malaria caused by parasites that matched the vaccine in the entire C-terminal region of the CSP (50.3%) compared with mismatched parasites (33.4%); in this population, < 10% of parasites matched the vaccine [13]. This indicates that the efficacy of RTS,S/AS01 is partially strain-specific and is influenced by the proportion of matching parasites circulating in the local parasite population.
Samples collected from participants of the RTS/AS01E trials have been used for in-depth interrogation of vaccine-induced immune responses which could enable the identification of immunological surrogates of protection and may be used to inform the development of optimised RTS,S vaccines and other CSP-based vaccines. RTS,S-induced protection is thought to be predominantly mediated by CSP-specific antibodies against the immunodominant central repeat region of the CSP, although T cells may also play a role. Anti-circumsporozoite antibody titres have been shown to be a surrogate of protection and may be used to predict vaccine efficacy over time [103]. While mechanisms of protective immunity are not completely understood, recent work suggests that functionality of the antibodies (e.g. Fc-receptor-mediated functions [104], inhibition of sporozoite invasion and complement fixation and activation [105]) is important. Interestingly, the observed decline in these functional antibodies within a year of vaccination mirrors the decline in vaccine efficacy over that same period [104, 105]. Although the role of CD4+ T cells has not been consistently demonstrated across different studies, they may contribute to protection by providing help to B cells for antibody production, maintenance, class switching and co-operating with other immune cells. Developing a more in-depth understanding of the precise mechanisms of vaccine-induced protective immunity is critical as this may facilitate further optimisation of RTS,S/AS01 to augment vaccine efficacy as well as inform the development of next-generation vaccines.
6.3.1.1 RTS,S/AS01—Beyond the Phase III Trial
Following completion of the phase III trials and a review of the quality, safety, and efficacy data for the vaccine, in 2015, the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) adopted a positive scientific opinion for RTS,S/AS01 for use outside of the European Union [106]. Later that year, two independent WHO advisory groups, the Strategic Advisory Group of Experts (SAGE) and the Malaria Policy Advisory Committee (MPAC), called for a pilot implementation programme in sub-Saharan Africa. Subsequently, WHO-coordinated trials were undertaken in Ghana, Kenya, and Malawi to evaluate the vaccine’s role in reducing mortality, its safety with routine use and the feasibility of delivering it within regular childhood services. In 2021, based on the results from the ongoing pilot programme and advice from SAGE and MPAC, the WHO recommended the use of RTS,S/AS01 for the prevention of P. falciparum malaria in children living in regions with moderate-high malaria transmission. Additionally, the WHO recommended that it should be provided as a four-dose regimen to children aged ≥ 5 months for the reduction of malarial disease. RTS,S/AS01 is the first vaccine against any parasitic disease to be recommended for routine use in humans. Efforts to further optimise the vaccine to improve its efficacy are ongoing and are discussed below.
6.3.1.2 RTS,S/AS01—Modification of the Regimen
As three full doses of RTS,S/AS01 was shown to provide only partial protection in phase III trials, a modified regimen including a fractional dose of RTS,S/AS01 was assessed in an effort to improve vaccine efficacy (Tables 4, 5). In malaria-naïve adults, vaccine efficacy against challenge with CHMI was 86.7% (26/30) in individuals who received a delayed, low, fractional boost for the third vaccination, whereas the efficacy in individuals who received the standard regimen was only 62.5% (10/16) [107]. Individuals who received delayed, fractional dosing had a more balanced antibody response to the repeat and C-terminal regions of CSP, whereas the standard regimen induced a more focused response to the repeat region [108]. Fractional boosting also resulted in expanded C-terminal Fc-mediated effector functions and increased antibody avidity [107, 108]. In a further study, five delayed fractional dose vaccine regimens were tested in malaria-naïve adults using different vaccine doses and schedules for RTS,S/AS01B and RTS,S/AS01E [109]. Following challenge with CHMI, all three-dose vaccine regimens with fractional doses demonstrated efficacy against challenge with CHMI ranging from 55 to 76%. For the adult formulation, the efficacy of the three-dose regimen (55%) was clearly superior to a two-dose regimen (29%). In an extension of this study, a proportion of vaccinees received a further fourth fractional dose (one-fifth dose volume) of RTS,SAS01E 12 months after their third vaccine dose. Three weeks later they were re-challenged by CHMI; vaccine efficacy was comparable between those previously protected against CHMI (52%) and those who were not (54%). This demonstrates that fractional RTS,S/AS01E boosters given 12 months after the primary vaccine regimen can both extend and induce protection against CHMI [110]. An interim analysis from an ongoing phase IIb trial in 5- to 17-month-old infants in Africa comparing the efficacy of the standard vaccine regimen (three full doses) against a regimen with a fractional third dose showed that vaccine efficacy against clinical malaria was similar in both groups over 12 months of follow-up and that there was no difference in antibody avidity [111]. Furthermore, this efficacy was not improved by delaying the fractional dose by 5 months. This study is continuing and will examine the impact of annual boosters on the efficacy of the different vaccine regimens over 50 months of follow-up.
