
Abstract
Objective
We aimed to evaluate the similarity of BAT2206 to its originator, ustekinumab, including pharmacokinetic profiles, immunogenicity, and safety in healthy Chinese male subjects.
Methods
This was a double-blinded, randomized, single-dose, parallel-group clinical trial, in which 270 healthy male subjects were enrolled to receive a single subcutaneous injection (45 mg) of either BAT2206 or ustekinumab (European Union or USA) at a 1:1:1 ratio. The pairwise pharmacokinetic similarities and the safety and immunogenicity of both drugs were evaluated and compared.
Results
The results showed that the 90% confidence interval of the geometric mean ratio for primary pharmacokinetic parameters (maximum plasma concentration and area under the plasma concentration–time curve from time zero to infinity) among BAT2206 and ustekinumab (USA or European Union sourced) groups were all within the predefined equivalent interval of 80–125%. Furthermore, all the groups had similar incidences of treatment-emergent adverse events, in which the majority of cases belonged to Common Terminology Criteria for the Classification of Adverse Events Grade 1 or 2. Anti-drug antibodies were detected in 54 (20.1%) subjects, namely 24 (26.7%), 13 (14.8%), and 17 (18.9%) patients in the BAT2206, ustekinumab (European Union), and ustekinumab (USA) groups, respectively. In contrast, the incidences of positive neutralizing antibodies were similar among the three groups.
Conclusions
Pharmacokinetic similarity between BAT2206 and ustekinumab (USA or European Union sourced) was confirmed. The three groups had similar safety profiles, and the investigational drugs were well tolerated by subjects.
Clinical Trial Registration
This study was registered with ClinicalTrials.gov (NCT04371185).
Similar content being viewed by others


Biosimilarity of BAT2206 and its originator ustekinumab (European Union or USA licensed) in pharmacokinetic profiles, safety, and immunogenicity was evaluated. |
Pharmacokinetic similarity between BAT2206 and ustekinumab (USA or European Union sourced) was confirmed. |
The incidence of anti-drug antibodies in the BAT2206 group was higher than those in the other two groups, whereas the incidences of positive neutralizing antibodies were similar among the three groups. |
The three groups had similar safety profiles and the investigational drugs were well tolerated by subjects. |
1 Introduction
Psoriasis is a chronic, immune-mediated inflammatory skin disease [1]. About 2.5% of European individuals, 0.05–3% of African individuals, and 0.1–0.5% of Asian individuals experience psoriasis [2,3,4]. Psoriasis is not curable to date and seriously affects a patient’s quality of life, even physical and mental health. Interleukin (IL)-12 and IL-23 are overexpressed in psoriasis plaques and are positively correlated with the severity of skin lesions [5]. Psoriatic arthritis is an inflammatory arthritis associated with psoriasis. About 1.3–34.7% of patients with psoriasis develop psoriatic arthritis. It has been shown in an animal model and clinical study that IL-12 and IL-23 are involved in symptom development and joint change of immune-related arthritis [6].
Ustekinumab (Stelara®) is a fully humanized, IL-12/IL-23 monoclonal antibody developed by Johnson & Johnson, Inc. Ustekinumab can disrupt the binding of IL-12 and IL-23 to IL-12Rβ1 through binding to the p40 subunit shared by IL-12 and IL-23 with high affinity and specificity, leading to the inhibition of IL-12-induced and IL-23-induced signaling and the cytokine cascade [7]. Ustekinumab was approved in succession in Canada, the USA, the European Union (EU), and Japan in 2008. Its clinical applications are the treatment of severe plaque psoriasis, active psoriatic arthritis, moderate-to-severe Crohn’s disease, and ulcerative colitis in adults. Currently, ustekinumab has been approved for the treatment of moderate-to-severe active Crohn’s disease and moderate-to-severe plaque psoriasis in China.
BAT2206 is a biosimilar drug to ustekinumab that was developed by Bio-Thera Solutions, Ltd. (Guangzhou, China). Using recombinant DNA technology, the expression plasmid of BAT2206 was constructed and expressed in a mammalian Chinese hamster ovary cell system. The monoclonal cell bank of BAT2206 underwent full inspection in accordance with the standard in Chinese pharmacopeia (2015). In a comparison evaluation on their structural characteristics and physicochemical properties using many analytical assays, BAT2206 was found to be highly similar to ustekinumab.
