
Abstract
Numerous toxins translocate to the cytosol in order to fulfil their function. This demonstrates the existence of routes for proteins from the extracellular space to the cytosol. Understanding these routes is relevant to multiple aspects related to therapeutic applications. These include the development of anti-toxin treatments, the potential use of toxins as shuttles for delivering macromolecular cargo to the cytosol or the use of drugs based on toxins. Compared with other strategies for delivery, such as chemicals as carriers for macromolecular delivery or physical methods like electroporation, toxin routes present paths into the cell that potentially cause less damage and can be specifically targeted. The efficiency of delivery via toxin routes is limited. However, low-delivery efficiencies can be entirely sufficient, if delivered cargoes possess an amplification effect or if very few molecules are sufficient for inducing the desired effects. This is known for example from RNA-based vaccines that have been developed during the coronavirus disease 2019 pandemic as well as for other approved RNA-based drugs, which elicited the desired effect despite their typically low delivery efficiencies. The different mechanisms by which toxins enter cells may have implications for their technological utility. We review the mechanistic principles of the translocation pathway of toxins from the extracellular space to the cytosol, the delivery efficiencies, and therapeutic strategies or applications that exploit toxin routes for intracellular delivery.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Intracellular delivery of macromolecules for therapy is promising and at the same time challenging, but toxins from plants or bacteria are able to specifically translocate their toxic components to the cytosol. |
Translocation routes used by toxins are of interest for delivery of macromolecules to the cytosol by toxin-based vehicles, for anti-toxin treatments, and for therapies directly using toxins as components of a drug. |
The exploitation of toxins and the translocation routes they employ for reaching their site of action (in short called “toxin routes” here) has contributed to the development of therapeutic strategies in the fields of cancer therapy, antivirals, bacterial infections with toxic effects and intoxications, protein misfolding diseases such as cystic fibrosis, and for extending the space that can be reached by protein drugs to the cytosol. |
1 Introduction
The number of US-approved (Food and Drug Administration-approved) proteinaceous drugs is constantly increasing [1]. Because membranes present barriers to macromolecules, the extracellular space is most easily accessible to biologics. The delivery of macromolecules to the cytosol would have enormous potential for many applications, but the implementation is challenging.
In particular, the delivery of proteins into cells offers specific opportunities, such as targeting different protein conformations, post-translational modifications, individual splice variants or a whole set of splice variants at once and different functional sites on the same protein, causing effects only in selected compartments while leaving other compartments unaffected and inducing effects on proteins independently of their half-life [2]. The intracellular delivery of antibodies as proteins would offer these opportunities in particular. Antibodies inside cells have so far mostly been used in the form of expressed binders, i.e. intracellular antibodies (intrabodies) expressed from nucleic acids [3, 4].
Many intracellular delivery methods for macromolecules are associated with some degree of membrane damage, for example, many chemicals that are used as carriers for macromolecule delivery or electroporation as a delivery method [5, 6]. Delivery of macromolecules in a way that does not result in damage to membranes might be less toxic for the cells. Toxins that need to reach the cytosol to fulfil their function demonstrate a natural path for proteins from the extracellular space to the cytosol. Depending on the toxin route, damaging the membrane may not be required for delivery to the cytosol.
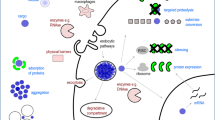
We discuss the opportunities and limitations for protein delivery to the cytosol via toxin routes, the properties that cargo molecules need to possess for successful delivery and therapeutic applications that have emerged from knowledge about toxin routes. The different mechanisms by which different toxins enter cells have implications for applications. Therefore, mechanistic principles are reviewed in addition to the efficiency of the delivery and to applications related to toxin routes. Types of toxin routes, barriers to the delivery via toxin routes and applications related to toxin routes are summarised in Fig. 1.

Toxin routes and their relevance for therapeutic strategies (blue and turquoise) and barriers to delivery via toxin routes (red). Delivery may occur directly from endosomes (B) or from the endoplasmic reticulum (ER) to the cytosol by hijacking the ER-associated degradation pathway [ERAD] (A). Barriers to the delivery of toxic or non-toxic cargoes can be due to immunogenicity of the toxin-based molecules and the elimination of toxins by an immune reaction. Cargo delivery can also be reduced by routing to lysosomes, by exocytosis or by degradation by the proteasome (C). Applications (D): toxin routes can be inhibited by antibodies or small molecules at various levels including cell-binding and intracellular transport, which can be applied for anti-toxin treatments (D). Toxins have been delivered to kill cells for therapeutic purposes and also non-toxic cargoes have been delivered via toxin routes (D). (E) summarises toxin routes, barriers to delivery via toxin routes (red) and applications related to toxin routes (blue and turquoise)
2 Toxins Acting Inside the Cell: Mechanistic Principles
Toxins that need to reach the cytosol to have an effect demonstrate how macromolecules can travel from the extracellular space to the cytosol. A detailed understanding of the mechanism by which these toxins reach their site of action is beneficial for judging the therapeutic potential of toxins as delivery vehicles for biological drugs, toxins as part of drugs or of anti-toxin treatments. Detailed knowledge about the mechanism of toxin entry into cells also helps with identifying which cargo molecules are suitable for delivery via toxin routes. In the following, they are separated into two categories, namely “based on translocation via the ER-associated degradation pathway (ERAD)” or “other mechanisms”.
2.1 Mechanistic Principles: Toxins That Exploit ERAD
Many toxins belong to the group of AB toxins, which consist of a component that binds to cells (“B” for binding to cells) and an enzymatic component that exerts the actual toxicity (“A” for active) [7]. If the target of the catalytic part of a toxin is localised in the cytosol, the active part of the toxin has to reach this compartment. One way to reach the cytosol involves hijacking components of the ERAD pathway. This pathway normally has the function of translocating misfolded proteins from the endoplasmic reticulum (ER) to the cytosol to allow their degradation by the proteasome [8,9,10,11,12]. Retrotranslocation from the ER to the cytosol may require the protein to unfold. Translocated proteins can subsequently be degraded in the cytosol by the proteasome [9, 13]. Pseudomonas exotoxin from Pseudomonas aeruginosa [14, 15], cholera toxin from Vibriae cholera [8, 14, 16], Shiga toxin from Shigella dysenteriae or Shiga-like toxin-producing bacteria (STEC) [10, 17,18,19] and ricin from the castor bean plant (Ricinus communis) [11, 14, 20] may exploit the ERAD pathway.
2.1.1 The Uptake
The first step for toxins in following this pathway is the uptake of the toxin by the cell. Interestingly, the binding affinity of the toxin to its cellular receptor is not always most decisive for toxicity, as has been found for the Shiga toxin. The intracellular part of the transport might play an important role as well [19, 21, 22]. Although a Shiga toxin 1 (Stx-1) had a higher affinity to its receptor than a Shiga toxin 2 (Stx-2) [22], Stx-2 was more toxic in mice compared with a Stx-1 [21]. Many toxins bind to carbohydrates of cellular receptors (see Table 1).
2.1.2 From Endosomes to Golgi
Endocytosed molecules can move to the lysosome and are degraded there. Toxins can avoid the route towards degradation by being transported from endosomes to the Golgi. Multiple routes lead from endosomes to the Golgi, including transport from early endosomes to the Golgi and transport from late endosomes to the Golgi. Influencing the route a toxin takes and rerouting toxins from an endosome-to-Golgi path to an endosome-to-lysosome path has been suggested as a potential anti-toxin strategy [23,24,25,26].
2.1.3 From Golgi to ER
Transport from the Golgi to the ER can follow multiple paths. Some toxins contain an ER retrieval sequence (ER retrieval sequence KDEL: amino acids Lys-Asp-Glu-Leu), like Pseudomonas exotoxin or cholera toxin [27,28,29,30]. Other toxins, like Shiga toxin or ricin, do not have a KDEL signal [30,31,32,33]. Transport to the ER can depend on the KDEL receptor or it can be independent from it. For example, cytotoxicity of Pseudomonas exotoxin A was reduced in cells that overexpressed lysozyme-KDEL, but overexpression of lysozyme-KDEL did not affect the cytotoxicity of diphtheria toxin or Shiga-like toxin I. This indicates the transport of Pseudomonas exotoxin A from the Golgi to the cytosol by means of the KDEL-retrieval system, while diphtheria toxin and Shiga-like toxin I were transported independently of it [34]. A Rab6-dependent path from the Golgi to the ER might be a KDEL-independent path used by Golgi enzymes and toxins [19, 30, 35]. Even for toxins that possess a KDEL motif, like cholera toxin, the KDEL motif accelerated the toxin’s effect, but was not essential for cytotoxicity [36].
2.1.4 From ER to Cytosol
The A1 fragment of Shiga toxin forms by separation from the A2 fragment. This occurs after cleavage by furin in endosomes and in the trans-Golgi network (TGN) and after disulphide reduction in the ER [19, 37, 38]. The Shiga toxin fragment A1 interacts with chaperones in the ER that are known to be involved in the process of retrotranslocation of proteins to the cytosol. Translocation to the cytosol may be mediated by Sec61 [19, 39]. A peptide domain at the C-terminus of the A1 fragment of Shiga toxin was found to be required for retrotranslocation. The mechanism induced by this peptide domain was assumed to include hydrophobic properties or misfolding [17]. Many toxins may be able to enter ERAD based on hydrophobic properties [40]. There is also evidence for ricin reaching the cytosol by using the ERAD pathway via the translocon Sec61 [14, 20, 41]. For cholera toxin, the factors that have been suggested to be involved in its retrotranslocation include gp78, Der1, Hrd1 and Sec61 [8, 42,43,44]. Pertussis toxin was previously thought to mimic a misfolded protein by a hydrophobic C-terminus, but the hydrophobic C-terminus was not required for translocation to the cytosol. The Pertussis toxin S1 subunit (PTS1) was eventually suggested to not mimic, but to actually be a misfolded protein because of its thermal instability [12, 45, 46]. Once in the ER, the intrinsic instability of PTS1 and its resulting “misfolding” make it a substrate for ERAD [12, 46, 47]. With ERAD-defective cell lines, the role of ERAD in allowing pertussis toxin to be toxic has been confirmed [12, 47].
Cytosolic factors such as chaperones might help with a one-directional translocation through the membrane channel into the cytosol [12, 48, 49]. For example, the A1 unit of cholera toxin (CTA1) is refolded by the cytosolic chaperone Hsp90. The already folded part at the cytosolic side cannot fit back through the pore anymore [12, 49].
2.1.5 Action in the Cytosol
Having reached the cytosol without being degraded, the catalytically active domain of ricin and Shiga toxin inhibit ribosomes [19, 50,51,52]. The Shiga toxin group causes cell damage in organs that express the receptor for Shiga toxin and can lead to a severe condition called haemolytic uremic syndrome [19]. The mechanism of toxicity of pertussis toxin and cholera toxin is based on chemically modifying certain regulators of the enzyme adenylate cyclase (chemical modification: ADP ribosylation, regulators: GTP-binding proteins) [8, 12, 53]. While pertussis toxin chemically modifies an inhibitory regulator and locks it in an inactive state, cholera toxin locks a stimulating regulator in an active state [54]. In both cases, cyclic adenosine monophosphate (cAMP) increases as a consequence and leads to derailed cell signalling [8, 12]. Effects of derailed signalling include insulin secretion in the case of pertussis toxin in pancreatic cells [12, 55, 56] or chloride secretion in intestinal cells that result in massive diarrhoea in the case of cholera toxin [8, 54].
2.1.6 How Quantity and Functionality of Molecules Travelling Along Toxin Routes Develops
For toxins, the transport to the ER is associated with losses. The majority of internalised pertussis toxin is degraded in lysosomes, therefore only a fraction of it reaches the ER [12, 57].
Because the ERAD pathway normally serves the degradation of proteins, the delivery of functional toxins to the cytosol via ERAD requires at least some deviation from this path. At least a fraction of the toxins that arrive in the cytosol need to evade proteasomal degradation. The majority of ricin has been reported to be degraded in the cytosol of yeast (with only ~ 20% not being degraded) [20]. The introduction of additional lysines into the ricin toxin A subunit (RTA) enhanced degradation [20, 58]. The PTS1 subunit of pertussis toxin has no lysine residues [59]. In most cases, ubiquitin is linked to lysine. Mutated versions of PTS1 in which arginine residues had been exchanged by lysine residues showed reduced effects in cells due to degradation [12, 60]. The cholera toxin A1 domain also contains only a few lysines. However, the presence of lysines alone is not ultimately decisive for ubiquitination. The folding state of the toxin or whether chaperones bind to the translocated toxin influences whether lysines become a substrate for ubiquitination or whether they are protected from ubiquitin ligases [61].
Although RTA was suggested to be not a good substrate for ubiquitination because of its low lysine content, ubiquitin-independent degradation may occur [20, 58]. The A1 chain of cholera toxin can be degraded by the proteasome, even without ubiquitination [8, 16, 62]. PTS1 contains no lysine that would be available for ubiquitination, but may also still be degraded in the proteasome by a ubiquitination-independent mechanism owing to its intrinsic instability [12, 46].
Toxin molecules eventually have to refold after translocation across the ER membrane to the cytosol in order to be functional. In addition to ubiquitination, unfolding is also an important factor for degradation by the proteasome. Therefore, quick refolding can be a mechanism by which proteasomal degradation is evaded [52, 63]. For example, refolding of RTA might be aided by cytosolic chaperones [64]. A subunit of the proteasome might even act as a chaperone on RTA and contribute to the recovery of its catalytic activity [65]. Several toxins may exploit the ERAD pathway to reach the cytosol (Table 1), including ricin [11, 14, 20], cholera toxin [8, 14, 16], pertussis toxin [12, 47], Shiga and Shiga-like toxins [10, 17,18,19] and Pseudomonas exotoxin [14, 15].
2.2 Mechanistic Principles: Toxins Reaching the Cytosol by Other Mechanisms
Various toxins reach their site of action by a different mechanism than those that exploit the ERAD pathway (Table 2). For example, toxins may reach the cytosol by translocating from endosomes to the cytosol, as suggested for the mechanisms of Clostridioides difficile toxins, diphtheria toxin or anthrax toxin [77,78,79,80,81,82].