Efforts to improve the efficacy of RTS,S/AS01 have also focused on using a heterologous prime-boost strategy in an attempt to increase the CD4+ T-cell responses and augment the antibody response (Table 4). Priming with the full-length CS-expressing replication-deficient recombinant human adenovirus 35 (Ad35.CS.01) followed by two doses of RTS,S/AS01B did not improve vaccine efficacy against challenge with CHMI when compared with three doses of RTS,S/AS01B [112]. Combining three doses of RTS,S/AS01B with a dose each of the viral vectors ChAd63 and MVA expressing ME-TRAP yielded a vaccine regimen that was more effective against challenge with CHMI than three doses of RTS,S/AS01B alone (vaccine efficacy: 82.4 vs 75%) [113]. A further study evaluated different vaccine regimens of RTS,S/AS01B alone (with or without a third fractional dose) or with concomitant administration of ChAd63 and MVA expressing ME-TRAP [114]. When both RTS,S/AS01B and the viral vectors were administered at the same time, they were also administered at the same site. Interestingly, the highest vaccine efficacy against challenge with CHMI was observed in the group that received two full and one fractional dose of RTS,S/AS01B (vaccine efficacy: 88.9%). Co-administering the RTS,SAS01B with the ChAd63 and MVA ME-TRAP led to reduced immunogenicity and a reduced efficacy of 55.6%. This effect could be attributed to immune interference and indicates that alternative vaccine schedules and different sites may be needed to improve vaccine efficacy.
6.3.1.3 RTS,S and Seasonal Malaria Chemoprevention
As RTS,S/AS01 has been shown to provide incomplete protection of limited duration, a novel approach to malaria control could involve combining seasonal RTS,S/AS01E vaccination with chemoprevention in areas of high seasonal malaria transmission. A recent trial undertaken in children 5–17 months of age in Burkina Faso and Mali [115] demonstrated that in areas of high seasonal malaria transmission, a combination of the RTS,S/AS01E vaccine with seasonal malaria chemoprophylaxis (SMC) (sulfadoxine-pyrimethamine + amodiaquine) had greater efficacy against uncomplicated malaria, severe malaria, and death from malaria than either intervention alone over the 3-year study period. The vaccine was delivered as three doses over three consecutive months immediately prior to the transmission season and one booster dose/year in the following 2 years. The SMC was delivered as four courses at monthly intervals over the malaria transmission season each year. Such an approach may also be relevant within the context of intermittent preventive treatment in infants and pregnancy and mass drug administration during the final stages of a malaria eradication programme [116].
6.3.2 R21
Like RTS,S, R21 also contains the HBsAg fused to truncated CSP, which self-assemble into VLPs when expressed in yeast. Unlike RTS,S, the R21 particles are formed exclusively from CSP-HBsAg chimeric fusion proteins and do not contain monomeric HBsAg; this results in a higher density of the CSP on the surface of the VLPs [117]. Following pre-clinical evaluation of R21 in combination with different adjuvants [117], the vaccine was progressed into clinical trials with Matrix-M, a saponin-based adjuvant that simulates antibody and cellular immune responses. In a phase IIb trial, Burkinabe infants aged 5–17 months received three doses of R21 (5 µg)/Matrix-M (MM) (25 or 50 µg). Vaccine efficacy against clinical malaria was 77% in the high-dose adjuvant group at 6 and 12 months following the final vaccine dose [118]. The higher dose of MM was associated with an 86% increase in CSP NANP-repeat-specific antibodies after the third vaccine dose, compared with the lower dose of MM. One year after the primary three-dose regimen was completed, participants received a booster vaccination and were followed for a further 12 months. Over this period, vaccine efficacy against clinical malaria was 80% in the high-dose adjuvant group and titres of CSP NANP-repeat-specific antibodies correlated positively with protection in both years of follow-up [119].
Although larger studies at multiple sites in areas of differing malaria endemicities will be required to fully evaluate the efficacy of R21/MM, so far the results are promising. They meet the WHO-stated goal of a vaccine with >75% efficacy against P. falciparum clinical malaria over 12 months of follow-up in the target population but fall short of the WHO’s PPC for efficacy against clinical malaria. R21 is currently undergoing evaluation in a phase III trial.