In a preclinical pharmacokinetic (PK) study using monkeys, the plasma exposure (area under the plasma concentration–time curve from time zero to 336 hours) and maximum plasma concentration (Cmax) of BAT2206 were considered to be bioequivalent to those of the reference drugs (EU and USA sourced) at the same dose, all of which were commercially available. Additionally, in two mice models, mice injected with recombinant human IL-12 and the ear swelling model induced by recombinant human IL-23, treatment with BAT2206 showed a similar anti-inflammatory effect as Stelara® at the same dose (unpublished data).
A high similarity between BAT2206 and Stelara® was found in all of these preclinical studies. To obtain marketing approval for BAT2206, the phase I trial was performed and the purpose of the trial was to investigate the PK similarity of BAT2206 and the originator ustekinumab (EU and USA sourced STELARA®) in Chinese healthy male subjects after a subcutaneous single dose, focusing on their PK profile, safety, and immunogenicity, leading to the collection of evidence for the phase III clinical study on the efficacy and safety in the indicated patients.
2 Materials and Methods
The Ethics Committee of Jilin University First Hospital Clinical Research Institute in Changchun City (Jilin Province, China) approved our protocol. All clinical procedures were performed in our hospital (Phase I Clinical Trial Unit, The First Hospital of Jilin University). This study was conducted in agreement with the Declaration of Helsinki and the Guidelines for Good Clinical Practice. The registration number of this study is NCT04371185 (https://www.clinicaltrials.gov/). All participants provided written informed consent for this study.
2.1 Drugs for Evaluation
BAT2206 injection (specification: 45 mg), a recombinant human IL-12 monoclonal antibody solution, was manufactured by Bio-Thera Solutions, Ltd. Reference drugs: EU-sourced ustekinumab and USA-sourced ustekinumab (Stelara®; specification: 45 mg) were purchased from Janssen-Cilag International NV (Schaffhausen, Switzerland) and Janssen Biotech, Inc. (Malvern, PA, USA), respectively. All subjects received BAT2206 or ustekinumab from a single lot.
2.2 Subjects
The eligibility of participants was healthy male individuals aged from 18 to 55 years with a body weight between 55 and 85 kg and body mass index values between 18 and 28 kg/m2. Subjects with the following conditions were excluded from this study: diagnosis of cardiovascular disease and diseases of the central nervous system, liver, kidney, or other organs; abnormalities in vital signs, electrocardiograms, clinical laboratory tests (blood routine, biochemical test, thyroid function, antinuclear antibodies, C-reactive protein, rheumatoid factors), or chest X-ray image and ultrasonography; active infections including acute and chronic infections and local infections, within 2 months before screening; had used ustekinumab (or its biosimilar) or any anti-IL-12/23, anti-IL-12, or anti-IL-23 agents or had received treatment with tumor necrosis factor-targeted or IL-17-targeted agents within 6 months prior to screening, or use of any biological product or monoclonal antibody within 3 months prior to screening; has severe allergies caused by food or drugs; had taken alcohol or alcohol-containing drinks within 24 hours prior to assignment of a test drug; had positive results from the tuberculosis enzyme-linked immune SPOT test; or had hepatitis B virus, hepatitis C virus, or human immunodeficiency virus.
2.3 Design of Study and Drug Administration
This was a randomized, double-blind, single-dose, parallel-group clinical trial for the evaluation of the pharmacokinetics, immunogenicity, and safety of BAT2206 injection compared to ustekinumab (EU) or ustekinumab (USA) in Chinese healthy male subjects. For ustekinumab, the inter-individual coefficient of variation of area under the plasma concentration–time curve from time zero to infinity (AUC0–inf) and Cmax did not exceed 36% [8, 9]. Given that the geometric mean ratio (GMR) of AUC0–inf and Cmax was equal to 0.95, we considered the power (1 – β) to be 90%, the individual coefficient of variation as 36%, and the one-sided test level α as 0.05. Thus, the 20% shedding rate was considered. Therefore, 270 subjects were enrolled (90 subjects for BAT2206, 90 subjects for ustekinumab [EU], and 90 for ustekinumab [USA]). The participants were randomly assigned at a 1:1:1 ratio to receive either BAT2206, ustekinumab (EU), or ustekinumab (USA) via a subcutaneous injection (45 mg/0.5 mL).