Diphtheria toxin is taken up into cells by receptor-mediated endocytosis [83]. A conformational change is induced upon acidification in endosomes, which leads to exposure of hydrophobic areas and insertion into the membrane. The catalytic domain subsequently transfers from endosomes to the cytosol [77, 78, 80, 84]. Diphtheria toxin has been shown to form channels in lipid bilayers based on conductance measurements [85, 86]. An open channel state was found not to be required for translocation of diphtheria toxin [87]. However, host factors might be relevant for cytosolic entry [88, 89]. Because the introduction of disulphide-bridges inhibited translocation of diphtheria toxin A (DTA) to the cytosol, unfolding might be required for translocation [90].
Anthrax toxin consists of several proteins (protective antigen; lethal factor; edema factor), which are non-toxic individually but can exert toxicity in combination [91, 92]. Upon acidification in endosomes, the protein called “protective antigen” forms pores, which allow delivery of other proteins belonging to the toxin to the cytosol (e.g. lethal factor) [82, 91, 93]. Passage of proteins through the pore requires unfolding [82, 93, 94].
3 Efficiency of Delivery Via Toxin Routes
Knowing toxin delivery efficiencies can be crucial for the success of potential applications related to toxin routes, such as the use of toxin routes for macromolecule delivery, antitoxin treatments or toxin-derived therapeutics. Many toxins possess high potency. A single molecule of some toxins in the cytosol can already be sufficient to kill a cell, such as for example, diphtheria toxin or ricin [110, 111]. Therefore, high delivery efficiency is not obligatory for a toxic effect. Lethal doses of plant-derived and bacterial toxins are often in the range of nanograms or micrograms per kilogram bodyweight [112]. Results on the efficiency with which molecules were delivered to their site of action (in short called “efficiency data” here) are therefore particularly relevant for applications using toxin routes for delivery.
3.1 Ricin Efficiency Data
Based on binding studies, approximately 107 binding sites for ricin have been estimated to be present on a single HeLa cell [113]. However, the number of molecules that reaches the cytosol is limited. For example, only about 5% of the internalised ricin co-localised with a protein in the Golgi after an hour of incubation. This corresponded to 6–8 × 104 molecules per cell in this experiment based on counting immunogold-labelled molecules in cryosections [114]. Already after 45–60 min, ricin reaches an equilibrium for entering and exiting cells. Within 15 min, ricin reaches the ER [115]. Most of the internalised ricin was found in endosomes and the lysosome. Only a fraction reaches the Golgi [114]. About 70–80% of the Golgi-associated ricin was found in the TGN [114]. Only a fraction eventually reaches the ER and the cytosol [64, 116]. Ricin has a molecular activity of about 1400 ribosomes per minute [117].
3.2 Pseudomonas Exotoxin Efficiency Data
The efficiency of cytosolic delivery of cargoes with components of Pseudomonas exotoxin was estimated by quantification of molecules by western blot based on a modification (biotinylation) that takes place in the cytosol. After 4 h and after 20 h of incubation, the cytosolic concentration of delivered molecules was similar with ~ 5 × 10−7 M molecules (4 h) or ~ 6 × 10−7 M molecules (20 h). This was calculated based on the simplified assumption of cells having a spherical shape with an average diameter of 13 µm [118]. A saturation of delivery had been observed at a concentration of 200 nM. Incubation of cells with an increased concentration of 2 µM for 4 h did not increase the cytosolic delivery further. The reasons why increased concentrations above this threshold did not increase cytosolic delivery are not entirely understood. The different levels at which cytosolic delivery are limited could in principle include receptor binding, retrograde transport to the ER (expression level of the KDEL receptor) or retrotranslocation to the cytosol (expression level of Sec61) [118].
Although a single Pseudomonas exotoxin molecule in the cytosol might be sufficient to cause cell death, usually cells need to be treated in vitro with ~ 1000 molecules per cell to kill the cell. In a mouse model, 400–750 molecules of a Pseudomonas exotoxin-based immunotoxin were required to be bound per cell for tumour remission. The majority of Pseudomonas exotoxin that has been applied might traffic to lysosomes and it has been estimated that less than 1% reach the cytosol. This estimate is based on observations from the experiments with immunotoxins, in which killing of a tumour cell typically required at least several hundred molecules, although a single molecule of Pseudomonas exotoxin in the cytosol may already be sufficient for killing the cell [119, 120].
3.3 Pertussis Toxin Efficiency Data
Most of the internalised pertussis toxin is also degraded in the lysosome and only a part reaches the Golgi and the ER [12, 57]. Only 3% of surface-associated PTS1 was delivered to the cytosol after 3 h of intoxication, representing an average of 38,000 molecules of PTS1 per cell in the cytosol according to Banerjee and colleagues [47]. Although it was possible to visualise pertussis toxin in the endocytic pathway and in the Golgi, it was not detected in the ER or cytosol by fluorescence microscopy by Plaut and Carbonetti [12, 121].
3.4 Diphtheria Toxin Efficiency Data
Diphtheria toxin has also been found to reach the cytosol as a small fraction of what is available to the cell in total. If a reduction of the disulphide bond between two fragments of the toxin is taken as a measure for cytosolic entry, then only 5–10% of the cell-bound toxin reached the cytosol in Vero cells [84, 122]. This corresponds to only 200–400 toxin molecules, if a number of approximately 4000 molecules bind to a Vero cell. Approximately this number can already cause an effect [122].
3.5 Cholera Toxin Efficiency Data
The translocation of a tagged version of CTA1 from the ER to the cytosol was investigated. The majority of the molecule that reached the cytosol was detected during a pulse chase experiment within the first hour of chase, while cytosolic levels dropped almost completely already after the second hour of chase [16]. Cholera toxin had reached the ER after 60–90 min [28]. After 1 h of chase, approximately 26–27% of the tagged version of CTA1 had translocated from the ER to the cytosol. Rapid degradation after 2–3 h was reduced in the presence of a proteasome inhibitor [16]. Tagged CTA1 versions had a half-life in the range of 71–85 min. A proteasome inhibitor increased the half-life to 120 min [16]. In an experiment that analysed the export of CTA1 from microsomes, an export of approximately 18% had been observed after 1 h [42].
3.6 Shiga Toxin Efficiency Data
As the analysis of the export of Shiga toxin (also termed verotoxin) from microsomes showed, only a fraction of the toxin molecules that are present in microsomes are translocated out of them [39]. Only 4% of the activated form of the verotoxin 1 A subunit reached the cytosol within 4 h of continuous exposure to the toxin [123].
3.7 Anthrax-Based Delivery System Efficiency Data
In experiments that used PA for delivering cargo molecules that had been linked to LF, an estimated number of 110,000 cargo molecules (construct “Lv5”: monobody HA4-7c12 linked to the N-terminal domain of LF, LFN) or 79,000 cargo molecules (construct “Lv6”: affibody ABRaf linked to LFN) had been delivered per cell [124]. The genome editing efficiency of CRISPR-associated endonuclease Cas9 (Cas9) linked to LF or other Cas9 versions was determined to be in the range of a low single-digit percentage for an anthrax-based delivery system in another study [125].
3.8 Efficiency Comparison with the TAT Peptide and Chemicals for Delivery
For comparison, the TAT peptide (residues 47–57 of the human immunodeficiency virus [HIV] TAT protein), which has often been used in the context of cellular delivery, reached the cytosol at 0.08%, 0.38% or 0.66%, if cells were incubated with concentrations of 0.2 µM, 10 µM and 20 µM, respectively [126]. Delivery of the enzymatic domain of DTA mediated by the TAT peptide was found to be 1000-fold less efficient than delivery of DTA by an anthrax toxin-based delivery system, as measured based on protein synthesis inhibition by delivered DTA. The delivery of an antibody that mimics the anthrax delivery system was also found to be more efficient than delivery by TAT [124]. A comparison of the delivery efficiencies of the cell-penetrating peptides TAT and Penetratin with delivery systems based on the toxins Pseudomonas exotoxin A, diphtheria toxin and anthrax toxin had been performed by Verdurmen and colleagues, including comparisons in four different cell lines. The number of cargo molecules detected in cells varied highly, with a range from ~ 4500 molecules to ~ 3,280,000 per cell with pronounced differences between cell lines. Cytosolic delivery efficiencies for most of the toxin-based delivery systems were higher than for cell-penetrating peptides in the comparison by Verdurmen and colleagues [127].
These results show how the efficiency of cytosolic delivery was rather limited for TAT. Cytosolic delivery methods based on chemicals are also known to have very limited delivery efficiency in the low single percentage range (1–2% reaching the cytosol [128], ~ 3.5% reaching the cytosol [129] and 1–5% of nucleic acids reaching the nucleus [130]), although delivery by chemicals is already used for approved drugs, such as coronavirus disease 2019 vaccines [6, 131,132,133,134,135]. Therefore, provided the low efficiency of delivery to the cytosol is sufficient for the application, delivery via toxin routes might be just as suitable for therapeutic strategies as approved drugs using chemicals for delivery.
3.9 Possibilities to Modulate Delivery Efficiencies
The dependence of delivery efficiency on toxicity may be low because of the high potency of toxins. Nevertheless, delivery efficiency influences toxicity. Delivery efficiency can even be increased to a certain degree. For example, the toxicity of some toxins has been observed to be increased with the ER retention signal KDEL [32, 41, 136]. Additionally, Golgi retention sequences (YQRL) may increase toxicity [136]. Delivery may be decreased if the KDEL receptor is blocked, as shown by lowered cytotoxic effects of Pseudomonas exotoxin if lysozyme-KDEL competed for binding to the KDEL receptor. Overexpression of the KDEL receptor led to increased toxic effects on cells [34]. Another example of a strategy to increase cytosolic delivery of toxins and toxin-containing drugs is inducing endosomal escape of toxins, for example by chemicals, which is often successful in vitro, but brings along challenges in view of clinical applicability [137]. This may not correspond to the natural entry path of toxins that reach the cytosol via ERAD, but may still increase the number of toxin molecules that reach the cytosol.
4 Toxins for Therapy
4.1 Therapy: Are Toxin Pathways Suitable as Delivery Routes?
Delivery of macromolecular drugs and biologicals to the cytosol remains a great challenge. Some proteinaceous toxins obviously achieve entering the cytosol, although they start from the extracellular space. Therefore, toxin routes are of interest as drug delivery routes for new therapeutic strategies. The idea of exploiting the mechanisms of toxins for delivering macromolecules has already been pursued in the 1990s [69]. For example, there have been first approaches in developing delivery systems based on diphtheria toxin [138] or with components based on Pseudomonas exotoxin A [139] for a delivery system for deoxyribonucleic acid (DNA). The systems described by Uherek et al. [138] and Fominaya and Wels [139] also contained Poly-l-Lysine as one of the components of the delivery system. This is relevant, because Poly-l-Lysine has also been used for the purpose of transfection.
Toxin routes are attractive as delivery routes for drugs because they may bypass the degradative lysosome by exploiting a retrograde pathway [30]. The use of toxin routes for drug delivery has to fulfil certain criteria to allow drug delivery to the cytosol to be effective. Important criteria include the relationship between the number of cargo molecules that can be typically delivered, the number of cargo molecules that is required for an effect and the requirement and ability of cargo molecules to refold. A criterion might also be the suitability of a cargo as an ERAD substrate. For example, misfolding of part of the toxin in the ER or properties that mimic a misfolded protein may be responsible for “labelling” the molecule as an ERAD substrate [10, 17, 46, 47, 62] and this might be required for translocation to the cytosol.
The formation of a pore by the component “protective antigen (PA)” of anthrax toxin allows delivery of the anthrax component “lethal factor (LF)”; therefore, this has been suggested as a mechanism that could be exploited for delivering chosen cargoes [69]. To use the pore originating from anthrax toxin for delivery, cargoes need to have a conformational state that allows passage through the narrow pore. For example, in a comparison by Rabideau et al., some small-molecule drugs were able to pass through the pore, while cyclic peptides and a small-molecule drug with a bigger size were not [140].
Because many toxins are highly potent, other cargo molecules have to either possess similar potency or the efficiency of delivery has to be sufficient for the lower potencies of the respective cargoes. Therefore, when using toxin routes for delivery, the combination of the delivered quantity and the cargo’s individual potency may be the decisive criterion for whether the threshold for causing an effect is reached. As a consequence, while some cargoes might not be suitable for delivery via toxin routes, the delivery via this path might be particularly suitable for other groups of cargo types. This is also valid for a potentially required ability of the cargo to refold in the cytosol.
4.1.1 Quantitative Analysis of Macromolecules Delivered Via Toxin Routes
Designed Ankyrin Repeat Proteins (DARPins) were used as a model cargo to develop an assay for quantitative detection of cytosolic delivery. The assay is based on a peptide sequence (avi-tag) that is fused to the cargo and biotinylated in the cytosol by the Escherichia coli-derived biotin ligase BirA, which was stably overexpressed in cells for this purpose. Cargoes that have successfully been delivered to the cytosol can consequently be quantified by determining the amount of biotinylated cargo. Detection can simply be achieved via SDS-PAGE and blotting, if biotinylation outside the cytosol due to mixing of contents from different compartments as a consequence of cell lysis can be excluded [118]. Because the avi-tag is not a substrate for all biotin ligases, but specifically biotinylated by the E. coli-derived biotin ligase BirA, cells that stably overexpress BirA were used for this assay [118]. The quantity of cargo that is delivered is important for any application beyond just toxin delivery. Therefore, it is of extraordinarily high value to quantify the amount of cargo that is typically delivered to the cytosol.
Delivery via the anthrax pathway was dependent on the cargo’s stability. Relatively high stability potentially lowers delivery efficiency to the cytosol [118]. Unfolding of the cargo is required for delivery through anthrax pores. The ability to translocate to the cytosol was compared for DARPins with different thermodynamic stabilities and as a conclusion, delivery through anthrax pores was suggested to be probably limited to molecules that unfold easily [141]. High stability was not limiting for delivery of Pseudomonas exotoxin A-based cargoes in the study by Verdurmen et al. from 2015 [118], although unfolding is also required for the route of Pseudomonas exotoxin A. As a potential explanation for this observation, authors suggested the unfolding machinery involved in the anthrax route to be less forceful than the machinery for translocation and unfolding of the cell, which Pseudomonas exotoxin A uses [118].