7 Conclusion
There is no doubt that decades of malaria vaccine research have yielded some major outcomes over recent years. The decision to recommend and then implement RTS,S/AS01 vaccination in young children at high risk of P. falciparum infection is a significant moment in the history of malaria vaccine research and control. RTS,S/AS01 is also the first anti-parasite vaccine to be recommended for routine use in humans. Although it is an imperfect vaccine, it is hoped that it will make a significant contribution to reducing malaria-related morbidity in areas of high endemicity when used in combination with other control measures such as SMC and insecticide-treated bed nets. Research continues with the aim of developing a more effective malaria vaccine. This could involve further optimising RTS,S/AS01 through alternative regimens as mentioned above or by including additional CSP alleles to address the limitations imposed on vaccine efficacy by the strain-specific nature of the protective immune response. It could also involve developing next-generation CSP-based vaccines such as R21, which has so far demonstrated a high level of efficacy against clinical malaria in a single phase IIb study in Africa, although this vaccine may also be impacted by the genetic diversity of the CSP and may need to include other variants. An additional sub-unit vaccine approach would be to use a partially effective pre-erythrocytic-stage vaccine like RTS,S/AS01 in combination with vaccine candidates that target the blood-stage of the parasite (asexual stage and transmission blocking vaccine candidates) to prevent the development of a blood-stage infection and transmission of the parasites to the mosquito vector. Whole-parasite vaccine approaches such as the PfSPZ Vaccine and PfSPZ-CVac have now been evaluated in the field and while further optimisation is required to maximise the protective efficacy of these vaccine candidates, the results are encouraging, and the trials are a significant milestone in the development of whole-parasite malaria vaccines. Placental malaria vaccines are urgently needed to prevent the severe outcomes associated with pregnancy-associated malaria for both mother and offspring. Further optimisation of the placental malaria vaccine candidates PRIMVAC and PAMVAC could improve their activity against heterologous parasite strains and recent structural studies have suggested how this may be achieved [120]. We should also not forget the need for a P. vivax vaccine, which is a more challenging proposition due to its latent hypnozoite form in the liver.
The methodologies, knowledge and infrastructure acquired throughout the many malaria vaccine trials that have been conducted so far will benefit and inform the development of the next generation of malaria vaccine candidates. While there is reason to be optimistic about the possibility of malaria vaccines playing a crucial role in reducing malaria-attributable disease and death and ultimately contributing to malaria eradication, it is important that we do not lose the momentum that we have built over decades of vaccine research and development.
References
World Health Organisation. World malaria report 2021. World Health Organisation, Editor. 2022.
Doolan D, Dobano C, Baird J. Acquired immunity to malaria. Clin Microbiol Rev. 2009;22(1):13–36.
Ranson H, Lissenden N. Insecticide resistance in African anopheles mosquitoes: a worsening situation that needs urgent action to maintain malaria control. Trends Parasitol. 2016;32(3):187–96.
Blasco B, Leroy D, Fidock DA. Antimalarial drug resistance: linking Plasmodium falciparum parasite biology to the clinic. Nat Med. 2017;23(8):917–28.
Phyo AP, et al. Declining efficacy of artemisinin combination therapy against P. falciparum malaria on the Thai-Myanmar border (2003–2013): the role of parasite genetic factors. Clin Infect Dis. 2016;63(6):784–91.
van der Pluijm RW, et al. Determinants of dihydroartemisinin-piperaquine treatment failure in Plasmodium falciparum malaria in Cambodia, Thailand, and Vietnam: a prospective clinical, pharmacological, and genetic study. Lancet Infect Dis. 2019;19(9):952–61.
Balikagala B, et al. Evidence of artemisinin-resistant malaria in Africa. N Engl J Med. 2021;385(13):1163–71.
Stanisic DI, Barry AE, Good MF. Escaping the immune system: How the malaria parasite makes vaccine development a challenge. Trends Parasitol. 2013;29(12):612–22.
Malaria Vaccine Funders Group. Malaria vaccine technology roadmap. 2013.
Malaria vaccines: preferred product characteristics and clinical development considerations. Geneva: World Health Organization; 2022. Licence: CC BY-NC-SA 3.0 IGO.
Bijker EM, et al. Protection against malaria after immunization by chloroquine prophylaxis and sporozoites is mediated by preerythrocytic immunity. Proc Natl Acad Sci USA. 2013;110(19):7862–7.
Barry AE, et al. Contrasting population structures of the genes encoding ten leading vaccine-candidate antigens of the human malaria parasite, Plasmodium falciparum. PLoS ONE. 2009;4(12): e8497.
Neafsey DE, et al. Genetic diversity and protective efficacy of the RTS, S/AS01 malaria vaccine. N Engl J Med. 2015;373(21):2025–37.
Genton B, et al. A recombinant blood-stage malaria vaccine reduces Plasmodium falciparum density and exerts selective pressure on parasite populations in a phase 1–2b trial in Papua New Guinea. J Infect Dis. 2002;185(6):820–7.
World Health Organisation. Malaria vaccines: WHO Position Paper—March 2022. 2022.
Stanisic DI, McCarthy JS, Good MF. Controlled human malaria infection: applications, advances, and challenges. Infect Immun. 2018;86(1):1–17.