In a previous report on the population PK profiles using data from patients with psoriasis, body weight was found to be the most significant covariate factor that affects the clearance of ustekinumab. Additionally, in patients with Crohn’s disease and ulcerative colitis, body weight was the main covariate factor that affects the volume of distribution of ustekinumab. Therefore, body weight may have a significant influence on PK parameters, according to an analysis by the Summary of Product Characteristics of Stelara® [9]. Randomization was based on body weight stratification factors (weight ≥ 55 kg and < 65 kg, weight ≥ 65 kg and < 75 kg, weight ≥ 75 kg and < 85 kg), which were assigned by a computer-generated randomization schedule. In this way, the comparable distribution of body weight in the three arms can be ensured, allowing for distinguishing the differences in PK profiles between the proposed drug and the reference drug. All the enrolled subjects received subcutaneous administration on day 1 and were then allowed to leave the clinical study site after completing the required safety monitoring for at least 9 days. After that, subjects were required to return to the clinical site for safety evaluations, and blood tests of PK profiles on days 10, 11, 13, 15, 22, 29, 43, 57, 71, 85, 99, and 113 (end of the study period).
2.4 Safety Evaluation
In this study, adverse events (AEs) were reported from initial drug administration to 113 days post-dose. The definition of AEs was according to the National Cancer Institute Common Terminology Criteria for the Classification of Adverse Events, version 5.0. Assessment of AEs included the following characteristics: the degree of severity (mild, moderate, and severe), duration of symptom severity, AE-related clinical outcomes, and association with the test drug. Additional parameters included physical examinations such as body temperature, sitting blood pressure, and heart rate, as well as electrocardiogram findings and clinical laboratory tests such as hematology, urinalysis, and biochemistry tests.
2.5 PK Assessment
To study the PK profile, the blood samples were collected at the following timepoints: 0 hours (pre-dose), 4 hours, 12 hours, and 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 13, 15, 22, 29, 43, 57, 71, 85, 99, and 113 days post-dose. After 0.5 hour at room temperature, blood samples were centrifuged at 2000g at 4 °C for 15 minutes. The serum samples were stored at − 60 °C until subsequent measurement of the drug concentration by a validated enzyme-linked immunosorbent assay (Covance Inc., Shanghai, China). The lower limits of quantification of the assay for serum BAT2206 or ustekinumab were 50.0 ng/mL, and the calibration range was 50.0–1600 ng/mL. The accuracy was − 2.5 to 3.2%, with the precision within the 3.9% coefficient of variation.
Pharmacokinetic evaluations included the following parameters: Cmax, time to Cmax, area under the plasma concentration–time curve from time 0 to 672 hours (AUC0–672), AUC over the dosing interval (AUC0–t), AUC0–inf, elimination half-life, first-order rate constant of drug associated with the terminal portion of the curve, apparent clearance (CL/F), and apparent volume of distribution. The calculation of PK parameters was performed using WinNonlin software (version 8.0; Pharsight Corporation, St. Louis, MO, USA) with the non-compartment model.
2.6 Immunogenicity Evaluation
Anti-drug antibody tests were performed at 0 hours (before drug administration) and on days 10, 15, 29, 57, 85, and 113. If the subjects had positive ADA results, a neutralizing antibody (Nab) was detected. An electrochemiluminescence assay was used for the quantification of ADAs and Nab that reacted to BAT2206 or ustekinumab [10, 11]. Anti-drug antibodies and Nab were analyzed by Covance Inc.
This electrochemiluminescence method was designed to detect the presence of anti- BAT2206, as well as the neutralizing antibodies of ustekinumab (USA) and ustekinumab (EU) in human serum. The control sample was prepared by adding an anti-BAT2206 positive control antibody to the mixed human serum. Samples were subjected to acid treatment and incubated with Sulfo tag-labeled BAT2206/ustekinumab (USA)/ustekinumab (EU) to form the complexes. After incubation, the antibody complex was added to the MSD plate coated with Pp40-Fc fusion protein. Pp40-Fc protein on the plate-bound to Sulfo Tag-labeled BAT2206/ustekinumab (USA)/ustekinumab (EU) would generate an electrochemiluminescence response. The plate was washed to remove any non-specific bound complexes; then a reading buffer (2X) was added to each well of the plate. The plate was read on the MSD Quickplex 120 or equivalent. The binding of free Sulfo tag-labeled BAT2206 to the Pp40-Fc protein could generate a maximal luminescent signal, meanwhile, the intensity of the signal will decrease in the presence of Nab.