Further limitations in delivery efficiency of an anthrax-based delivery system were observed and overcome by Becker et al. by reengineering the system. The anthrax delivery system was re-engineered for preventing the toxic premature pore formation at the cell surface and to limit the occurrence of pore formation to endosomes. The reengineered anthrax delivery system allowed using higher concentrations of the constructs without increased toxicity, thus allowing higher amounts of cargoes to be delivered to the cytosol [142].
4.1.2 Assessing the Functionality of Macromolecules After Delivery Via Toxin Routes
In addition to the amount of cargo that reaches the cytosol, the portion of functional cargo is essential for therapeutic applicability. Assays using biotinylation as an indicator of cytosolic localisation may require additional analyses that provide information on how many of the molecules that reached the cytosol are also functional. Because of the often required step of unfolding, refolding for delivery and potentially escape from degradation in the cytosol, it is crucial to evaluate the functionality of delivered cargo.
Functionality can for example be evaluated in the form of gene modification upon successful delivery of functional Cas9 molecules to their site of action. An engineered version of anthrax toxin was proposed as a delivery system by Hirschenberger et al. and delivery of functional Cas9 was evaluated by monitoring the knockout of green fluorescent protein in a 293T cell line that stably expressed green fluorescent protein. It was evaluated as well by monitoring the knockout of a gene in a colon cancer cell line [125]. Briefly, the system described in this study comprises transfection of cells with plasmids for guide RNA expression and subsequent treatment of cells with a 63-kDa fragment of protective antigen from anthrax toxin and versions of the cargo molecule Cas9. Plasmids encoding guide RNAs were delivered to cells by means of chemicals for nucleic acid transfection, which were the lipid-based transfection reagent Lipofectamine 2000 or the proprietary formulation TransIT-LT1 containing “a lipid and protein/polyamine mixture” according to the manufacturer [125, 143]. Two versions of the cargo Cas9 had been applied to cells, a histidine-tagged Cas9 (His-Cas9) and Cas9 fused to the recognition domain of lethal factor for PA of anthrax toxin (LFN-Cas9). Knockouts were observed both after treating cells with His-Cas9 and a 63-kDa fragment of protective antigen or with LFN-Cas9 and a 63-kDa fragment of protective antigen [125]. The cell culture medium had been renewed after transfection of the colon cancer cell line with guide RNA-encoding plasmids [125]. Removal of transfection reagents might be a relevant detail in this context because the delivery of proteins by certain chemicals has been demonstrated [6, 144, 145]. In addition, Cas9 together with nucleic acids have been delivered to cells by various chemicals [6, 146,147,148,149]. Furthermore, histidine residues have been suggested to potentially enhance cytosolic delivery [150,151,152]. Therefore, in case of any potential delivery-enhancing effects originating from chemicals or histidines, cargoes could in such a case be imagined to possibly reach the cytosol by a mixed mechanism.
4.1.3 Suitability of Molecules for Delivery by Toxin Routes and Examples for Cargo Molecules
While some cargo molecules may not be able to refold sufficiently in the cytosol (as for example expected for antibodies), other cargo molecules might either refold readily or not even require refolding for being functional (e.g. peptides). An example for a peptide that is functional in the cytosol and has been delivered to the cytosol via a strategy that was based on a toxin route is the NF-kappaB Essential Modifier-binding peptide. The NF-kappaB Essential Modifier-binding peptide modulated intracellular signalling and ameliorated rheumatoid arthritis in murine models [153,154,155]. Another peptide that was delivered using a similar strategy consisted of seven amino acids and contained the interaction site between the cytosolic portion of a transmembrane immunoreceptor and its cytosolic adaptor. In murine models, delivery of this peptide showed effects on inflammatory disorders [156].
Molecules that are suitable for refolding, molecules that do not require folding for functionality (e.g. peptides) as well as molecules with high potency (e.g. enzymes) might be particularly suitable for delivery to the cytosol via toxin routes, but the feasibility has to be assessed for each molecule individually. A molecule type that combines the advantage of being independent from folding for functionality and possessing high potency are small interfering RNA (siRNA). An attenuated version of diphtheria toxin was used by Arnold et al. for the delivery of siRNAs to silence a survival gene and a gene that is involved in invasion and metastasis of cancer cells [157]. The attenuated diphtheria toxin had been conjugated to the siRNA. Non-targeting siRNA conjugated to the attenuated toxin or “siRNA only” were used as controls. As a positive control, siRNA with lipofectamine was used [157]. Delivery of siRNA with attenuated diphtheria toxin was performed with two genetic targets. Functional effects in the form of reduced invasion for one downregulated target or cell death for the other were observed [157]. These in vitro results can spur further in vivo studies, which may require adaptation of the strategy to in vivo conditions (e.g. chemical stabilisation of siRNAs for preventing degradation by nucleases in the in vivo environment or addressing potential issues with immunogenicity), but may bear high potential to further advance RNA interference-based therapies. Compared with approved siRNA drugs that are based on a chemical carrier for delivery, such as patisiran [131], or that are based on receptor-mediated internalisation for delivery, such as givosiran [158], the different intracellular mechanism of toxin routes might result in different delivery characteristics. It might be of interest in the future to compare quantitative delivery efficiencies and further aspects such as toxicity between the different delivery mechanisms in more detail. Small interfering RNA can be considered a “potent” cargo because gene silencing is a catalytic process. DNA is also a potent cargo, because a single or a few DNA molecules may be sufficient for an effect owing to amplification by transcription and translation [159,160,161]. Therefore, it is a suitable cargo for delivery routes with limited efficiency. Examples for delivery systems based on toxins are given in Table 3.
4.1.4 Potential Side Effects of Exploiting Toxin Routes
“ER stress” with accumulation of misfolded proteins in the ER can trigger an unfolded protein response (UPR). Unfolded or misfolded proteins can be translocated to the cytosol via ERAD for degradation. Prolonged ER stress can eventually lead to apoptosis [168]. For example, ER stress has been observed as a potential effect of Shiga toxins, also eventually apoptosis [19, 67, 169].
4.1.5 Conclusion for Toxin Routes as Delivery Routes
Because there are proteinaceous toxins that obviously translocate to the cytosol from the extracellular space, there is proof for this type of delivery route. Several macromolecules have furthermore been delivered via toxin routes. Therefore, drug delivery via toxin routes is an option to address the persistent challenge of delivering macromolecules from the extracellular space to the cytosol, but this strategy is not generally applicable to all molecules and has to be assessed individually.
Although feasible in principle, the approach is limited to a selection of molecules that have to fulfil multiple criteria that are essential for an effect after passing toxin routes, for example, certain properties of cargo molecules as discussed previously [141]. This considerably narrows down the choice of cargo molecules. Importantly, the assessment of true delivery to the cytosol requires thorough scrutiny [145, 170]. Toxin routes may not be a universally applicable strategy for delivering macromolecules into the cell, but if a small selection of cargo types is suitable for this delivery route, delivery via toxin routes may offer considerable advantages compared to other delivery strategies. In contrast to delivery via membrane-damaging chemicals, delivery via toxin routes does not necessarily result in membrane damage. In contrast to chemical carriers that can dissociate from their cargoes or form aggregate sizes that are difficult to control with effects on in vivo usage, there is no risk of dissociation if the cargo and carrier are two proteins that are covalently linked to each other. Furthermore, specific targeting to cell types would be possible.
Promising cargoes might be molecules that are able to refold and molecules with an inherent amplification effect. Molecules that do not need to be in excess to their targets might be promising. Competitive inhibitors may only be worth considering as cargoes in case of very low-target molecule concentrations. Molecules that amplify their function (similar to nucleic acids or molecules with catalytic activity), with very few molecules being sufficient for an effect, are particularly promising, as the number of translocated molecules can be low via toxin routes.
4.2 Therapy: Implications for Toxicity and Anti-Toxin Treatments
Any potential efficiency issues that might limit the delivery via toxin routes can be a therapeutic advantage for treating diseases caused by toxins. Strategies for anti-toxin treatments can for example include the prevention of toxins from reaching their site of action by blocking the routes toxins take, or the inhibition of the toxin’s enzymatic activity. Blocking toxin routes may include blocking attachment of toxins to cells or interfering with intracellular transport [171,172,173,174]. Antibodies may also interfere with oligomerisation of toxin components or conversion of pre-pores to pores for toxins that require this step as part of their intoxication process [175,176,177]. Genome-wide screens can help with identifying host factors, which might be suitable targets for blocking toxins from entering cells [178, 179]. Alternatively, the source of toxins can be targeted, for example by eliminating toxin-producing bacteria with antibiotics or preventing infections with vaccinations [19, 180].
In particular, post-exposure antitoxins are needed because treatments for neutralising toxins outside the cell may not be effective any longer as soon as toxins are inside the cell [181]. Additionally, antibiotics can be ineffective against even lethal consequences, if the effect against bacteria alone cannot reverse the effect of already produced toxins (e.g. anthrax toxin) [92, 182]. Even an increased toxin release can be caused by lytic antibiotics [19, 183, 184].
Antibodies have been generated against various toxins [185, 186], including for example diphtheria toxin [187], Clostridioides difficile toxin [188, 189] or botulinum toxin [190,191,192]. An antibody that binds the pore-forming protein of anthrax toxin has been developed and approved as a therapeutic (raxibacumab; Abthrax) [193].
4.2.1 The Extracellular Space as a Target for Antitoxin Treatments
Translocation of toxins to their site of action can be interfered with at different levels. At the extracellular level, toxins can be prevented from binding to cells [173, 174, 194]. For example, chemical receptor analogues such as an oligosaccharide receptor analogue of the receptor Gb3 have been used as a therapeutic strategy for the Shiga toxin-hijacked receptor Gb3. Although effective in vitro, Gb3 receptor analogues were not sufficiently effective in vivo [195]. Further carbohydrate-based inhibitors have been evaluated and may be further optimised [196]. A mechanism for blocking the attachment of the toxin to the cell can, for example, be steric hindrance by an antibody, which occurs if a suitable epitope on the toxin is bound [173, 174]. For example, ricin toxin subunit B-specific antibodies that block attachment of ricin to the cell have been described [173, 174].
Preventing attachment of toxins to cells depends on multiple factors. The number of toxin receptors on cells can be high [113, 174], some toxins have more than one binding site for their receptor and a single antibody may sterically not be able to cover all binding sites simultaneously [174, 197]. Furthermore, toxins may bind promiscuously to several receptors [11, 12, 113, 198]. For example, Shiga toxin STx 2e may bind to more than one receptor [19, 199]. Additionally, pertussis toxin binds promiscuously to various glycoproteins and might enter the cell via multiple endocytosis routes [12, 198]. Ricin is known to have multiple uptake pathways because it binds to several glycoproteins or glycolipids [11, 52, 113]. Therefore, it can be challenging to completely block the uptake of these toxins.
4.2.2 The Intracellular Space as a Target for Anti-Toxin Treatments
The transport from the cell surface via the TGN to the ER can be interfered with. For example, conjugating ricin to gold particles or to horseradish peroxidase can already affect intracellular routing [200]. Inside cells, endocytosis and sorting of toxins can be interfered with at multiple individual steps of the transport process. Inhibitors have been described for preventing endocytic uptake, sorting to the Golgi apparatus, Golgi-to-ER transport and for interference with translocation from ER to cytosol (an overview of Shiga toxin uptake steps and compounds for interfering with them is provided in Table 1 and Figure 3 of the review by Kavaliauskiene et al.) [201].
Small molecules have been used to inhibit intracellular trafficking. For example, small molecules have been used to block the transport of ricin and Shiga toxin from the endosomes to the Golgi. A compound protected mice from the toxic effects of ricin [202, 203]. Targets involved in endosome-to-Golgi transport have been suggested and a range of small-molecule inhibitors for toxin trafficking have been described and suggested as a potential therapeutic strategy to protect from the effects of Shiga toxins, as reviewed by Li et al. [25].
Blocking retrotranslocation with small molecules has been described as a further strategy to prevent the effects of toxins. For example, a chemical library was screened for interference with RTA retrotranslocation using an attenuated and enhanced green fluorescent protein-labelled RTA [204]. A strategy to prevent retrotranslocation can also be based on preventing misfolding by using chemical chaperones. For example, PTS1 is an ERAD substrate because it unfolds. If unfolding is prevented by chemical chaperones that are known from therapeutic strategies against protein misfolding diseases, PTS1 loses its toxicity [12, 47].
Examples for compounds that have been suggested for inhibiting toxin trafficking include compound 134, retro-2 and manganese [25, 201, 203, 205]. Manganese has been reported to protect 3800-fold against Shiga toxin in vitro or even showed complete protection of mice to a normally lethal challenge [205].
Transport to the ER has been blocked by an antibody specific to Shiga toxin subunit A, which was presumably caused by recycling of the antibody-toxin complex back to the cell surface [172, 174]. Additionally, monoclonal antibodies have been used to prevent the retrograde transport of Shiga toxin, resulting in accumulation of the toxin in endosomes. In mice, this antibody had also protected from the effects of Shiga toxin [19, 206, 207].
An antibody specific to ricin has been reported to travel with the toxin inside the cell and to delay trafficking inside cells [115]. A bispecific antibody with specificity to two epitopes on the toxin ricin reduced the retrograde transport of ricin and resulted in endosomal accumulation instead [208]. Neutralising antibodies specific for ricin toxin subunit B as well as RTA have been reported [174, 209, 210]. Toxin-specific antibodies can be neutralising, non-neutralising or toxin enhancing [174, 189, 209, 211, 212]. Antibodies can be associated with increased uptake of the toxin ricin into cells, as has been observed for mab 24B11. This antibody does not block uptake and increases uptake instead, but still acts against toxicity because 24B11-toxin-complexes being transported to the lysosome instead of reaching the TGN [213]. In contrast to mab 24B11, which increases uptake but still has an anti-toxic effect, there are also antibodies that lead to enhanced toxicity. The mechanism of toxin-enhancing antibodies is not sufficiently understood [174, 209].