Murphy SC, et al. PfSPZ-CVac efficacy against malaria increases from 0% to 75% when administered in the absence of erythrocyte stage parasitemia: a randomized, placebo-controlled trial with controlled human malaria infection. PLoS Pathog. 2021;17(5): e1009594.
Studniberg SI, et al. Molecular profiling reveals features of clinical immunity and immunosuppression in asymptomatic P falciparum malaria. Mol Syst Biol. 2022;18(4):e10824.
Stanisic DI, Good MF. Whole parasite blood stage vaccines. In: Kremsner PG, Krishna S, editors. Encyclopedia of malaria. New York: Springer, New York; 2018.
Stanisic DI, et al. Vaccination with chemically attenuated Plasmodium falciparum asexual blood-stage parasites induces parasite-specific cellular immune responses in malaria-naive volunteers: a pilot study. BMC Med. 2018;16(1):184.
Good MF, et al. Cross-species malaria immunity induced by chemically attenuated parasites. J Clin Invest. 2013;123:3353–62.
Webster R, et al. Safety, infectivity and immunogenicity of a genetically attenuated blood-stage malaria vaccine. BMC Med. 2021;19(1):293.
Ogutu BR, et al. Blood stage malaria vaccine eliciting high antigen-specific antibody concentrations confers no protection to young children in Western Kenya. PLoS ONE. 2009;4(3): e4708.
Sagara I, et al. A randomized controlled phase 2 trial of the blood stage AMA1-C1/Alhydrogel malaria vaccine in children in Mali. Vaccine. 2009;27(23):3090–8.
Thera MA, et al. A field trial to assess a blood-stage malaria vaccine. N Engl J Med. 2011;365(11):1004–13.
Payne RO, et al. Demonstration of the blood-stage Plasmodium falciparum controlled human malaria infection model to assess efficacy of the P. falciparum apical membrane antigen 1 vaccine, FMP2.1/AS01. J Infect Dis. 2016;213(11):1743–51.
Sirima SB, Cousens S, Druilhe P. Protection against malaria by MSP3 candidate vaccine. N Engl J Med. 2011;365(11):1062–4.
Palacpac NM, et al. Phase 1b randomized trial and follow-up study in Uganda of the blood-stage malaria vaccine candidate BK-SE36. PLoS ONE. 2013;8(5): e64073.
Minassian AM, et al. Reduced blood-stage malaria growth and immune correlates in humans following RH5 vaccination. Med (NY). 2021;2(6):701-719 e19.
Sirima SB, et al. A phase 2b randomized, controlled trial of the efficacy of the GMZ2 malaria vaccine in African children. Vaccine. 2016;34(38):4536–42.
Dejon-Agobe JC, et al. Controlled human malaria infection of healthy adults with lifelong malaria exposure to assess safety, immunogenicity, and efficacy of the asexual blood stage malaria vaccine candidate GMZ2. Clin Infect Dis. 2019;69(8):1377–84.
O’Neil-Dunne I, et al. Gravidity-dependent production of antibodies that inhibit binding of Plasmodium falciparum-infected erythrocytes to placental chondroitin sulfate proteoglycan during pregnancy. Infect Immun. 2001;69(12):7487–92.
Fried M, et al. Maternal antibodies block malaria. Nature. 1998;395(6705):851–2.
Fried M, Duffy PE. Adherence of Plasmodium falciparum to chondroitin sulfate A in the human placenta. Science. 1996;272(5267):1502–4.
Mordmuller B, et al. First-in-human, randomized, double-blind clinical trial of differentially adjuvanted PAMVAC, a vaccine candidate to prevent pregnancy-associated malaria. Clin Infect Dis. 2019;69(9):1509–16.
Sirima SB, et al. PRIMVAC vaccine adjuvanted with Alhydrogel or GLA-SE to prevent placental malaria: a first-in-human, randomised, double-blind, placebo-controlled study. Lancet Infect Dis. 2020;20(5):585–97.
Chichester JA, et al. Safety and immunogenicity of a plant-produced Pfs25 virus-like particle as a transmission blocking vaccine against malaria: a Phase 1 dose-escalation study in healthy adults. Vaccine. 2018;36(39):5865–71.
Talaat KR, et al. Safety and immunogenicity of Pfs25-EPA/Alhydrogel(R), a transmission blocking vaccine against Plasmodium falciparum: an open label study in malaria naive adults. PLoS ONE. 2016;11(10): e0163144.
Sagara I, et al. Safety and immunogenicity of Pfs25H-EPA/Alhydrogel, a transmission-blocking vaccine against Plasmodium falciparum: a randomised, double-blind, comparator-controlled, dose-escalation study in healthy Malian adults. Lancet Infect Dis. 2018;18(9):969–82.
Healy SA, et al. Pfs230 yields higher malaria transmission-blocking vaccine activity than Pfs25 in humans but not mice. J Clin Invest. 2021;131(7): e146221.