2.7 Statistical Analyses
The serum concentration versus time profile of three drugs (BAT2206, ustekinumab [EU], or ustekinumab [USA]) was determined by calculations using WinNonlin software with the non-compartment model. SAS Enterprise Guide 9.4 software was used for data analysis. Analysis of variance was used to compare the differences in PK parameters between the two drugs, including primary PK parameters (Cmax and AUC0–inf) and secondary PK parameters (AUC0–t, AUC0–672 h), after logarithmic transformation. In the model, the experimental groups were used as fixed effects, whereas body weight stratification (weight ≥ 55 kg and < 65 kg, weight ≥ 65 kg and < 75 kg, weight ≥ 75 kg and < 85 kg) was used as covariables. The standard bioequivalence margin was used for setting the criteria of PK similarity. The PK similarity of BAT2206 and its reference ustekinumab (EU or USA) could be determined by evaluating the mean log-transformed value of the 90% confidence interval (CI) for the GMR of test/reference. If the ratio of the values of two drugs was close, within the range of 0.8–1.25, the pairwise PK similarity of BAT2206 with ustekinumab (EU or USA) could be considered, according to the Guidelines for Bioequivalence Studies of Generic Products [12].
3 Results
3.1 Demographics
A total of 270 healthy male subjects were enrolled in the study, of whom 266 (98.5%) completed the study, in the treatment groups of BAT2206 (n = 90), ustekinumab (EU) (n = 88), and ustekinumab (USA) (n = 88) (Fig. 1). The summary of the demographic characteristics with the baseline is shown in Table 1. Age, height, and body mass index were similar across treatment groups. The number of subjects in the same weight stratification was evenly distributed among the groups.

Scheme for the clinical trial with the reasons for premature withdrawal before dosing. EU European Union, US USA
3.2 Assessment of Drug Tolerability
All subjects dosed were included in the safety analysis. There were no deaths, serious AEs, or discontinuations due to treatment-emergent AEs (TEAEs). A total of 425 clinical TEAEs were reported in 186 (69.1%) subjects with medication administration, including 63 (70.0%) subjects in the BAT2206 group, 63 (70.8%) subjects in the ustekinumab (EU) group, and 60 (66.7%) subjects in the ustekinumab (USA) group. The TEAE incidence was similar among the three treatment groups. Among all TEAEs, only 13 were Grade 3 or above (Common Terminology Criteria for the Classification of Adverse Events version 5.0), which were observed in ten (3.7%) subjects, including three (3.3%) subjects in the BAT2206 group, three (3.4%) in the ustekinumab (EU) group, and four (4.4%) in the ustekinumab (USA) group. Among the 13 cases, two cases of hypertriglyceridemia were drug related and were reported in two subjects (one in the BAT2206 group and the other in the ustekinumab (EU) group). The most frequently (≥ 5%) occurring drug-related TEAEs in any of the three groups (BAT2206 vs ustekinumab [EU] vs ustekinumab [USA]) were hypertriglyceridemia (11.1% vs 11.2% vs 7.8%), hyperglycemia (5.6% vs 3.4% vs 4.4%), elevated alanine aminotransferase (7.8% vs 5.6% vs 6.7%), elevated neutrophil count (5.6% vs 2.2% vs 3.3%), decreased neutrophil count (5.6% vs 2.2% vs 3.3%), elevated white blood cell count (5.6% vs 3.4% vs 4.4%), elevated hemobilirubin (5.6% vs 12.4% vs 5.6%), and hematuria (7.8% vs 1.1% vs 2.2%). The most common AEs (> 5%) are shown in Table 2.