Competition for binding to the KDEL receptor has been shown to reduce the toxicity of Pseudomonas exotoxin A [34]. Therefore, the KDEL receptor could also be a potential target for interference, if short-term blockage does not affect ER homeostasis too severely and if it prevents lethal effects of the toxin.
4.2.3 Inhibiting the Enzymatic Activity of Toxins
A different strategy is the inhibition of the enzymatic activity of toxins [171, 214,215,216]. For example, small-molecule inhibitors have been described and chemical libraries have been screened for small-molecule inhibitors of ricin and Shiga toxin [171, 214, 216]. In addition, substrate analogues that inhibit the activity of the toxin by binding to its active site and which are derived from nucleic acid-based molecules have been described for ribosome inactivating proteins (RIPs) [215, 217]. Even antibodies that interfere with enzymatic activity of toxins have been described, such as the inhibition of the enzymatic activity of a Clostridioides difficile toxin [218] or the partial reduction of enzymatic activity of RTA [212], although the therapeutic applicability is difficult to imagine in case it depended on retrotranslocation of the antibody to the cytosol. However, the neutralising effect of antibodies that bind to the enzymatically active part of toxins may also originate from interference with intracellular transport. For example, antibodies specific to RTA interfered with intracellular toxin transport and probably led to degradation of the toxin in lysosomes [219].
4.2.4 Eliminating the Source of Toxin Production
A further strategy to prevent damaging effects caused by toxins is to target toxin-producing microorganisms. Antibiotics to eliminate toxin-producing bacteria or eliminating infection with these pathogens, for example by vaccinating cattle against bacteria, prevent damaging effects from toxins. However, treatment is only applicable with certain antibiotics because several antibiotics have been found to even upregulate the production of Shiga toxin and to increase severity of haemolytic uremic syndrome [19, 180, 220].
4.3 Therapy: Toxins as Drugs
Toxins and their engineered versions have been suggested for therapeutic use for achieving antiviral effects, treating protein misfolding diseases and for killing cells such as cancer cells [69, 221,222,223,224]. Suicide gene therapy, for example with diphtheria toxin expression under control of tissue-specific promoters to kill cancer cells, is another therapeutic strategy for using toxins therapeutically [80], but delivery of genes for this therapeutic strategy usually does not involve delivery via toxin routes, therefore it is not further discussed here.
4.3.1 Protein Misfolding Diseases as an Application Area for Toxins as Therapeutics
A potential application area of toxin-derived therapeutics is protein-misfolding diseases. If proteins are partially misfolded due to a mutation, they might be degraded although they might have some remaining functionality if they were not degraded. Mutant but still partly functional proteins can be rescued by temporarily saturating the ERAD pathway with toxin-derived proteins. Preventing degradation of the mutated protein via ERAD can ameliorate the pathological phenotype [69, 225,226,227]. Furthermore, ERAD inhibition might enhance folding of the mutated proteins by prolonging ER retention [225].
Cell surface expression of a mutated version of the chloride channel cystic fibrosis transmembrane conductance regulator, which leads to cystic fibrosis, was increased by inactivated cholera toxin and Shiga toxin. Inactivated toxins increased cell surface expression of the channel 20-fold and transport of chloride two-fold [226]. In addition to cystic fibrosis, treatment of further protein misfolding diseases including lysosomal storage diseases such as Gaucher disease, Krabbe disease, Fabry disease or Tay Sachs disease could potentially benefit from this therapeutic strategy. Lysosomal storage diseases are characterised by deficiencies in enzymatic activity and accumulation of metabolites [225,226,227,228,229].
The induction of the UPR might depend on the individual mechanism of interference with ERAD, but the induction of the UPR can be low if proteins are rescued by competing ERAD substrates under appropriate conditions [225, 226]. In contrast, the UPR was induced by CTB with a KDEL motif and resulted in an effect with therapeutic relevance, which was the induction of wound healing [230].
4.3.2 Vaccinations Supported by Toxins
Because the receptor for Shiga toxin (Gb3) is also expressed on dendritic cells, the subunit B of Shiga toxin has been suggested as a vehicle to internalise antigens into dendritic cells for vaccination purposes [68, 231]. Tumour antigens linked to the B subunit of Shiga toxin were able to induce antigen-specific cytotoxic T lymphocytes, although the simultaneous application of the B subunit of Shiga toxin and the tumour antigen as separate entities did not induce cytotoxic T lymphocytes [232]. In mice, the B subunit of Shiga toxin linked to an antigen of the human papillomavirus 16 protected mice from tumour cells that expressed this antigen [233]. Because linkage of antigens to the B subunit of Shiga toxin was found to allow targeting dendritic cells and eliciting specific cytotoxic T lymphocytes, this has been suggested as a vaccination strategy. While part of the molecules of the Shiga toxin B subunit was transported to lysosomes, another part underwent retrograde transport to the ER in this system, as confirmed by co-localisation with a Golgi marker and glycosylation by an enzyme in the ER [231].
4.3.3 Antiviral Immunotoxins
Immunotoxins have been developed mostly for applications in cancer therapy, but they also have been employed as an antiviral strategy. Antiviral effects of some toxins have been observed and suggested to be one of the natural functions of these toxins, for example, in plants [234, 235]. For example, the ribosome inactivating protein Pokeweed antiviral protein has been suggested to have a defence function in plants against pathogens [217, 234, 235]. The physiological role of RIPs from plants has also been suggested to be a defence mechanism of plants against being consumed by animals [236].
In plants, RIPs have been found in various compartments including vacuoles and the extracellular space [237, 238]. Wounding of plant cells might allow RIPs to enter the cytoplasm and in case breakage of the cell wall allows viral entry, protein synthesis inhibition might be a potential mechanism that confines viral spreading [224, 237, 239].
Antiviral mechanisms of RIPs include protein synthesis inhibition due to ribosome inactivation or inactivation of viral genomes by depurination due to adenine polynucleotide glycosylase activity. Therefore, viral translation, replication and transcription can be inhibited by RIPs [224, 234, 240, 241].
Antiviral activity of RIPs against a range of different types of viruses has been reported, like double-stranded DNA viruses (e.g. hepatitis B virus, HBV [242]), retroviruses (e.g. HIV [243]), positive-sense single stranded RNA viruses (e.g. Dengue virus [244]) or negative-sense single stranded RNA viruses (lymphocytic choriomeningitis virus [245]) [224]. Uptake of RIPs into cells might be aided by the endocytic uptake of viruses, which might explain why the inhibition of protein synthesis was observed to be more pronounced in infected cells [224, 246]. Activity of Shiga toxin against bovine immunodeficiency virus [247, 248], bovine leukaemia virus [248, 249] and HIV [250] has been reported.
A critical parameter of antiviral agents is a sufficient difference between concentrations with an antiviral effect and severely toxic concentrations, to ensure an effect with tolerable side effects. For some RIPs, antiviral concentrations differ substantially from their toxic concentrations [224]. For example, an antiviral effect against HIV in the form of inhibition of p24 expression and viral reverse transcriptase activity by 98% and 87% was induced by a RIP at a concentration of ~30 nM [251]. Cellular DNA and protein synthesis was not inhibited at this concentration, suggesting the absence of in vitro cytotoxicity at effective concentrations [251].
A strategy to further improve the therapeutic index of toxins is the use of immunotoxins. Immunotoxins employ targeting moieties to target toxins towards selected cells. This can be applied in antiviral therapy for selectively targeting virus-infected cells [222]. An important mechanism of action of antiviral immunotoxins is based on the eradication of infected cell populations [222, 252].
An advantage of antiviral immunotoxins compared with immunotoxins for cancer therapy is the chance to better discriminate between diseased and healthy cells. Antiviral immunotoxins allow the targeting of viral molecules that are foreign to the host cells, while targets on tumour cells are normally also expressed on healthy cells, even if expression levels are lower. Targeting viral proteins is therefore advantageous for reducing side effects [222] and may offer the potential for an improved therapeutic index of antiviral immunotoxins compared with anticancer immunotoxins.
For example, viral GPCRs (vGPCRs) such as a vGPCR from human cytomegalovirus have been targeted by immunotoxins. The immunotoxin consisted of a chemokine ligand for the vGPCR fused to the toxic domain of Pseudomonas exotoxin A. Side effects might be low because the vGPCR was targeted by a chemokine that is rarely acting as a ligand for host GPCRs (it binds to only a single host chemokine receptor) [222, 252]. This immunotoxin killed vGPCR-expressing cells and the antiviral effectiveness of the immunotoxin was superior to the virostatic small-molecule drug ganciclovir [252]. An example for a further viral factor that has been suggested as a promising target is ORF74 from Kaposi sarcoma-associated herpes virus. This factor is associated with proinflammatory and proliferative effects as well as angiogenic effects. Inhibiting each effect individually might be difficult. An immunotoxin that targets ORF74 was suggested as a promising approach to inhibit oncogenic signalling by killing KHSV-infected cells and thereby inhibiting all of these effects at once [222].
Although targeting a viral factor is advantageous to reduce side effects, host factors that are involved in viral infection could also be targeted by immunotoxins. For example, the host receptor EBI2, which is upregulated by Epstein–Barr virus [253], could be targeted by immunotoxins to counteract virus-associated diseases [222]. An advantage of targeting host proteins is their “sequence stability” compared with some more rapidly mutating virus genes. This lowers the risk for emerging drug resistance (e.g. loss of binding of immunotoxins to a mutated target).
Another relevant aspect for the choice of the target is whether it can be rapidly internalised, to allow delivery of the immunotoxin into the cell [222]. Eventually, targets are ideally of viral origin and internalise to efficiently deliver immunotoxins. Targets are ideally expressed in lytically and latently infected cells [222]. An immunotoxin allowed killing KHSV (HHV-8)-infected cells in the lytic phase in vitro, which could be of interest as a therapeutic approach for the treatment of diseases that are associated with acute infections with this virus [222, 254]. Particularly valuable would also be effective treatments for eradicating persistent reservoirs that are difficult to attack with standard treatments. For example, latently infected cell reservoirs that harbour HIV, which cannot be eradicated by the otherwise successful antiretroviral therapy, pose a requirement for further therapeutic strategies [255]. Even though immunotoxins so far have not been sufficiently effective as a monotherapy for treating HIV [255, 256], antiviral immunotoxins could still be of interest as a component of combination therapies [255, 257]. The rationale of such combination therapies is to complement the effect of suppressing new rounds of infections originating from an infected cell by antiretroviral therapy with killing infected cells by immunotoxins [255].
Examples for antiviral immunotoxins include immunotoxins directed against HIV [257, 258], herpes simplex virus 2 [259], rabies virus [260], Ebola virus [261] or human cytomegalovirus [222, 252, 262].
Many immunotoxins contain affinity reagents as their targeting moiety in contrast to ligands as targeting moieties. For example, an affinity reagent that binds a protein on the surface of herpes simplex virus 2 was combined with Pseudomonas exotoxin A to form an immunotoxin that killed virus-producing cells. As an application of this immunotoxin, the prevention of infection of other persons by the herpes simplex virus 2-positive person was proposed [259].
Challenges in the development of antiviral immunotoxins are similar to those for immunotoxins in cancer therapy, such as immunogenicity of immunotoxins. To reduce immunogenicity of toxins that are derived from non-human organisms, PEGylation or genetic engineering has been employed [258, 263, 264]. The stimulation of an immune response by immunotoxins has also been discussed as a potential benefit. There are data that suggest the development of anti-tumour immunity after treatment with immunotoxins. Anti-tumour immunity was hypothesised to be based on the ability of the immunotoxin to stimulate a strong immune activation [265]. Various mechanisms of resistance to immunotoxins have also been described, including mechanisms at the level of binding and internalisation, processing and trafficking and protein synthesis inhibition [266].
Regarding side effects, trichosanthin has been reported to be mostly well tolerated in patients with acquired immunodeficiency syndrome. However, compared with immunotoxins that are applied for cancer therapy, there are fewer clinical trials for RIPs in the context of antiviral therapy [222, 224].
4.3.4 Cancer-Targeting Immunotoxins
Toxin routes exemplify a concept that is useful as a therapeutic strategy for cancer treatment. The concept of delivering a toxic payload to a cell after specifically binding to a cellular receptor has been implemented in the form of immunotoxins that target cancer cells. Delivery has been adapted for this purpose by replacing the natural binding domain of the toxin by a binding moiety, which specifically binds to molecules that preferentially occur in cancer cells.
Cancer-targeting immunotoxins have advanced to clinical trials or have already been approved as a drug. They include immunotoxins based on P. aeruginosa exotoxin, Shiga toxin or diphtheria toxin [267,268,269,270]. An immunotoxin based on the cytokine interleukin-2 and on diphtheria toxin with the name denileukin diftitox has been approved in the USA for recurrent or persistent cutaneous T-cell lymphoma [271]. An immunotoxin that was approved for the treatment of relapsed or refractory hairy cell leukaemia is moxetumomab pasudotox (Lumoxiti; AstraZeneca AB, Cambridge, UK) [268]. Moxetumomab pasudotox is an anti-CD22 directed immunotoxin based on Pseudomonas exotoxin A [221, 268]. Cancer-targeting immunotoxins are reviewed more in depth elsewhere [221, 223, 270, 272,273,274,275].
5 Conclusions
Targeting the cytosol with macromolecules is difficult and toxins demonstrate one potential approach to how proteins can translocate to the cytosol from the extracellular space. Other methods to deliver macromolecules to the cytosol include strategies based on chemicals. In contrast to chemicals that disturb membrane barriers (like endosomal membranes) to allow cytosolic delivery of macromolecules, no membrane disruption is required if proteins are translocated from the ER to the cytosol via the ERAD pathway. Compared to many chemicals that are used for cytosolic delivery, using toxin routes could therefore avoid damage to the cell.
The type of molecules that are normally delivered to the cytosol via toxin routes is proteinaceous in nature. Cargoes that can be delivered via toxin routes might therefore be limited to molecules such as proteins or peptides with similar properties, potentially narrowing applications down to this class of molecules. Nucleic acids are not the type of cargo that normally travels via the toxin routes described here, different to commonly used chemicals for cytosolic delivery of macromolecules (e.g. lipid- or polymer-based reagents). Chemicals may therefore allow a slightly more versatile choice of cargoes, having been applied for both nucleic acid delivery and the direct delivery of proteins, although mainly applied for nucleic acid delivery [6, 145, 276]. However, nucleic acids have also already been described as a potential cargo when delivering via toxin routes [277, 278].