Clyde DF, et al. Immunization of man against sporozite-induced falciparum malaria. Am J Med Sci. 1973;266(3):169–77.
Clyde DF, et al. Specificity of protection of man immunized against sporozoite-induced falciparum malaria. Am J Med Sci. 1973;266(6):398–403.
Hoffman SL, et al. Protection of humans against malaria by immunization with radiation-attenuated Plasmodium falciparum sporozoites. J Infect Dis. 2002;185(8):1155–64.
Nussenzweig RS, et al. Protective immunity produced by the injection of x-irradiated sporozoites of plasmodium berghei. Nature. 1967;216(5111):160–2.
Hoffman SL, et al. Development of a metabolically active, non-replicating sporozoite vaccine to prevent Plasmodium falciparum malaria. Hum Vaccin. 2010;6(1):97–106.
Epstein JE, et al. Live attenuated malaria vaccine designed to protect through hepatic CD8+ T cell immunity. Science. 2011;334(6055):475–80.
Seder RA, et al. Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science. 2013;341(6152):1359–65.
Sissoko MS, et al. Safety and efficacy of PfSPZ vaccine against Plasmodium falciparum via direct venous inoculation in healthy malaria-exposed adults in Mali: a randomised, double-blind phase 1 trial. Lancet Infect Dis. 2017;17(5):498–509.
Sissoko MS, et al. Safety and efficacy of a three-dose regimen of Plasmodium falciparum sporozoite vaccine in adults during an intense malaria transmission season in Mali: a randomised, controlled phase 1 trial. Lancet Infect Dis. 2022;22(3):377–89.
Oneko M, et al. Safety, immunogenicity and efficacy of PfSPZ Vaccine against malaria in infants in western Kenya: a double-blind, randomized, placebo-controlled phase 2 trial. Nat Med. 2021;27(9):1636–45.
Mordmuller B, et al. A PfSPZ vaccine immunization regimen equally protective against homologous and heterologous controlled human malaria infection. NPJ Vaccines. 2022;7(1):100.
Jongo SA, et al. Multi-dose priming regimens of PfSPZ Vaccine: safety and efficacy against controlled human malaria infection in Equatoguinean adults. Am J Trop Med Hyg. 2022;106(4):1215–26.
Nevagi RJ, Good MF, Stanisic DI. Plasmodium infection and drug cure for malaria vaccine development. Expert Rev Vaccines. 2021;20(2):163–83.
Roestenberg M, et al. Protection against a malaria challenge by sporozoite inoculation. N Engl J Med. 2009;361(5):468–77.
Mwakingwe-Omari A, et al. Two chemoattenuated PfSPZ malaria vaccines induce sterile hepatic immunity. Nature. 2021;595(7866):289–94.
Jongo SA, et al. Immunogenicity and protective efficacy of radiation-attenuated and chemo-attenuated PfSPZ vaccines in equatoguinean adults. Am J Trop Med Hyg. 2021;104(1):283–93.
Coulibaly D, et al. PfSPZ-CVac malaria vaccine demonstrates safety among malaria-experienced adults: a randomized, controlled phase 1 trial. EClinicalMedicine. 2022;52: 101579.
Sulyok Z, et al. Heterologous protection against malaria by a simple chemoattenuated PfSPZ vaccine regimen in a randomized trial. Nat Commun. 2021;12(1):2518.
Murphy SC, et al. A genetically engineered Plasmodium falciparum parasite vaccine provides protection from controlled human malaria infection. Sci Transl Med. 2022;14(659):eabn9709.
Roestenberg M, et al. A double-blind, placebo-controlled phase 1/2a trial of the genetically attenuated malaria vaccine PfSPZ-GA1. Sci Transl Med. 2020;12(544):eaaz5629.
Butler NS, et al. Superior antimalarial immunity after vaccination with late liver stage-arresting genetically attenuated parasites. Cell Host Microbe. 2011;9(6):451–62.
Goswami D, et al. A replication-competent late liver stage-attenuated human malaria parasite. JCI Insight. 2020;5(13): e135589.
Franke-Fayard B, et al. Creation and preclinical evaluation of genetically attenuated malaria parasites arresting growth late in the liver. NPJ Vaccines. 2022;7(1):139.
Eappen AG, et al. In vitro production of infectious Plasmodium falciparum sporozoites. Nature. 2022;612(7940):534–9.
Ogwang C, et al. Prime-boost vaccination with chimpanzee adenovirus and modified vaccinia Ankara encoding TRAP provides partial protection against Plasmodium falciparum infection in Kenyan adults. Sci Transl Med. 2015;7(286):286re5.
Mensah VA, et al. Safety, immunogenicity and efficacy of prime-boost vaccination with ChAd63 and MVA encoding ME-TRAP against Plasmodium falciparum infection in adults in Senegal. PLoS ONE. 2016;11(12): e0167951.