3.3 PK Analysis
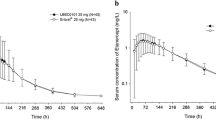
As shown in Fig. 2, the mean serum concentration–time profile between the BAT2206 and ustekinumab groups (EU/USA) exhibited a similar trend. Pharmacokinetic parameters including Cmax, AUC0–t, and AUC0–inf were comparable among the three treatment groups, median time to Cmax was 6–7 days after dosing, and similar values of CL/F and apparent volume of distribution were found. Other PK parameters (AUC0–672h, elimination half-life, first-order rate constant of drug associated with the terminal portion of the curve, and CL/F) were not significantly different between the two drugs (p > 0.05; Table 3). Assessment of the PK profiles between the two drugs is shown in Table 4. For the primary PK parameters, Cmax and AUC0–inf, after a single subcutaneous of BAT2206 or ustekinumab (EU or USA), the 90% CIs for the ratios of GMs were constrained within 0.80–1.25, supporting the pairwise bioequivalence between the two drugs.

Mean serum concentration–time profiles after a single 45-mg subcutaneous administration of BAT2206, ustekinumab (European Union [EU]) and ustekinumab (USA [US]). h hours
3.4 Assessment of Drug Immunogenicity
The lower detectable limit of the ADA assay used in the study was 4.97 ng/mL for the screen test and 5.47 ng/mL for the confirmation test. Overall, 54 (20.1%) subjects had positive results from the ADA test. Among them, 24 subjects (26.7%) were in the BAT2206 group, while 13 subjects (14.8%) were in the ustekinumab (EU) group, and 17 (18.9%) were in the ustekinumab (USA) group. Only one subject (S0606) in the EU-sourced ustekinumab group was ADA positive at baseline, and the others were ADA positive after dosing. The median time of the first detection of ADA positivity in the BAT2206, ustekinumab (EU), and ustekinumab (USA) groups was around 85, 57, and 57 days post-injection, respectively. The number of NAb-positive subjects in the BAT2206, ustekinumab (EU), and ustekinumab (USA) groups was eight (8.9%), five (5.7%), and six (6.7%), respectively. A descriptive summary of the effects of positive ADA on PK parameters is shown in Table 5. There were more ADA-positive subjects in the BAT2206 group than in the ustekinumab group. Furthermore, there was no significant difference in mean serum concentration–time curves for the ADA-positive and total participants among the three treatment groups (Fig. 3). If the ADA-positive and ADA-negative subjects were included in the analysis, the 90% CIs for Cmax and AUC0–inf were within 0.8–1.25. Anti-drug antibody-positive subjects were excluded for the sensitivity analysis. The results showed that after the exclusion of ADA-positive subjects, the 90% CIs for Cmax and AUC0–inf were still within 0.8–1.25, which is an acceptable range of PK similarity.

Mean serum concentration (mean ± standard deviation)–time curves in anti-drug antibody (ADA)-positive subjects/ADA-negative subjects/all subjects (linear and log-linear data are presented in the left-handed and right-handed panels, respectively). A BAT2206 injection versus ustekinumab (European Union). B BAT2206 injection versus ustekinumab (USA). C Ustekinumab (European Union) versus ustekinumab (USA). h hours
4 Discussion
This is a parallel, randomized, phase I clinical trial evaluating the PK profiles between BAT2206 and its reference drug ustekinumab (EU) and ustekinumab (USA), which was conducted with a single subcutaneous injection (45 mg). Analyses of primary PK parameters (Cmax and AUC0–inf) in the three treatment groups (BAT2206 and ustekinumab [EU and USA]) revealed that the 90% CIs for the GMRs were constrained within the range of 0.80–1.25, matching the predetermined PK bioequivalence range.
The incidence of ADA in the BAT2206 group was higher than those in the other two groups, whereas the incidences of positive NAb were similar among the three groups. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including Nab) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, the timing of sample collection, concomitant medications, and underlying disease [9]. After being treated with ustekinumab, approximately 6–12.4% of the patients with psoriasis and psoriatic arthritis and 2.9% or 4.6% of the patients with Crohn’s disease or ulcerative colitis may develop antibodies against ustekinumab [9]. Based on the information as mentioned above, it may not be reasonable to directly compare the incidence of ADA among the groups or with the reference data. Nevertheless, the immunogenicity of BAT2206 and ustekinumab is to be further evaluated in a comparative study of efficacy.