Potential effects of using toxin routes might include ER stress with UPR induction or immunogenicity of the bacteria- or plant-derived components. However, it might be application dependent whether such effects are disturbing or may even support the desired outcome, for example, if the aim is to kill cancer cells. In case immunogenicity is an unwanted effect, strategies for deimmunising toxin components by engineering or modification have been described. Furthermore, the induction of the UPR may be low if appropriate conditions are chosen [225, 226, 258, 263,264,265] or might not even apply depending on the entry mechanism of the toxin. The low quantitative efficiency of many delivery pathways via toxin routes is a potential limitation because only cargo molecules with high potency might be suitable for delivery via these routes. However, the development of RNA-based vaccines during the coronavirus disease 2019 pandemic as well as other RNA-based drugs have demonstrated how even delivery by routes that are expected to have low delivery efficiency can be sufficient for the desired therapeutic effect and for becoming approved as a drug [6, 131, 134, 135, 279].
Furthermore, every challenge for delivery of cargo via toxin routes can present a chance for therapy in the context of anti-toxin treatments and vice versa. Because a toxin often promiscuously binds to various host receptors, antitoxin strategies that aim at blocking toxin entry into cells can be challenging [11, 12, 19, 113, 174, 198, 199]. Therefore, toxin routes are promising targets for antitoxin treatments, as has been confirmed by antibodies that interfered with intracellular trafficking of toxins [19, 172, 174, 206, 207].
Finally, the development of therapeutics is already advanced in the field of immunotoxins. While antiviral immunotoxins have been explored and their application may have specific advantages concerning side effects over anti-cancer immunotoxins, the application of anti-cancer immunotoxins is so far most advanced, having resulted in approved drugs [221, 222, 268, 270, 273].
References
de la Torre BG, Albericio F. The pharmaceutical industry in 2020: an analysis of FDA drug approvals from the perspective of molecules. Molecules. 2021;26:627.
Zhang C, Ötjengerdes RM, Roewe J, Mejias R, Marschall ALJ. Applying antibodies inside cells: principles and recent advances in neurobiology, virology and oncology. BioDrugs. 2020. https://doi.org/10.1007/s40259-020-00419-w.
Marschall ALJ, Dübel S, Böldicke T. Specific in vivo knockdown of protein function by intrabodies. MAbs. 2015;7:1010–35.
Marschall ALJ, Dübel S. Antibodies inside of a cell can change its outside: can intrabodies provide a new therapeutic paradigm? Comput Struct Biotechnol J. 2016;14:304–8.
Stewart MP, Langer R, Jensen KF. Intracellular delivery by membrane disruption: mechanisms, strategies, and concepts. Chem Rev. 2018;118:7409–531.
Marschall ALJ. Targeting the inside of cells with biologicals: chemicals as a delivery strategy. BioDrugs Clin Immunother Biopharm Gene Ther. 2021;35:1–29.
Guillard S, Minter RR, Jackson RH. Engineering therapeutic proteins for cell entry: the natural approach. Trends Biotechnol. 2015;33:163–71.
Wernick NLB, Chinnapen DJ-F, Cho JA, Lencer WI. Cholera toxin: an intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins. 2010;2:310–25.
Bagola K, Mehnert M, Jarosch E, Sommer T. Protein dislocation from the ER. Spec Sect Protein Translocat Inser Membr. 2011;1808:925–36.
Spooner RA, Lord JM. How ricin and Shiga toxin reach the cytosol of target cells: retrotranslocation from the endoplasmic reticulum. Curr Top Microbiol Immunol. 2012;357:19–40. https://doi.org/10.1007/82_2011_154.
Spooner RA, Lord JM. Ricin trafficking in cells. Toxins. 2015;7:49–65.
Teter K. Intracellular trafficking and translocation of pertussis toxin. Toxins. 2019;11:437.
Ruggiano A, Foresti O, Carvalho P. ER-associated degradation: protein quality control and beyond. J Cell Biol. 2014;204:869–79.
Teter K, Holmes RK. Inhibition of endoplasmic reticulum-associated degradation in CHO cells resistant to cholera toxin, Pseudomonas aeruginosa exotoxin A, and ricin. Infect Immun. 2002;70:6172–9.
Michalska M, Wolf P. Pseudomonas exotoxin A: optimized by evolution for effective killing. Front Microbiol. 2015;6:963–963.
Teter K, Allyn RL, Jobling MG, Holmes RK. Transfer of the cholera toxin A1 polypeptide from the endoplasmic reticulum to the cytosol is a rapid process facilitated by the endoplasmic reticulum-associated degradation pathway. Infect Immun. 2002;70:6166–71.
LaPointe P, Wei X, Gariépy J. A role for the protease-sensitive loop region of Shiga-like toxin 1 in the retrotranslocation of its A1 domain from the endoplasmic reticulum lumen*. J Biol Chem. 2005;280:23310–8.
Li S, Spooner RA, Hampton RY, Lord JM, Roberts LM. Cytosolic entry of Shiga-like toxin A chain from the yeast endoplasmic reticulum requires catalytically active Hrd1p. PLoS ONE. 2012;7: e41119.
Chan YS, Ng TB. Shiga toxins: from structure and mechanism to applications. Appl Microbiol Biotechnol. 2016;100:1597–610.
Simpson JC, Roberts LM, Romisch K, Davey J, Wolf DH, Lord JM. Ricin A chain utilises the endoplasmic reticulum-associated protein degradation pathway to enter the cytosol of yeast. FEBS Lett. 1999;459:80–4.
Tesh VL, Burris JA, Owens JW, Gordon VM, Wadolkowski EA, O’Brien AD, et al. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect Immun. 1993;61:3392–402.
Itoh K, Tezuka T, Inoue K, Tada H, Suzuki T. Different binding property of verotoxin-1 and verotoxin-2 against their glycolipid receptor, globotriaosylceramide. Tohoku J Exp Med. 2001;195:237–43.
Sandvig K, van Deurs B. Transport of protein toxins into cells: pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett. 2002;529:49–53.
Pfeffer SR. Multiple routes of protein transport from endosomes to the trans Golgi network. Golgi Complex. 2009;583:3811–6.
Li D, Selyunin A, Mukhopadhyay S. Targeting the early endosome-to-Golgi transport of Shiga toxins as a therapeutic strategy. Toxins. 2020;12:342.
Sandvig K, Kavaliauskiene S, Skotland T. The protein toxins ricin and Shiga toxin as tools to explore cellular mechanisms of internalization and intracellular transport. Toxins. 2021;13:377.
Chaudhary VK, Jinno Y, FitzGerald D, Pastan I. Pseudomonas exotoxin contains a specific sequence at the carboxyl terminus that is required for cytotoxicity. Proc Natl Acad Sci. 1990;87:308–12.
Majoul IV, Bastiaens PI, Söling HD. Transport of an external Lys-Asp-Glu-Leu (KDEL) protein from the plasma membrane to the endoplasmic reticulum: studies with cholera toxin in Vero cells. J Cell Biol. 1996;133:777–89.
Hessler JL, Kreitman RJ. An early step in Pseudomonas exotoxin action is removal of the terminal lysine residue, which allows binding to the KDEL receptor. Biochemistry. 1997;36:14577–82.
Tarragó-Trani MT, Storrie B. Alternate routes for drug delivery to the cell interior: pathways to the Golgi apparatus and endoplasmic reticulum. Adv Drug Deliv Rev. 2007;59:782–97.
Wales R, Chaddock JA, Roberts LM, Lord JM. Addition of an ER retention signal to the ricin A chain increases the cytotoxicity of the holotoxin. Exp Cell Res. 1992;203:1–4.
Wales R, Roberts LM, Lord JM. Addition of an endoplasmic reticulum retrieval sequence to ricin A chain significantly increases its cytotoxicity to mammalian cells. J Biol Chem. 1993;268:23986–90.
Johannes L, Tenza D, Antony C, Goud B. Retrograde transport of KDEL-bearing B-fragment of Shiga toxin. J Biol Chem. 1997;272:19554–61.
Jackson ME, Simpson J, Girod A, Pepperkok R, Roberts L, Lord JM. The KDEL retrieval system is exploited by Pseudomonas exotoxin A, but not by Shiga-like toxin-1, during retrograde transport from the Golgi complex to the endoplasmic reticulum. J Cell Sci. 1999;112(Pt 4):467–75.
Girod A, Storrie B, Simpson J, Johannes L, Roberts L, Lord J, et al. Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat Cell Biol. 1999;1:423–30.
Lencer WI, Constable C, Moe S, Jobling MG, Webb HM, Ruston S, et al. Targeting of cholera toxin and Escherichia coli heat labile toxin in polarized epithelia: role of COOH-terminal KDEL. J Cell Biol. 1995;131:951–62.
Garred Ø, van Deurs B, Sandvig K. Furin-induced cleavage and activation of Shiga toxin*. J Biol Chem. 1995;270:10817–21.
Garred Ø, Dubinina E, Polesskaya A, Olsnes S, Kozlov J, Sandvig K. Role of the disulfide bond in Shiga toxin A-chain for toxin entry into cells *. J Biol Chem. 1997;272:11414–9.
Yu M, Haslam DB. Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERdj3. Infect Immun. 2005;73:2524–32.
Hazes B, Read RJ. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry. 1997;36:11051–4.
Wesche J, Rapak A, Olsnes S. Dependence of ricin toxicity on translocation of the toxin A-chain from the endoplasmic reticulum to the cytosol *. J Biol Chem. 1999;274:34443–9.
Schmitz A, Herrgen H, Winkeler A, Herzog V. Cholera toxin is exported from microsomes by the Sec61p complex. J Cell Biol. 2000;148:1203–12.
Bernardi KM, Forster ML, Lencer WI, Tsai B. Derlin-1 facilitates the retro-translocation of cholera toxin. Mol Biol Cell. 2008;19:877–84.
Bernardi KM, Williams JM, Kikkert M, van Voorden S, Wiertz EJ, Ye Y, et al. The E3 ubiquitin ligases Hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol Biol Cell. 2010;21:140–51.
Veithen A, Raze D, Locht C. Intracellular trafficking and membrane translocation of pertussis toxin into host cells. Int J Med Microbiol. 2000;290:409–13.
Pande AH, Moe D, Jamnadas M, Tatulian SA, Teter K. The pertussis toxin S1 subunit is a thermally unstable protein susceptible to degradation by the 20S proteasome. Biochemistry. 2006;45:13734–40.
Banerjee T, Cilenti L, Taylor M, Showman A, Tatulian SA, Teter K. Thermal unfolding of the pertussis toxin S1 subunit facilitates toxin translocation to the cytosol by the mechanism of endoplasmic reticulum-associated degradation. Infect Immun. 2016;84:3388–98.
Peskin CS, Odell GM, Oster GF. Cellular motions and thermal fluctuations: the Brownian ratchet. Biophys J. 1993;65:316–24.
Burress H, Taylor M, Banerjee T, Tatulian SA, Teter K. Co- and post-translocation roles for HSP90 in cholera intoxication*. J Biol Chem. 2014;289:33644–54.
Olsnes S, Pihl A. Ricin: a potent inhibitor of protein synthesis. FEBS Lett. 1972;20:327–9.
Brown JE, Rothman SW, Doctor BP. Inhibition of protein synthesis in intact HeLa cells by Shigella dysenteriae 1 toxin. Infect Immun. 1980;29:98–107.
Sowa-Rogozińska N, Sominka H, Nowakowska-Gołacka J, Sandvig K, Słomińska-Wojewódzka M. Intracellular transport and cytotoxicity of the protein toxin ricin. Toxins. 2019;11:350.
Hsia JA, Tsai SC, Adamik R, Yost DA, Hewlett EL, Moss J. Amino acid-specific ADP-ribosylation. Sensitivity to hydroxylamine of [cysteine(ADP-ribose)]protein and [arginine(ADP-ribose)]protein linkages. J Biol Chem. 1985;260:16187–91.
Alberts B, Johnson A, Lewis J. Signaling through G-protein-linked cell-surface receptors. Mol Biol Cell. 4th edition. New York: Garland Science; 2002. Available from: https://www.ncbi.nlm.nih.gov/books/NBK26912/. Accessed 2 Jul 2022.
Katada T, Ui M. Slow interaction of islet-activating protein with pancreatic islets during primary culture to cause reversal of alpha-adrenergic inhibition of insulin secretion. J Biol Chem. 1980;255:9580–8.
Mangmool S, Kurose H. Gi/o protein-dependent and -independent actions of Pertussis toxin (PTX). Toxins. 2011;3:884.
Kügler S, Böcker K, Heusipp G, Greune L, Kim KS, Schmidt MA. Pertussis toxin transiently affects barrier integrity, organelle organization and transmigration of monocytes in a human brain microvascular endothelial cell barrier model. Cell Microbiol. 2007;9:619–32.
Deeks ED, Cook JP, Day PJ, Smith DC, Roberts LM, Lord JM. The low lysine content of ricin A chain reduces the risk of proteolytic degradation after translocation from the endoplasmic reticulum to the cytosol. Biochemistry. 2002;41:3405–13.
Nicosia A, Perugini M, Franzini C, Casagli MC, Borri MG, Antoni G, et al. Cloning and sequencing of the pertussis toxin genes: operon structure and gene duplication. Proc Natl Acad Sci. 1986;83:4631–5.
Worthington ZEV, Carbonetti NH. Evading the proteasome: absence of lysine residues contributes to pertussis toxin activity by evasion of proteasome degradation. Infect Immun. 2007;75:2946–53.
Wernick NLB, De Luca H, Kam WR, Lencer WI. N-terminal extension of the cholera toxin A1-chain causes rapid degradation after retrotranslocation from endoplasmic reticulum to cytosol. J Biol Chem. 2010;285:6145–52.
Pande AH, Scaglione P, Taylor M, Nemec KN, Tuthill S, Moe D, et al. Conformational instability of the cholera toxin A1 polypeptide. J Mol Biol. 2007;374:1114–28.
Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102.
Spooner RA, Hart PJ, Cook JP, Pietroni P, Rogon C, Höhfeld J, et al. Cytosolic chaperones influence the fate of a toxin dislocated from the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2008;105:17408–13.
Pietroni P, Vasisht N, Cook JP, Roberts DM, Lord JM, Hartmann-Petersen R, et al. The proteasome cap RPT5/Rpt5p subunit prevents aggregation of unfolded ricin A chain. Biochem J. 2013;453:435–45.
Endo Y, Mitsui K, Motizuki M, Tsurugi K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes: the site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J Biol Chem. 1987;262:5908–12.
Tesh VL. Activation of cell stress response pathways by Shiga toxins. Cell Microbiol. 2012;14:1–9.
Liu Y, Tian S, Thaker H, Dong M. Shiga toxins: an update on host factors and biomedical applications. Toxins. 2021;13:222.
Piot N, van der Goot FG, Sergeeva OA. Harnessing the membrane translocation properties of AB toxins for therapeutic applications. Toxins. 2021;13:36.
Tamura M, Nogimori K, Murai S, Yajima M, Ito K, Katada T, et al. Subunit structure of islet-activating protein, pertussis toxin, in conformity with the A-B model. Biochemistry. 1982;21:5516–22.
Stein PE, Boodhoo A, Armstrong GD, Heerze LD, Cockle SA, Klein MH, et al. Structure of a pertussis toxin-sugar complex as a model for receptor binding. Nat Struct Biol. 1994;1:591–6.
Locht C, Antoine R. The history of pertussis toxin. Toxins. MDPI; 2021;13:623.
Kounnas MZ, Morris RE, Thompson MR, FitzGerald DJ, Strickland DK, Saelinger CB. The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J Biol Chem. 1992;267:12420–3.
Ogata M, Fryling CM, Pastan I, FitzGerald DJ. Cell-mediated cleavage of Pseudomonas exotoxin between Arg279 and Gly280 generates the enzymatically active fragment which translocates to the cytosol. J Biol Chem. 1992;267:25396–401.
Eshraghi A, Maldonado-Arocho FJ, Gargi A, Cardwell MM, Prouty MG, Blanke SR, et al. Cytolethal distending toxin family members are differentially affected by alterations in host glycans and membrane cholesterol *. J Biol Chem. 2010;285:18199–207.
Scuron MD, Boesze-Battaglia K, Dlakić M, Shenker BJ. The cytolethal distending toxin contributes to microbial virulence and disease pathogenesis by acting as a tri-perditious toxin. Front Cell Infect Microbiol. 2016. https://doi.org/10.3389/fcimb.2016.00168.
Lemichez E, Bomsel M, Devilliers G, van der Spek J, Murphy JR, Lukianov EV, et al. Membrane translocation of diphtheria toxin fragment A exploits early to late endosome trafficking machinery. Mol Microbiol. 1997;23:445–57.
Chenal A, Nizard P, Gillet D. Structure and function of diphtheria toxin: from pathology to engineering. Toxin Rev. 2002;21:321–59.
Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18:247–63.
Shapira A, Benhar I. Toxin-based therapeutic approaches. Toxins. 2010;2:2519–83.
Gerding DN, Johnson S, Rupnik M, Aktories K. Clostridium difficile binary toxin CDT. Gut Microb. 2014;5:15–27.
Friebe S, Van der Goot FG, Bürgi J. The ins and outs of anthrax toxin. Toxins. 2016;8:69.
Morris RE, Gerstein AS, Bonventre PF, Saelinger CB. Receptor-mediated entry of diphtheria toxin into monkey kidney (Vero) cells: electron microscopic evaluation. Infect Immun. 1985;50:721–7.
Moskaug JO, Sandvig K, Olsnes S. Low pH-induced release of diphtheria toxin A-fragment in Vero cells Biochemical evidence for transfer to the cytosol. J Biol Chem. 1988;263:2518–25.
Donovan JJ, Simon MI, Draper RK, Montal M. Diphtheria toxin forms transmembrane channels in planar lipid bilayers. Proc Natl Acad Sci. 1981;78:172–6.
Kagan BL, Finkelstein A, Colombini M. Diphtheria toxin fragment forms large pores in phospholipid bilayer membranes. Proc Natl Acad Sci. 1981;78:4950–4.
Ladokhin AS, Vargas-Uribe M, Rodnin MV, Ghatak C, Sharma O. Cellular entry of the diphtheria toxin does not require the formation of the open-channel state by its translocation domain. Toxins. MDPI. 2017;9:299.
Ratts R, Zeng H, Berg EA, Blue C, McComb ME, Costello CE, et al. The cytosolic entry of diphtheria toxin catalytic domain requires a host cell cytosolic translocation factor complex. J Cell Biol. 2003;160:1139–50.
Schuster M, Schnell L, Feigl P, Birkhofer C, Mohr K, Roeder M, et al. The Hsp90 machinery facilitates the transport of diphtheria toxin into human cells. Sci Rep. 2017;7:613.
Falnes PO, Choe S, Madshus IH, Wilson BA, Olsnes S. Inhibition of membrane translocation of diphtheria toxin A-fragment by internal disulfide bridges. J Biol Chem. 1994;269:8402–7.
Young J, Collier R. Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annu Rev Biochem. 2007;76:243–65.
Gurnev PA, Nestorovich EM. Channel-forming bacterial toxins in biosensing and macromolecule delivery. Toxins. 2014;6.
Collier RJ. Membrane translocation by anthrax toxin. Anthrax. 2009;30:413–22.
Feld GK, Thoren KL, Kintzer AF, Sterling HJ, Tang II, Greenberg SG, et al. Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat Struct Mol Biol. 2010;17:1383–90.
Duesbery N, Webb C, Leppla S, Gordon V, Klimpel K, Copeland T, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–7.
Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JAT. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–9.
Scobie Heather M., Rainey G. Jonah A., Bradley Kenneth A., Young John A. T. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci. 2003;100:5170–4.
van der Goot G, Young JAT. Receptors of anthrax toxin and cell entry. Anthrax. 2009;30:406–12.
Tucker KD, Wilkins TD. Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect Immun. 1991;59:73–8.
Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 1995;375:500–3.
Gülke I, Pfeifer G, Liese J, Fritz M, Hofmann F, Aktories K, et al. Characterization of the enzymatic component of the ADP-ribosyltransferase toxin CDTa from Clostridium difficile. Infect Immun. 2001;69:6004–11.
Nottrott S, Schoentaube J, Genth H, Just I, Gerhard R. Clostridium difficile toxin A-induced apoptosis is p53-independent but depends on glucosylation of Rho GTPases. Apoptosis. 2007;12:1443–53.
Panagiotis P, Carette JE, Bell GW, Schwan C, Guttenberg G, Brummelkamp TR, et al. Lipolysis-stimulated lipoprotein receptor (LSR) is the host receptor for the binary toxin Clostridium difficile transferase (CDT). Proc Natl Acad Sci. 2011;108:16422–7.
Hartley-Tassell Lauren E, Awad Milena M, Seib Kate L, Scarselli M, Savino S, Tiralongo J, et al. Lectin activity of the TcdA and TcdB toxins of Clostridium difficile. Infect Immun. 2019;87:e00676-e718.
Martínez-Meléndez A, Cruz-López F, Morfin-Otero R, Maldonado-Garza HJ, Garza-González E. An update on Clostridioides difficile binary toxin. Toxins. 2022;14:305.
Moskaug JO, Stenmark H, Olsnes S. Insertion of diphtheria toxin B-fragment into the plasma membrane at low pH: characterization and topology of inserted regions. J Biol Chem. 1991;266:2652–9.
Mitamura T, Higashiyama S, Taniguchi N, Klagsbrun M, Mekada E. Diphtheria toxin binds to the epidermal growth factor (EGF)-like domain of human heparin-binding EGF-like growth factor/diphtheria toxin receptor and inhibits specifically its mitogenic activity. J Biol Chem. 1995;270:1015–9.
Holmes RK. Biology and molecular epidemiology of diphtheria toxin and the tox gene. J Infect Dis. 2000;181:S156–67.
Simon NC, Aktories K, Barbieri JT. Novel bacterial ADP-ribosylating toxins: structure and function. Nat Rev Microbiol. 2014;12:599–611.
Yamaizumi M, Mekada E, Uchida T, Okada Y. One molecule of diphtheria toxin fragment a introduced into a cell can kill the cell. Cell. 1978;15:245–50.
Eiklid K, Olsnes S, Pihl A. Entry of lethal doses of abrin, ricin and modeccin into the cytosol of HeLa cells. Exp Cell Res. 1980;126:321–6.
Gill DM. Bacterial toxins: a table of lethal amounts. Microbiol Rev. 1982;46:86–94.
Sandvig K, Olsnes S, Pihl A. Kinetics of binding of the toxic lectins abrin and ricin to surface receptors of human cells. J Biol Chem. 1976;251:3977–84.
van Deurs B, Sandvig K, Petersen OW, Olsnes S, Simons K, Griffiths G. Estimation of the amount of internalized ricin that reaches the trans-Golgi network. J Cell Biol. 1988;106:253–67.
Song K, Mize RR, Marrero L, Corti M, Kirk JM, Pincus SH. Antibody to ricin a chain hinders intracellular routing of toxin and protects cells even after toxin has been internalized. PLoS ONE. 2013;8: e62417.
Rapak A, Falnes PØ, Olsnes S. Retrograde transport of mutant ricin to the endoplasmic reticulum with subsequent translocation to cytosol. Proc Natl Acad Sci. 1997;94:3783.
Olsnes S, Fernandez-Puentes C, Carrasco L, Vazquez D. Ribosome inactivation by the toxic lectins abrin and ricin. Eur J Biochem. 1975;60:281–8.
Verdurmen WPR, Luginbühl M, Honegger A, Plückthun A. Efficient cell-specific uptake of binding proteins into the cytoplasm through engineered modular transport systems. J Control Release. 2015;200:13–22.
Kreitman RJ, Pastan I. Accumulation of a recombinant immunotoxin in a tumor in vivo: fewer than 1000 molecules per cell are sufficient for complete responses. Cancer Res. 1998;58:968–75.
Weldon JE, Pastan I. A guide to taming a toxin: recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011;278:4683–700.
Plaut RD, Carbonetti NH. Retrograde transport of pertussis toxin in the mammalian cell. Cell Microbiol. 2008;10:1130–9.
Moskaug JO, Sandvig K, Olsnes S. Cell-mediated reduction of the interfragment disulfide in nicked diphtheria toxin: a new system to study toxin entry at low pH. J Biol Chem. 1987;262:10339–45.
Tam P, Lingwood C. Membrane-cytosolic translocation of Verotoxin A1-subunit in target cells. Microbiol Read Engl. 2007;153:2700–10.
Liao X, Rabideau AE, Pentelute BL. Delivery of antibody mimics into mammalian cells via anthrax toxin protective antigen. Chembiochem Eur J Chem Biol. 2014;15:2458–66.
Hirschenberger M, Stadler N, Fellermann M, Sparrer KMJ, Kirchhoff F, Barth H, et al. CRISPA: a non-viral, transient Cas9 delivery system based on reengineered anthrax toxin. Front Pharmacol. 2021;12: 770283.
Lucchino M, Billet A, Bai S-K, Dransart E, Hadjerci J, Schmidt F, et al. Absolute quantification of drug‐vector delivery to the cytosol. Angew Chem Int Ed. 2021;60.
Verdurmen WPR, Mazlami M, Plückthun A. A quantitative comparison of cytosolic delivery via different protein uptake systems. Sci Rep. 2017;7:13194.
Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31:638–46.
Wittrup A, Ai A, Liu X, Hamar P, Trifonova R, Charisse K, et al. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat Biotechnol. 2015;33:870–6.
Cohen RN, van der Aa MAEM, Macaraeg N, Lee AP, Szoka FC Jr. Quantification of plasmid DNA copies in the nucleus after lipoplex and polyplex transfection. J Control Rel. 2009;135:166–74.
Kristen AV, Ajroud-Driss S, Conceição I, Gorevic P, Kyriakides T, Obici L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener Dis Manag. 2019;9:5–23.
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383:2603–15.
Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2021;384:403–16.
Verbeke R, Lentacker I, De Smedt SC, Dewitte H. The dawn of mRNA vaccines: the COVID-19 case. J Control Release. 2021;333:511–20.
Schoenmaker L, Witzigmann D, Kulkarni JA, Verbeke R, Kersten G, Jiskoot W, et al. mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int J Pharm. 2021;601: 120586.
Zhan J, Stayton P, Press OW. Modification of ricin A chain, by addition of endoplasmic reticulum (KDEL) or Golgi (YQRL) retention sequences, enhances its cytotoxicity and translocation. Cancer Immunol Immunother CII Germany. 1998;46:55–60.
Fuchs H, Weng A, Gilabert-Oriol R. Augmenting the efficacy of immunotoxins and other targeted protein toxins by endosomal escape enhancers. Toxins. 2016;8:200.
Uherek C, Fominaya J, Wels W. A modular DNA carrier protein based on the structure of diphtheria toxin mediates target cell-specific gene delivery*. J Biol Chem. 1998;273:8835–41.
Fominaya J, Wels W. Target cell-specific DNA transfer mediated by a chimeric multidomain protein: novel non-viral gene delivery system∗. J Biol Chem. 1996;271:10560–8.
Rabideau AE, Liao X, Akçay G, Pentelute BL. Translocation of non-canonical polypeptides into cells using protective antigen. Sci Rep. 2015;5:11944.
Becker L, Singh Badwal J, Brandl F, Verdurmen WPR, Plückthun A. Thermodynamic stability Is a strong predictor for the delivery of DARPins to the cytosol via anthrax toxin. Pharmaceutics. 2021;13:1285.
Becker L, Verdurmen WPR, Plückthun A. Reengineering anthrax toxin protective antigen for improved receptor-specific protein delivery. BMC Biol. 2020;18:100–100.
Mirus. Frequently asked questions: TransIT®-LT1 transfection reagent. 2022. https://www.mirusbio.com/tech-resources/faqs/transit-lt1-faqs. Accessed 8 Jul 2022.