Tiono AB, et al. First field efficacy trial of the ChAd63 MVA ME-TRAP vectored malaria vaccine candidate in 5–17 months old infants and children. PLoS ONE. 2018;13(12): e0208328.
Kemp DJ, et al. Expression of Plasmodium falciparum blood-stage antigens in Escherichia coli: detection with antibodies from immune humans. Proc Natl Acad Sci USA. 1983;80(12):3787–91.
Ellis J, et al. Cloning and expression in E. coli of the malarial sporozoite surface antigen gene from Plasmodium knowlesi. Nature. 1983;302(5908):536–8.
Enea V, et al. DNA cloning of Plasmodium falciparum circumsporozoite gene: amino acid sequence of repetitive epitope. Science. 1984;225(4662):628–30.
Friedman-Klabanoff DJ, et al. Low dose recombinant full-length circumsporozoite protein-based Plasmodium falciparum vaccine is well-tolerated and highly immunogenic in phase 1 first-in-human clinical testing. Vaccine. 2021;39(8):1195–200.
Chuang I, et al. DNA prime/Adenovirus boost malaria vaccine encoding P. falciparum CSP and AMA1 induces sterile protection associated with cell-mediated immunity. PLoS ONE. 2013;8(2): e55571.
Gordon DM, et al. Safety, immunogenicity, and efficacy of a recombinantly produced Plasmodium falciparum circumsporozoite protein-hepatitis B surface antigen subunit vaccine. J Infect Dis. 1995;171(6):1576–85.
Stoute JA, et al. A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. RTS, S Malaria Vaccine Evaluation Group. N Engl J Med. 1997;336(2):86–91.
Kester KE, et al. Efficacy of recombinant circumsporozoite protein vaccine regimens against experimental Plasmodium falciparum malaria. J Infect Dis. 2001;183(4):640–7.
Kester KE, et al. Phase 2a trial of 0, 1, and 3 month and 0, 7, and 28 day immunization schedules of malaria vaccine RTS, S/AS02 in malaria-naive adults at the Walter Reed Army Institute of Research. Vaccine. 2008;26(18):2191–202.
Kester KE, et al. A phase I/IIa safety, immunogenicity, and efficacy bridging randomized study of a two-dose regimen of liquid and lyophilized formulations of the candidate malaria vaccine RTS, S/AS02A in malaria-naive adults. Vaccine. 2007;25(29):5359–66.
Epstein JE, et al. Safety, tolerability, and antibody responses in humans after sequential immunization with a PfCSP DNA vaccine followed by the recombinant protein vaccine RTS, S/AS02A. Vaccine. 2004;22(13–14):1592–603.
Wang R, et al. Induction in humans of CD8+ and CD4+ T cell and antibody responses by sequential immunization with malaria DNA and recombinant protein. J Immunol. 2004;172(9):5561–9.
Dunachie SJ, et al. A clinical trial of prime-boost immunisation with the candidate malaria vaccines RTS, S/AS02A and MVA-CS. Vaccine. 2006;24(15):2850–9.
Bojang KA, et al. Efficacy of RTS, S/AS02 malaria vaccine against Plasmodium falciparum infection in semi-immune adult men in The Gambia: a randomised trial. Lancet. 2001;358(9297):1927–34.
Bojang KA, et al. Safety and immunogenicty of RTS, S/AS02A candidate malaria vaccine in Gambian children. Vaccine. 2005;23(32):4148–57.
Alonso PL, et al. Efficacy of the RTS, S/AS02A vaccine against Plasmodium falciparum infection and disease in young African children: randomised controlled trial. Lancet. 2004;364(9443):1411–20.
Alonso P, et al. Duration of protection with RTS, S/AS02A malaria vaccine in prevention of Plasmodium falciparum disease in Mozambican children: single-blind extended follow-up of a randomised control trial. Lancet. 2005;366:2012–8.
Sacarlal J, et al. Long-term safety and efficacy of the RTS, S/AS02A malaria vaccine in Mozambican children. J Infect Dis. 2009;200(3):329–36.
Guinovart C, et al. Insights into long-lasting protection induced by RTS, S/AS02A malaria vaccine: further results from a phase IIb trial in Mozambican children. PLoS ONE. 2009;4(4): e5165.
Macete EV, et al. Evaluation of two formulations of adjuvanted RTS, S malaria vaccine in children aged 3 to 5 years living in a malaria-endemic region of Mozambique: a Phase I/IIb randomized double-blind bridging trial. Trials. 2007;8:11.
Aponte JJ, et al. Safety of the RTS, S/AS02D candidate malaria vaccine in infants living in a highly endemic area of Mozambique: a double blind randomised controlled phase I/IIb trial. Lancet. 2007;370(9598):1543–51.