Regarding the association of the development of antibodies against ustekinumab with safety and pharmacokinetics, no significant influence of positive ADA was found on safety, and only very small differences in PK profiles were observed between ADA-positive and ADA-negative subjects. As shown in Fig. 3, there was a difference in the mean plasma concentration [ng/mL] (mean ± SD)–time curves of ADA-positive/ADA-negative/all subjects between the BAT2206 injection group and the ustekinumab (EU and USA) group. The mean plasma concentration–time curves of ADA-positive subjects, ADA-negative subjects, and all subjects were highly similar among the three treatment groups. In addition, at the end of curves, plasma concentrations of ADA-positive subjects tended to be slightly lower than those of ADA-negative subjects and all subjects in the three treatment groups. The primary PK parameters of ADA-positive/ADA-negative/all subjects were comparable (Table 5). A sensitivity analysis excluding the ADA-positive subjects further demonstrated that BAT2206 and ustekinumab (USA) and ustekinumab (EU) were still pairwise similar, indicating that positive ADA had little effect on the primary PK parameters (data not shown). In all three groups, the elimination half-life of the ADA-positive subjects was slightly lower than that of ADA-negative subjects and all subjects, and the first-order rate constant of drug associated with the terminal portion of the curve and CL/F were slightly higher than those of ADA-negative subjects and all subjects, indicating that the terminal elimination of ADA-positive subjects was slightly faster. The primary PK parameters of ADA-positive/ADA-negative/all subjects were comparable, and positive ADA had little effect on the primary PK parameters.
Anti-drug antibody positivity had no significant impact on safety, and no subjects had clinically significant hypersensitivity, severe hypersensitivity, rapid anaphylaxis, or injection site-related reactions as a result of ADA. The incidence of ADA in the BAT2206 injection group was higher than that in the other two groups, and the first detection time was longer, which needs to be further evaluated in the efficacy comparison study.
Twenty-nine subjects withdrew prematurely (n = 4) or missed visits (n = 25). The sensitivity analysis of the PK similarity was conducted, after the removal of AUC0–t, AUC0–inf, and AUC0–67 2h data of the above-mentioned subjects. The pairwise comparisons for GMR among the three treatment groups showed that 90% CIs were all within the equivalent interval (80.00–125.00), demonstrating that the BAT2206 injection was still pharmacokinetically similar to ustekinumab (EU sourced and US sourced) after excluding the above data.
The assessment of the safety of the three drugs revealed that there were no severe AEs in all three treatment groups, indicating good tolerance of those three drugs in healthy Chinese subjects. Two Grade 3 AEs of hypertriglyceridemia were reported in two subjects (one in the BAT2206 group and the other in the EU ustekinumab group). They were considered drug related by the investigators, but long-term hospitalization or a high-fat diet outside the hospital cannot be ruled out because they may be the cause. A few TEAEs related to infection and infestations were also reported, such as upper respiratory tract infection, urinary tract infection, and body tinea. Incidences of those symptoms were lower than 5%. For example, there were a total of seven cases of upper respiratory tract infection, including two cases in the BAT2206 group, two cases in the ustekinumab (EU) group, and three cases in the ustekinumab (USA) group. The incidence of upper respiratory tract infection was 2.6%. Therefore, the incidences of TEAEs related to infection were considered low.
Because of coronavirus disease 2019, 47 subjects had 92 visits missing or out of the window from days 43 to 113. This was not considered a significant protocol deviation and did not affect the trial conclusion.
One limitation of our study was that it only included male subjects. However, the PK differences between test drugs and reference drugs are better reflected by using a single sex to minimize the effects of physiological differences on the results. Additionally, because of the small sample size in each group and only a single dose used in this study for a short period of observation, the immunogenicity of the treatment with the corresponding drugs may not be fully detected. Therefore, the immunogenicity of BAT2206 and ustekinumab will be further evaluated in a subsequent comparison study of efficacy.
5 Conclusions
Our study confirmed the similar PK characteristics of a 45-mg single subcutaneous injection of BAT2206 in Chinese healthy subjects, compared with ustekinumab (EU or USA sourced). The incidence of ADA in the BAT2206 group was higher than those in the other two groups, whereas the incidences of positive NAb were similar among the three groups, and no significant influence of positive ADA on safety or pharmacokinetics was observed. The three groups had similar safety profiles, low immunogenicity, and all were well tolerated by the healthy Chinese male subjects, suggesting that BAT2206 is a high-potential biosimilar of ustekinumab in clinical application.
References
Tam A, Geier KA. Psoriatic arthritis. Orthop Nurs. 2004;23(5):311–4.