Zelphati O, Wang Y, Kitada S, Reed JC, Felgner PL, Corbeil J. Intracellular delivery of proteins with a new lipid-mediated delivery system. J Biol Chem. 2001;276:35103–10.
Marschall ALJ, Zhang C, Frenzel A, Schirrmann T, Hust M, Perez F, et al. Delivery of antibodies to the cytosol: debunking the myths. MAbs. 2014;6:943–56.
Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33:73–80.
Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng. 2017;1:889–901.
Lee B, Lee K, Panda S, Gonzales-Rojas R, Chong A, Bugay V, et al. Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours. Nat Biomed Eng. 2018;2:497–507.
Wei T, Cheng Q, Min Y-L, Olson EN, Siegwart DJ. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat Commun. 2020;11:3232.
Midoux P, Monsigny M. Efficient gene transfer by histidylated polylysine/pDNA complexes. Bioconjug Chem. 1999;10:406–11.
Lo SL, Wang S. An endosomolytic Tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials. 2008;29:2408–14.
El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11:13–22.
Sehnert B, Burkhardt H, Wessels JT, Schroder A, May MJ, Vestweber D, et al. NF-kappaB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-kappaB in immune-mediated diseases. Proc Natl Acad Sci USA. 2013;110:16556–61.
Sehnert B, Burkhardt H, Finzel S, Dübel S, Voll RE. The sneaking ligand approach for cell type-specific modulation of intracellular signalling pathways. Clin Immunol. 2018;186:14–20.
Sehnert B, Burkhardt H, Dübel S, Voll RE. Cell-type targeted NF-kappaB inhibition for the treatment of inflammatory diseases. Cells. 2020;9:1627.
Gibot S, Jolly L, Lemarié J, Carrasco K, Derive M, Boufenzer A. Triggering receptor expressed on myeloid cells-1 inhibitor targeted to endothelium decreases cell activation. Front Immunol. 2019;10:2314.
Arnold AE, Smith LJ, Beilhartz GL, Bahlmann LC, Jameson E, Melnyk RA, et al. Attenuated diphtheria toxin mediates siRNA delivery. Sci Adv. 2020;6:4848.
Dammes N, Peer D. Paving the road for RNA therapeutics. Trends Pharmacol Sci. 2020;41:755–75.
Schwake G, Youssef S, Kuhr J-T, Gude S, David MP, Mendoza E, et al. Predictive modeling of non-viral gene transfer. Biotechnol Bioeng. 2010;105:805–13.
Scholz C, Wagner E. Therapeutic plasmid DNA versus siRNA delivery: common and different tasks for synthetic carriers. Drug Deliv Res Eur. 2012;161:554–65.
Liu J, Carmell M, Rivas F, Marsden C, Thomson J, Song J-J, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–41.
Wesche J, Elliott JL, Falnes PØ, Olsnes S, Collier RJ. Characterization of membrane translocation by anthrax protective antigen. Biochemistry. 1998;37:15737–46.
Chen C, Przedpelski A, Tepp WH, Pellett S, Johnson EA, Barbieri JT. Heat-labile enterotoxin IIa, a platform to deliver heterologous proteins into neurons. mBio. 2015;6:e00734.
Beer L-A, Tatge H, Schneider C, Ruschig M, Hust M, Barton J, et al. The binary toxin CDT of Clostridium difficile as a tool for intracellular delivery of bacterial glucosyltransferase domains. Toxins. 2018;10.
Clemons NC, Bannai Y, Haywood EE, Xu Y, Buschbach JD, Ho M, et al. Cytosolic delivery of multidomain cargos by the N terminus of Pasteurella multocida yoxin. Infect Immun. 2018;86.
Schmit NE, Neopane K, Hantschel O. Targeted protein degradation through cytosolic delivery of monobody binders using bacterial toxins. ACS Chem Biol. 2019;14:916–24.
Tian S, Liu Y, Appleton E, Wang H, Church GM, Dong M. Targeted intracellular delivery of Cas13 and Cas9 nucleases using bacterial toxin-based platforms. Cell Rep. 2022. https://doi.org/10.1016/j.celrep.2022.110476.
Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69:169–81.
Lee S-Y, Lee M-S, Cherla RP, Tesh VL. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. 2008;10:770–80.
Marschall ALJ, Zhang C, Dübel S. Evaluating the delivery of proteins to the cytosol of mammalian cells. In: Kasid U, Clarke R, editors. Cancer Gene Netw. 2017;201–8. https://doi.org/10.1007/978-1-4939-6539-7_14.
Miller DJ, Ravikumar K, Shen H, Suh J-K, Kerwin SM, Robertus JD. Structure-based esign and characterization of novel platforms for ricin and Shiga toxin inhibition. J Med Chem. 2002;45:90–8.
Krautz-Peterson G, Chapman-Bonofiglio S, Boisvert K, Feng H, Herman IM, Tzipori S, et al. Intracellular neutralization of Shiga toxin 2 by an a subunit-specific human monoclonal antibody. Infect Immun. 2008;76:1931–9.
Yermakova A, Mantis NJ. Protective immunity to ricin toxin conferred by antibodies against the toxin’s binding subunit (RTB). Vaccine. 2011;29:7925–35.
O’Hara JM, Yermakova A, Mantis NJ. Immunity to ricin: fundamental insights into toxin-antibody interactions. Curr Top Microbiol Immunol. 2012;357:209–41.
Oganesyan V, Peng L, Damschroder MM, Cheng L, Sadowska A, Tkaczyk C, et al. Mechanisms of neutralization of a human anti-α-toxin antibody. J Biol Chem. 2014;289:29874–80.
Moayeri M, Leysath CE, Tremblay JM, Vrentas C, Crown D, Leppla SH, et al. A heterodimer of a VHH (variable domains of camelid heavy chain-only) antibody that inhibits anthrax toxin cell binding linked to a VHH antibody that blocks oligomer formation is highly protective in an anthrax spore challenge model. J Biol Chem. 2015;290:6584–95.
Xiong S, Zhou T, Zheng F, Liang X, Cao Y, Wang C, et al. Different mechanisms of two anti-anthrax protective antigen antibodies and function comparison between them. BMC Infect Dis. 2019;19:940.
Pacheco AR, Lazarus JE, Sit B, Schmieder S, Lencer WI, Blondel CJ, et al. CRISPR screen reveals that EHEC’s T3SS and Shiga toxin rely on shared host factors for infection. mBio. 2018;9:e01003-18.
Tian S, Muneeruddin K, Choi MY, Tao L, Bhuiyan RH, Ohmi Y, et al. Genome-wide CRISPR screens for Shiga toxins and ricin reveal Golgi proteins critical for glycosylation. PLOS Biol. 2018;16: e2006951.
Matthews L, Reeve R, Gally DL, Low JC, Woolhouse Mark EJ, McAteer SP, et al. Predicting the public health benefit of vaccinating cattle against Escherichia coli O157. Proc Natl Acad Sci. 2013;110:16265–70.
Wilson BA, Ho M. Cargo-delivery platforms for targeted delivery of inhibitor cargos against botulism. Curr Top Med Chem. 2014;14:2081–93.
Brazil M. Anthrax attacked! Nat Rev Drug Discov. 2004;3:13–13.
Spreer A, Kerstan H, Böttcher T, Gerber J, Siemer A, Zysk G, et al. Reduced release of pneumolysin by Streptococcus pneumoniae in vitro and in vivo after treatment with nonbacteriolytic antibiotics in comparison to ceftriaxone. Antimicrob Agents Chemother. 2003;47:2649–54.
Brown LA, Mitchell AM, Mitchell TJ. Streptococcus pneumoniae and lytic antibiotic therapy: are we adding insult to injury during invasive pneumococcal disease and sepsis? J Med Microbiol. 2017;66:1253–6.
Kuhn P, Fühner V, Unkauf T, Moreira GMSG, Frenzel A, Miethe S, et al. Recombinant antibodies for diagnostics and therapy against pathogens and toxins generated by phage display. Proteom Clin Appl. 2016;10:922–48.
Roth KDR, Wenzel EV, Ruschig M, Steinke S, Langreder N, Heine PA, et al. Developing recombinant antibodies by phage display against infectious diseases and toxins for diagnostics and therapy. Front Cell Infect Microbiol. 2021;11: 697876.
Wenzel EV, Bosnak M, Tierney R, Schubert M, Brown J, Dübel S, et al. Human antibodies neutralizing diphtheria toxin in vitro and in vivo. Sci Rep. 2020;10:571.
LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bezlotoxumab. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012. https://www.ncbi.nlm.nih.gov/books/NBK548863/. Accessed 10 Jul 2022.
Fühner V, Heine PA, Helmsing S, Goy S, Heidepriem J, Loeffler FF, et al. Development of neutralizing and non-neutralizing antibodies targeting known and novel epitopes of TcdB of Clostridioides difficile. Front Microbiol. 2018;9:2908.
Miethe S, Mazuet C, Liu Y, Tierney R, Rasetti-Escargueil C, Avril A, et al. Development of germline-humanized antibodies neutralizing botulinum neurotoxin A and B. PLoS ONE. 2016;11: e0161446.
Derman Y, Selby K, Miethe S, Frenzel A, Liu Y, Rasetti-Escargueil C, et al. Neutralization of botulinum neurotoxin type E by a humanized antibody. Toxins. 2016;8:257.
Rasetti-Escargueil C, Avril A, Miethe S, Mazuet C, Derman Y, Selby K, et al. The European AntibotABE Framework Program and its update: development of innovative botulinum antibodies. Toxins. 2017;9:309.
Tsai C-W, Morris S. Approval of raxibacumab for the treatment of inhalation anthrax under the US Food and Drug Administration “Animal Rule.” Front Microbiol. 2015;6:1320.
Murphy K, Janeway CA, Travers P, Walport M. Antibodies protect against extracellular pathogens and their toxic products. In: Janeways Immunobiology. 8th ed. London and New York: Garland Science, Taylor & Francis Group; 2012. p. 27.
Lingwood C. Verotoxin receptor-based pathology and therapies. Front Cell Infect Microbiol. 2020;10:123.
Haksar D, Asadpoor M, Heise T, Shi J, Braber S, Folkerts G, et al. Fighting shigella by blocking its disease-causing toxin. J Med Chem. 2021;64:6059–69.
Sphyris N, Lord JM, Wales R, Roberts LM. Mutational analysis of the Ricinus lectin B-chains: galactose-binding ability of the 2γ subdomain of Ricinus communis agglutinin B-cchain (∗). J Biol Chem. 1995;270:20292–7.
el Bayâ A, Brückener K, Schmidt MA. Nonrestricted differential intoxication of cells by pertussis toxin. Infect Immun. 1999;67:433–5.
Müthing J, Meisen I, Zhang W, Bielaszewska M, Mormann M, Bauerfeind R, et al. Promiscuous Shiga toxin 2e and its intimate relationship to Forssman. Glycobiology. 2012;22:849–62.
van Deurs B, Tønnessen TI, Petersen OW, Sandvig K, Olsnes S. Routing of internalized ricin and ricin conjugates to the Golgi complex. J Cell Biol. 1986;102:37–47.
Kavaliauskiene S, Dyve Lingelem AB, Skotland T, Sandvig K. Protection against Shiga toxins. Toxins. 2017;9:44.
Seaman MNJ, Peden AA. Ricin toxin hits a retrograde roadblock. Cell. 2010;141:222–4.
Stechmann B, Bai S-K, Gobbo E, Lopez R, Merer G, Pinchard S, et al. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell. 2010;141:231–42.
Redmann V, Gardner T, Lau Z, Morohashi K, Felsenfeld D, Tortorella D. Novel class of potential therapeutics that target ricin retrograde translocation. Toxins. 2013;6:33–53.
Mukhopadhyay S, Linstedt AD. Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science. 2012;335:332–5.
Smith MJ, Melton-Celsa AR, Sinclair JF, Carvalho HM, Robinson CM, O’Brien AD. Monoclonal antibody 11E10, which neutralizes shiga toxin type 2 (Stx2), recognizes three regions on the Stx2 A subunit, blocks the enzymatic action of the toxin in vitro, and alters the overall cellular distribution of the toxin. Infect Immun. 2009;77:2730–40.
Russo LM, Melton-Celsa AR, Smith MA, Smith MJ, O’Brien AD. Oral intoxication of mice with Shiga toxin type 2a (Stx2a) and protection by anti-Stx2a monoclonal antibody 11E10. Infect Immun. 2014;82:1213–21.
Herrera C, Klokk TI, Cole R, Sandvig K, Mantis NJ. A bispecific antibody promotes aggregation of ricin toxin on cell surfaces and alters dynamics of toxin internalization and trafficking. PLoS ONE. 2016;11: e0156893.
Maddaloni M, Cooke C, Wilkinson R, Stout AV, Eng L, Pincus SH. Immunological characteristics associated with the protective efficacy of antibodies to ricin. J Immunol. 2004;172:6221.
Prigent J, Panigai L, Lamourette P, Sauvaire D, Devilliers K, Plaisance M, et al. Neutralising antibodies against ricin toxin. PLoS ONE. 2011;6: e20166.
Mohamed N, Li J, Ferreira CS, Little SF, Friedlander AM, Spitalny GL, et al. Enhancement of anthrax lethal toxin cytotoxicity: a subset of monoclonal antibodies against protective antigen increases lethal toxin-mediated killing of murine macrophages. Infect Immun. 2004;72:3276–83.
O’Hara JM, Neal LM, McCarthy EA, Kasten-Jolly JA, Brey RN, Mantis NJ. Folding domains within the ricin toxin A subunit as targets of protective antibodies. Vaccine. 2010;28:7035–46.
Yermakova A, Klokk TI, Cole R, Sandvig K, Mantis NJ. Antibody-mediated inhibition of ricin toxin retrograde transport. mBio. 2014;5:e00995.
Yan X, Hollis T, Svinth M, Day P, Monzingo AF, Milne GWA, et al. Structure-based identification of a ricin inhibitor. J Mol Biol. 1997;266:1043–9.
Sturm MB, Roday S, Schramm VL. Circular DNA and DNA/RNA hybrid molecules as scaffolds for ricin inhibitor design. J Am Chem Soc. 2007;129:5544–50.
