
Abstract
Angiopoietin-like protein 3 (ANGPTL3) and apolipoprotein C-III (apoC-III) are novel metabolic targets for correcting hypertriglyceridaemia (HTG). As a background to their potential clinical use, we review the metabolic aetiology of HTG, particular abnormalities in triglyceride-rich lipoproteins (TRLs) and their role in atherosclerotic cardiovascular disease (ASCVD) and acute pancreatitis. Molecular and cardiometabolic aspects of ANGPTL3 and apoC-III, as well as inhibition of these targets with monoclonal antibody and nucleic acid therapies, are summarized as background information to descriptions and analyses of recent clinical trials. These studies suggest that ANGPTL3 and apoC-III inhibitors are equally potent in lowering elevated plasma triglycerides and TRLs across a wide range of concentrations, with possibly greater efficacy with inhibition of apoC-III. ANGPTL3 inhibition may, however, have the advantage of greater lowering of plasma LDL cholesterol and could specifically address elevated LDL cholesterol in familial hypercholesterolaemia refractory to standard drug therapies. Large clinical outcome trials in relevant populations are still required to confirm the long-term efficacy, safety and cost effectiveness of these potent agents for mitigating the complications of HTG. Beyond targeting severe chylomicronaemia in the prevention of acute pancreatitis, both agents could be useful in addressing residual risk of ASCVD due to TRLs in patients receiving best standard of care, including behavioural modifications, statins, ezetimibe, fibrates and proprotein convertase subtilisin/kexin type 9 inhibitors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Hypertriglyceridaemia, specifically due to elevated triglyceride-rich lipoproteins (TRLs), is associated with increased risk of atherosclerotic cardiovascular disease; severe chylomicronaemia can also cause acute pancreatitis. |
Treatments that target the production and clearance of TRLs mitigate these complications. |
Nucleic acid-based therapies that target ANGPTL3 or apoC-III offer a new approach to lowering elevated TRLs, with preliminary studies testifying to their utility in clinical practice. |
ANGPTL3 inhibition also lowers LDL particle concentration and may therefore have an overall advantage as a lipid-regulating agent. |
Large clinical outcome trials are needed to confirm the efficacy, safety, cost effectiveness and clinical value of these new agents. |
1 Introduction
Beyond the significant causal role of elevated low-density lipoprotein (LDL) cholesterol, epidemiological and Mendelian randomization studies have demonstrated that elevated plasma concentrations of triglycerides, which reflect the accumulation of triglyceride-rich lipoproteins (TRLs), are predictive of atherosclerotic cardiovascular disease (ASCVD) [1, 2]. However, treatments that lower plasma triglycerides have not been consistently shown to reduce cardiovascular outcomes. Angiopoietin-like protein 3 (ANGPTL3) and apolipoprotein C-III (apoC-III) are two new metabolic targets for the specific treatment of hypertriglyceridaemia (HTG). This article summarizes the aetiology and current treatment of HTG and reviews the recent clinical trials that may elucidate the therapeutic role of inhibitors of ANGPTL3 and apoC-III.
2 Metabolism of Triglyceride-Rich Lipoproteins (TRLs)
TRLs are composed of both exogenous and endogenous triglycerides. Triglycerides that originate from dietary fat are absorbed following a meal by enterocytes, where they combine with apolipoprotein (apo) B-48 to form chylomicrons, entering the circulation and picking up apoC-II, apoC-III and apoE [3, 4]. Within the circulation, chylomicrons are hydrolysed by lipoprotein lipase (LPL) to produce free fatty acids and chylomicron remnants; LPL activity is highly regulated by various proteins including apoC-II, apoC-III, apoA-V, ANGPTL3 and ANGPTL4 [5, 6]. The released free fatty acids are then used by various cell types as a fuel source or resynthesized with glycerol into triglycerides and stored. Chylomicron remnants, now rich in cholesterol esters and apoE, are removed from the circulation via the LDL receptor or LDL receptor-related proteins in the liver [7].