Chandran V, Raychaudhuri SP. Geoepidemiology and environmental factors of psoriasis and psoriatic arthritis. J Autoimmun. 2010;34(3):J314–21.
Ding X, Wang T, Shen Y, Wang X, Zhou C, Tian S, et al. Prevalence of psoriasis in China: a population-based study in six cities. Eur J Dermatol. 2012;22(5):663–7.
Gelfand JM, Stern RS, Nijsten T, Feldman SR, Thomas J, Kist J, et al. The prevalence of psoriasis in African Americans: results from a population-based study. J Am Acad Dermatol. 2005;52(1):23–6.
Wu Y, Chen J, Li YH, Ma GZ, Chen JZ, Gao XH, et al. Treatment of psoriasis with interleukin-12/23 monoclonal antibody: a systematic review. Eur J Dermatol. 2012;22(1):72–82.
Perrotta FM, Delle Sedie A, Scriffignano S, Volpe P, Cordisco E, Milano N, et al. Remission, low disease activity and improvement of pain and function in psoriatic arthritis patients treated with IL-12/23 and IL-17 inhibitors: a multicenter prospective study. Reumatismo. 2020;72(1):52–9.
Benson JM, Peritt D, Scallon BJ, Heavner GA, Shealy DJ, Giles-Komar JM, et al. Discovery and mechanism of ustekinumab: a human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. MAbs. 2011;3(6):535–45.
Gao L, Li Q, Zhang H, Wu M, Fang M, Yang L, et al. Biosimilarity study between QX001S and ustekinumab in healthy Chinese male subjects. Front Pharmacol. 2021;18(12):675358. https://doi.org/10.3389/fphar.2021.675358 (eCollection 2021).
Centocor Ortho Biotech Inc. Stelara™ (ustekinumab): prescribing information. 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125261s154,761044s006lbl.pdf. Accessed Dec 2020.
Marchese RD, Puchalski D, Miller P, Antonello J, Hammond O, Green T, et al. Optimization and validation of a multiplex, electrochemiluminescence-based detection assay for the quantitation of immunoglobulin G serotype-specific antipneumococcal antibodies in human serum. Clin Vaccine Immunol. 2009;16(3):387–96.
Blackburn GF, Shah HP, Kenten JH, Leland J, Kamin RA, Link J, et al. Electrochemiluminescence detection for development of immunoassays and DNA probe assays for clinical diagnostics. Clin Chem. 1991;37(9):1534–9.
Pharmaceuticals and Medical Devices Agency, Tokyo, Japan. Guidelines for bioequivalence studies of generic products. 2015. http://www.pmda.go.jp/files/000157415.pdf. Accessed 12 Jun 2015.
Acknowledgments
The authors appreciate the participation of the volunteers and the contribution of the staff to this trial.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was financially supported by the Capital Construction Funds within the provincial budget in 2020 (innovation capacity construction, Project No. 2020C038-1).
Conflicts of Interest/Competing Interests
YQ, JY, XY, ZW, and CG are employees of Bio-Thera Solutions, Ltd. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the article. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Ethics Approval
The Ethics Committee of Jilin University First Hospital Clinical Research Institute in Changchun City (Jilin Province, China) approved the protocols of our study.
Consent to Participate
All participants provided written informed consent for this study.
Consent for Publication
Not applicable.
Availability of Data and Material
Data and material are available from the corresponding author upon reasonable request.
Code Availability
The code is available from the corresponding author upon reasonable request.
Authors’ Contributions
MW and YD performed a review of the topic, and wrote and revised the manuscript. HZ and DY took part in analyzing the pharmacokinetic data. YQ, JY, and XL drafted part of the manuscript. YQ, JY, XY, ZW, and CG helped with the analysis and interpretation of the data, and prepared all the figures and tables. YD contributed to the writing of the manuscript and provided critical revisions. All authors approved the final version of the manuscript and agreed to be accountable for all aspects of the work.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, M., Li, X., Yang, D. et al. Comparison of Pharmacokinetic Similarity, Immunogenicity, and Safety of Ustekinumab and BAT2206 in Healthy Chinese Male Subjects in a Double-Blind, Randomized, Single-Dose, Parallel-Group Phase I Trial. BioDrugs 37, 89–98 (2023). https://doi.org/10.1007/s40259-022-00563-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-022-00563-5