Wahome PG, Bai Y, Neal LM, Robertus JD, Mantis NJ. Identification of small-molecule inhibitors of ricin and shiga toxin using a cell-based high-throughput screen. Toxicon. 2010;56:313–23.
Domashevskiy AV, Goss DJ. Pokeweed antiviral protein, a ribosome inactivating protein: activity, inhibition and prospects. Toxins. 2015;7:274–98.
Unger M, Eichhoff AM, Schumacher L, Strysio M, Menzel S, Schwan C, et al. Selection of nanobodies that block the enzymatic and cytotoxic activities of the binary Clostridium difficile toxin CDT. Sci Rep. 2015;5:7850.
Yermakova A, Klokk TI, O’Hara JM, Cole R, Sandvig K, Mantis NJ. Neutralizing monoclonal antibodies against disparate epitopes on ricin toxin’s enzymatic subunit interfere with intracellular toxin transport. Sci Rep. 2016;6:22721.
Ichinohe N, Ohara-Nemoto Y, Nemoto T, Kimura S, Ichinohe S. Effects of fosfomycin on Shiga toxin-producing Escherichia coli: quantification of copy numbers of Shiga toxin-encoding genes and their expression levels using real-time PCR. J Med Microbiol. 2009;58:971–3.
Alewine C, Hassan R, Pastan I. Advances in anticancer immunotoxin therapy. Oncologist. 2015;20:176–85.
Spiess K, Jakobsen MH, Kledal TN, Rosenkilde MM. The future of antiviral immunotoxins. J Leukoc Biol. 2016;99:911–25.
Zahaf N-I, Schmidt G. Bacterial toxins for cancer therapy. Toxins. 2017;9:236.
Citores L, Iglesias R, Ferreras JM. Antiviral activity of ribosome-inactivating proteins. Toxins. 2021;13:80.
Wang F, Song W, Brancati G, Segatori L. Inhibition of endoplasmic reticulum-associated degradation rescues native folding in loss of function protein misfolding diseases. J Biol Chem. 2011;286:43454–64.
Adnan H, Zhang Z, Park H-J, Tailor C, Che C, Kamani M, et al. Endoplasmic reticulum-targeted subunit toxins provide a new approach to rescue misfolded mutant proteins and revert cell models of genetic diseases. PLoS ONE. 2016;11: e0166948.
Lingwood C. Therapeutic uses of bacterial subunit toxins. Toxins. 2021;13:378.
Ferreira CR, Gahl WA. Lysosomal storage diseases. Transl Sci Rare Dis. 2017;2:1–71.
Rajkumar V, Dumpa V. Lysosomal Storage Disease. Treasure Island (FL): StatPearls Publishing; 2022. https://www.ncbi.nlm.nih.gov/books/NBK563270/?report=classic. Accessed 11 Jul 2022.
Royal J, Oh YJ, Grey M, Lencer W, Ronquillo N, Galandiuk S, et al. A modified cholera toxin B subunit containing an ER retention motif enhances colon epithelial repair via an unfolded protein response. FASEB J. 2019;33:fj.201901255R.
Haicheur N, Benchetrit F, Amessou M, Leclerc C, Falguières T, Fayolle C, et al. The B subunit of Shiga toxin coupled to full-size antigenic protein elicits humoral and cell-mediated immune responses associated with a Th1-dominant polarization. Int Immunol. 2003;15:1161–71.
Haicheur N, Bismuth E, Bosset S, Adotevi O, Warnier G, Lacabanne V, et al. The B subunit of Shiga toxin fused to a tumor antigen elicits CTL and targets dendritic cells to allow MHC class I-restricted presentation of peptides derived from exogenous antigens. J Immunol. 2000;165:3301.
Vingert B, Adotevi O, Patin D, Jung S, Shrikant P, Freyburger L, et al. The Shiga toxin B-subunit targets antigen in vivo to dendritic cells and elicits anti-tumor immunity. Eur J Immunol. 2006;36:1124–35.
Picard D, Kao CC, Hudak KA. Pokeweed antiviral protein inhibits Brome mosaic virus replication in plant cells. J Biol Chem. 2005;280:20069–75.
Di R, Tumer NE. Pokeweed antiviral protein: its cytotoxicity mechanism and applications in plant disease resistance. Toxins. 2015;7:755–72.
Damme E, Peumans W, Barre A, Rougé P. Plant lectins: a composite of several distinct families of structurally and evolutionary related proteins with diverse biological roles. Crit Rev Plant Sci. 1998;17:575–692.
Ready MP, Brown DT, Robertus JD. Extracellular localization of pokeweed antiviral protein. Proc Natl Acad Sci USA. 1986;83:5053–6.
Carzaniga R, Sinclair L, Fordham-Skelton AP, Harris N, Croy RRD. Cellular and subcellular distribution of saporins, type-1 ribosome-inactivating proteins, in soapwort (Saponaria officinalis L.). Planta. 1994;194:461–70.
Bonness MS, Ready MP, Irvin JD, Mabry TJ. Pokeweed antiviral protein inactivates pokeweed ribosomes; implications for the antiviral mechanism. Plant J. 1994;5:173–83.
Montanaro L, Sperti S, Stirpe F. Inhibition by ricin of protein synthesis in vitro. Ribosomes as the target of the toxin. Biochem J. 1973;136:677–83.
Barbieri L, Valbonesi P, Bonora E, Gorini P, Bolognesi A, Stirpe F. Polynucleotide:adenosine glycosidase activity of ribosome-inactivating proteins: effect on DNA, RNA and poly(A). Nucleic Acids Res. 1997;25:518–22.
He Y-W, Guo C-X, Pan Y-F, Peng C, Weng Z-H. Inhibition of hepatitis B virus replication by pokeweed antiviral protein in vitro. World J Gastroenterol. 2008;14:1592–7.
Kaur I, Puri M, Ahmed Z, Blanchet FP, Mangeat B, Piguet V. Inhibition of HIV-1 replication by balsamin, a ribosome inactivating protein of Momordica balsamina. PLoS ONE. 2013;8: e73780.
Rothan HA, Bahrani H, Mohamed Z, Abd Rahman N, Yusof R. Fusion of protegrin-1 and plectasin to MAP30 shows significant inhibition activity against Dengue virus replication. PLoS ONE. 2014;9: e94561.
Uckun FM, Rustamova L, Vassilev AO, Tibbles HE, Petkevich AS. CNS activity of pokeweed anti-viral protein (PAP) in mice infected with lymphocytic choriomeningitis virus (LCMV). BMC Infect Dis. 2005;5:9.
Foà-Tomasi L, Campadelli-Fiume G, Barbieri L, Stirpe F. Effect of ribosome-inactivating proteins on virus-infected cells: inhibition of virus multiplication and of protein synthesis. Arch Virol. 1982;71:323–32.
Ferens WA, Hovde CJ. The non-toxic A subunit of Shiga toxin type 1 prevents replication of bovine immunodeficiency virus in infected cells. Virus Res. 2007;125:29–41.
Ferens WA, Halver M, Gustin KE, Ott T, Hovde CJ. Differential sensitivity of viruses to the antiviral activity of Shiga toxin 1 A subunit. Virus Res. 2007;125:104–8.
Ferens WA, Hovde CJ. Antiviral activity of Shiga toxin 1: suppression of bovine leukemia virus-related spontaneous lymphocyte proliferation. Infect Immun. 2000;68:4462–9.
Shi PL, Binnington B, Sakac D, Katsman Y, Ramkumar S, Gariepy J, et al. Verotoxin A subunit protects lymphocytes and T cell lines against X4 HIV infection in vitro. Toxins. 2012;4:1517–34.
Lee-Huang S, Huang PL, Nara PL, Chen H-C, Kung H, Huang P, et al. MAP 30: a new inhibitor of HIV-1 infection and replication. FEBS Lett. 1990;272:12–8.
Spiess K, Jeppesen M, Malmgaard-Clausen M, Krzywkowski K, Dulal K, Cheng T, et al. Rationally designed chemokine-based toxin targeting the viral G protein-coupled receptor US28 potently inhibits cytomegalovirus infection in vivo. Proc Natl Acad Sci USA. 2015;112.
Birkenbach M, Josefsen K, Yalamanchili R, Lenoir G, Kieff E. Epstein-Barr virus-induced genes: first lymphocyte-specific G protein-coupled peptide receptors. J Virol. 1993;67:2209–20.
Chatterjee D, Chandran B, Berger EA. Selective killing of Kaposi’s sarcoma-associated herpesvirus lytically infected cells with a recombinant immunotoxin targeting the viral gpK8.1A envelope glycoprotein. mAbs. 2012;4:233–42.
Berger EA, Pastan I. Immunotoxin complementation of HAART to deplete persisting HIV-infected cell reservoirs. PLOS Pathog. 2010;6: e1000803.
Ananworanich J. What will it take to cure HIV? Top Antivir Med. 2015;23:80–4.
Denton PW, Long JM, Wietgrefe SW, Sykes C, Spagnuolo RA, Snyder OD, et al. Targeted cytotoxic therapy kills persisting HIV infected cells during ART. PLOS Pathog. 2014;10: e1003872.
Pincus SH, Song K, Maresh GA, Frank A, Worthylake D, Chung H-K, et al. Design and in vivo characterization of immunoconjugates targeting HIV gp160. J Virol. 2017;91:e01360-e1416.
Geoghegan EM, Zhang H, Desai PJ, Biragyn A, Markham RB. Antiviral activity of a single-domain antibody immunotoxin binding to glycoprotein D of herpes simplex virus 2. Antimicrob Agents Chemother. 2015;59:527–35.
Mareeva T, Wanjalla C, Schnell MJ, Sykulev Y. A novel composite immunotoxin that suppresses rabies virus production by the infected cells. J Immunol Methods. 2010;353:78–86.
Cai Y, Yu S, Chi X, Radoshitzky SR, Kuhn JH, Berger EA. An immunotoxin targeting Ebola virus glycoprotein inhibits Ebola virus production from infected cells. PLoS ONE. 2021;16: e0245024.
Spiess K, Jeppesen MG, Malmgaard-Clausen M, Krzywkowski K, Kledal TN, Rosenkilde MM. Novel chemokine-based immunotoxins for potent and selective targeting of cytomegalovirus infected cells. J Immunol Res. 2017;2017:4069260.
Mazor R, King EM, Pastan I. Strategies to reduce the immunogenicity of recombinant immunotoxins. Am J Pathol. 2018;188:1736–43.
Vallera DA, Kreitman RJ. Immunotoxins targeting B cell malignancy: progress and problems with immunogenicity. Biomedicines. 2019;7.
Leshem Y, Pastan I. Pseudomonas exotoxin immunotoxins and anti-tumor immunity: from observations at the patient’s bedside to evaluation in preclinical models. Toxins. 2019;11.
Dieffenbach M, Pastan I. Mechanisms of resistance to immunotoxins containing Pseudomonas exotoxin A in cancer therapy. Biomolecules. 2020;10.
Frankel AE, Woo JH, Ahn C, Foss FM, Duvic M, Neville PH, et al. Resimmune, an anti-CD3ε recombinant immunotoxin, induces durable remissions in patients with cutaneous T-cell lymphoma. Haematologica. 2015;100:794–800.
Dhillon S. Moxetumomab pasudotox: first global approval. Drugs. 2018;78:1763–7.
Persky DO, Musteata V, Zodelava M, Perekhrestenko T, Diaz AE, Guthrie TH, et al. A phase 2 study of MT-3724 to evaluate safety, pharmacodynamics and efficacy of MT-3724 for the treatment of patients with relapsed or refractory diffuse large B-cell lymphoma. Blood. 2019;134:5324.
Silver AB, Leonard EK, Gould JR, Spangler JB. Engineered antibody fusion proteins for targeted disease therapy. Trends Pharmacol Sci. 2021;42:1064–81.
LiverTox Clin Res Inf Drug-Induc Liver In. Denileukin diftitox. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012. https://www.ncbi.nlm.nih.gov/books/NBK548436/?report=classic. Accessed 16 Jul 2022.
Rust A, Partridge LJ, Davletov B, Hautbergue GM. The use of plant-derived ribosome inactivating proteins in immunotoxin development: past, present and future generations. Toxins. 2017;9:344.
Kreitman RJ, Pastan I. Development of recombinant immunotoxins for hairy cell leukemia. Biomolecules. 2020;10:1140.
Antignani A, Ho ECH, Bilotta MT, Qiu R, Sarnvosky R, FitzGerald DJ. Targeting receptors on cancer cells with protein toxins. Biomolecules. 2020;10:1331.
Knödler M, Buyel JF. Plant-made immunotoxin building blocks: a roadmap for producing therapeutic antibody-toxin fusions. Biotechnol Adv. 2021;47: 107683.
Marschall ALJ, Frenzel A, Schirrmann T, Schüngel M, Dubel S. Targeting antibodies to the cytoplasm. MAbs. 2011;3:3–16.
Dyer PDR, Shepherd TR, Gollings AS, Shorter SA, Gorringe-Pattrick MAM, Tang C-K, et al. Disarmed anthrax toxin delivers antisense oligonucleotides and siRNA with high efficiency and low toxicity. J Control Rel. 2015;220:316–28.
Shorter SA, Gollings AS, Gorringe-Pattrick MAM, Coakley JE, Dyer PDR, Richardson SCW. The potential of toxin-based drug delivery systems for enhanced nucleic acid therapeutic delivery. Expert Opin Drug Deliv. 2017;14:685–96.
Thapar M, Rudnick S, Bonkovsky HL. Givosiran, a novel treatment for acute hepatic porphyrias. Expert Rev Precis Med Drug Dev. 2021;6:9–18.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL.
Conflicts of interest/competing interests
Maximilian Ruschig and Andrea L. J. Marschall have no conflicts of interest that are directly relevant to the content of this article.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors’ contributions
ALJM conceived the project. ALJM and MR wrote the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ruschig, M., Marschall, A.L.J. Targeting the Inside of Cells with Biologicals: Toxin Routes in a Therapeutic Context. BioDrugs 37, 181–203 (2023). https://doi.org/10.1007/s40259-023-00580-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-023-00580-y