Aide P, et al. Safety, immunogenicity and duration of protection of the RTS, S/AS02(D) malaria vaccine: one year follow-up of a randomized controlled phase I/IIb trial. PLoS ONE. 2010;5(11): e13838.
Abdulla S, et al. Safety and immunogenicity of RTS, S/AS02D malaria vaccine in infants. N Engl J Med. 2008;359(24):2533–44.
Kester KE, et al. Randomized, double-blind, phase 2a trial of falciparum malaria vaccines RTS, S/AS01B and RTS, S/AS02A in malaria-naive adults: safety, efficacy, and immunologic associates of protection. J Infect Dis. 2009;200(3):337–46.
Polhemus ME, et al. Evaluation of RTS, S/AS02A and RTS, S/AS01B in adults in a high malaria transmission area. PLoS ONE. 2009;4(7): e6465.
Bejon P, et al. Efficacy of RTS, S/AS01E vaccine against malaria in children 5 to 17 months of age. N Engl J Med. 2008;359(24):2521–32.
Olotu A, et al. Efficacy of RTS, S/AS01E malaria vaccine and exploratory analysis on anti-circumsporozoite antibody titres and protection in children aged 5–17 months in Kenya and Tanzania: a randomised controlled trial. Lancet Infect Dis. 2011;11(2):102–9.
Olotu A, et al. Four-year efficacy of RTS, S/AS01E and its interaction with malaria exposure. N Engl J Med. 2013;368(12):1111–20.
Olotu A, et al. Seven-year efficacy of RTS, S/AS01 malaria vaccine among young African children. N Engl J Med. 2016;374(26):2519–29.
Asante KP, et al. Safety and efficacy of the RTS, S/AS01(E) candidate malaria vaccine given with expanded-programme-on-immunisation vaccines: 19 month follow-up of a randomised, open-label, phase 2 trial. Lancet Infect Dis. 2011;11(10):741–9.
Bejon P, et al. Efficacy of RTS, S malaria vaccines: individual-participant pooled analysis of phase 2 data. Lancet Infect Dis. 2013;13(4):319–27.
Rts SCTP, et al. First results of phase 3 trial of RTS, S/AS01 malaria vaccine in African children. N Engl J Med. 2011;365(20):1863–75.
Rts SCTP, et al. A phase 3 trial of RTS, S/AS01 malaria vaccine in African infants. N Engl J Med. 2012;367(24):2284–95.
Rts SCTP. Efficacy and safety of the RTS, S/AS01 malaria vaccine during 18 months after vaccination: a phase 3 randomized, controlled trial in children and young infants at 11 African sites. PLoS Med. 2014;11(7): e1001685.
Rts SCTP. Efficacy and safety of RTS, S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet. 2015;386(9988):31–45.
White MT, et al. Immunogenicity of the RTS, S/AS01 malaria vaccine and implications for duration of vaccine efficacy: secondary analysis of data from a phase 3 randomised controlled trial. Lancet Infect Dis. 2015;15(12):1450–8.
Feng G, et al. Induction, decay, and determinants of functional antibodies following vaccination with the RTS, S malaria vaccine in young children. BMC Med. 2022;20(1):289.
Kurtovic L, et al. Induction and decay of functional complement-fixing antibodies by the RTS, S malaria vaccine in children, and a negative impact of malaria exposure. BMC Med. 2019;17(1):45.
Europtean Medicines Agency, CHMP. Assessment report Mosquirix. 2015.
Regules JA, et al. Fractional third and fourth dose of RTS, S/AS01 malaria candidate vaccine: a Phase 2a controlled human malaria parasite infection and immunogenicity study. J Infect Dis. 2016;214(5):762–71.
Das J, et al. Delayed fractional dosing with RTS, S/AS01 improves humoral immunity to malaria via a balance of polyfunctional NANP6- and Pf16-specific antibodies. Med (NY). 2021;2(11):1269-1286 e9.
Moon JE, et al. A phase IIa controlled human malaria infection and immunogenicity study of RTS, S/AS01E and RTS, S/AS01B delayed fractional dose regimens in malaria-naive adults. J Infect Dis. 2020;222(10):1681–91.
Moon JE, et al. A phase IIA extension study evaluating the effect of booster vaccination with a fractional dose of RTS, S/AS01E in a controlled human malaria infection challenge. Vaccine. 2021;39(43):6398–406.
Samuels AM, et al. Efficacy of RTS, S/AS01(E) malaria vaccine administered according to different full, fractional, and delayed third or early fourth dose regimens in children aged 5–17 months in Ghana and Kenya: an open-label, phase 2b, randomised controlled trial. Lancet Infect Dis. 2022;22(9):1329–42.
Ockenhouse CF, et al. Ad35.CS.01-RTS, S/AS01 heterologous prime boost vaccine efficacy against sporozoite challenge in healthy malaria-naive adults. PLoS ONE. 2015;10(7):e0131571.
Rampling T, et al. Safety and high level efficacy of the combination malaria vaccine regimen of RTS, S/as01b with chimpanzee adenovirus 63 and modified vaccinia ankara vectored vaccines expressing ME-TRAP. J Infect Dis. 2016;214(5):772–81.
Rampling T, et al. Safety and efficacy of novel malaria vaccine regimens of RTS, S/AS01B alone, or with concomitant ChAd63-MVA-vectored vaccines expressing ME-TRAP. NPJ Vaccines. 2018;3:49.
Chandramohan D, et al. Seasonal malaria vaccination with or without seasonal malaria chemoprevention. N Engl J Med. 2021;385(11):1005–17.
Greenwood B, et al. Combining malaria vaccination with chemoprevention: a promising new approach to malaria control. Malar J. 2021;20(1):361.
Collins KA, et al. Enhancing protective immunity to malaria with a highly immunogenic virus-like particle vaccine. Sci Rep. 2017;7:46621.
Datoo MS, et al. Efficacy of a low-dose candidate malaria vaccine, R21 in adjuvant Matrix-M, with seasonal administration to children in Burkina Faso: a randomised controlled trial. Lancet. 2021;397(10287):1809–18.
Datoo MS, et al. Efficacy and immunogenicity of R21/Matrix-M vaccine against clinical malaria after 2 years’ follow-up in children in Burkina Faso: a phase 1/2b randomised controlled trial. Lancet Infect Dis. 2022;22(12):1728–36.
Ma R, et al. Structural basis for placental malaria mediated by Plasmodium falciparum VAR2CSA. Nat Microbiol. 2021;6(3):380–91.
Lyke KE, et al. Attenuated PfSPZ Vaccine induces strain-transcending T cells and durable protection against heterologous controlled human malaria infection. Proc Natl Acad Sci USA. 2017;114(10):2711–6.
Ishizuka AS, et al. Protection against malaria at 1 year and immune correlates following PfSPZ vaccination. Nat Med. 2016;22(6):614–23.
Epstein JE, et al. Protection against Plasmodium falciparum malaria by PfSPZ Vaccine. JCI Insight. 2017;2(1): e89154.
Jongo SA, et al. Safety, immunogenicity, and protective efficacy against controlled human malaria infection of Plasmodium falciparum sporozoite vaccine in Tanzanian adults. Am J Trop Med Hyg. 2018;99(2):338–49.
Jongo SA, et al. Increase of dose associated with decrease in protection against controlled human malaria infection by PfSPZ Vaccine in Tanzanian adults. Clin Infect Dis. 2020;71(11):2849–57.
Lyke KE, et al. Multidose priming and delayed boosting improve Plasmodium falciparum sporozoite vaccine efficacy against heterologous P. falciparum controlled human malaria infection. Clin Infect Dis. 2021;73(7):e2424–35.
Bastiaens GJH, et al. Safety, immunogenicity, and protective efficacy of intradermal immunization with aseptic, purified, cryopreserved Plasmodium falciparum sporozoites in volunteers under chloroquine prophylaxis: a randomized controlled trial. Am J Trop Med Hyg. 2016;94(3):663–73.
Mordmüller B, et al. Sterile protection against human malaria by chemoattenuated PfSPZ vaccine. Nature. 2017;542(7642):445–9.
Sirima SB, et al. A randomized controlled trial showing safety and efficacy of a whole sporozoite vaccine against endemic malaria. Sci Transl Med. 2022;14(674):eabj3776.
Stoute J, et al. Long-term efficacy and immune responses following immunization with the RTS, S malaria vaccine. J Infect Dis. 1998;178:1139–44.
Kester KE, et al. Sequential Phase 1 and Phase 2 randomized, controlled trials of the safety, immunogenicity and efficacy of combined pre-erythrocytic vaccine antigens RTS, S and TRAP formulated with AS02 Adjuvant System in healthy, malaria naive adults. Vaccine. 2014;32(49):6683–91.
Abdulla S, et al. Randomized, controlled trial of the long term safety, immunogenicity and efficacy of RTS, S/AS02(D) malaria vaccine in infants living in a malaria-endemic region. Malar J. 2013;12:11.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. The authors acknowledge grant support from a National Health and Medical Research Council Australia Investigator Grant and Program Grant.
Conflicts of interest
The authors are inventors on patents relating to whole-parasite blood-stage malaria vaccines.
Ethics approval
Not applicable.
Patient consent to participate/publish
Not applicable.
Availability of data and material
All data are derived from published journal articles.
Code availability
Not applicable.
Author contributions
Both authors contributed to the conception, design and writing of the manuscript. Both authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Stanisic, D.I., Good, M.F. Malaria Vaccines: Progress to Date. BioDrugs 37, 737–756 (2023). https://doi.org/10.1007/s40259-023-00623-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-023-00623-4