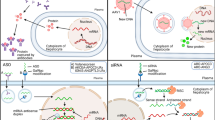
Triglycerides synthesized in the liver from free fatty acids and glycerol can form very low-density lipoprotein (VLDL) particles in association with apoB-100. During secretion, apoC-I, apoC-II, apoC-III and apoE are added to the surface of VLDL and the particles are then hydrolysed in the circulation by LPL, producing progressively smaller VLDL particles and then intermediate-density lipoproteins (IDLs); IDLs are taken up by the liver and catabolized or further catabolized by LPL and triglyceride lipase to form LDL particles (Fig. 1) [3, 4]. Significantly, ANGPTL3 and apoC-III are negative regulators of the clearance of all TRLs from the circulation at the level of LPL and hepatic receptors.

Schematic representation of the putative mode of action of angiopoietin-like protein 3 (ANGPTL3) and apolipoprotein C-III (apoC-III) inhibition on lipoprotein transport in the circulation in humans
3 Hypertriglyceridaemia
Elevated plasma triglycerides or HTG may be defined as borderline (mild), moderately elevated, severe and extreme, according to progressive gradations in plasma triglyceride concentrations (Table 1). A commonly agreed global definition of HTG is a plasma triglyceride concentration persistently ≥ 1.7 mmol/L (≥ 150 mg/dL) [8]. Over 95% of people susceptible to HTG carry multiple genes that interact with non-genetic factors and perturb the metabolism of TRLs [1, 9,10,11,12].
A subset of severe HTG is a rare form (1–10 per million) of monogenic HTG, called familial chylomicronaemia syndrome (FCS) due to mutations in one of at least six genes (LPL, APOC2, LMF1, GPHIHBP1, APOA5, G3PDH1) [9, 10]. Homozygous or compound heterozygous mutations in these genes markedly impair chylomicron clearance, leading to severe HTG that can manifest in youth as eruptive xanthomata, lipaemia retinalis and acute pancreatitis. Multifactorial chylomicronaemia syndrome (MCS) has a prevalence of 1 in 600 of the population, resulting from a combination of heterozygous variants in the six FCS genes and/or accumulated common small-effect triglyceride-raising polymorphisms identified in genome-wide association studies (GWAS), such as APOA1-C3-A4-A5, TRIB1, LPL, MLXIPL, GCKR, FADS1-2-3, NCAN, APOB, PLTP and ANGPTL3 [9, 10].
E2E2 homozygosity (prevalence 1%) is a necessary but not sufficient cause of type III dysbetalipoproteinaemia, which is rare (1 in 10,000) and causes HTG and premature atherosclerosis due to accumulation of TRL remnants that are not cleared by the liver [13].
Mild-to-moderate HTG is usually a result of multiple common allelic gene variants interacting with an unhealthy diet and lifestyle that impair TRL metabolism [12]. The most common forms of genetic HTG are familial combined hyperlipidaemia (FCHL) and familial hypertriglyceridaemia [14, 15]. FCHL can present as elevated total and LDL cholesterol levels with mild triglycerides, elevated triglycerides alone, or predominately elevated triglycerides and slightly elevated LD. FCHL is characterized by an increase in apoB and small dense LDL particles and is often seen in patients with metabolic syndrome, central obesity, type 2 diabetes mellitus (T2DM) and a family history of premature coronary artery disease (CAD). FCHL results from hepatic overproduction of VLDL apoB particles that may be variably enriched in triglycerides and impair clearance of apoB-containing particles from the circulation [16]. Familial hypertriglyceridaemia (type IV hyperlipoproteinaemia), also a multigenic condition [15], is due to an increase in triglyceride content in VLDL particles with no concomitant elevation of apo B concentration. The precise kinetic defect remains unclear but may be caused by increased triglyceride synthesis and secretion of very large triglyceride-enriched VLDL particles, and/or impaired clearance of VLDL-TGs [17, 18].
Secondary causes of HTG can also include T2DM, hypothyroidism, pregnancy, hepatosteatosis, significant weight re-gain after weight loss, nephrotic syndrome and some medications [1, 4, 12]. As reviewed by others [3, 4, 6, 12, 19], increased availability of free fatty acids (FFAs) from de novo lipogenesis (DNL) from glucose and the adipose tissue through increased lipolysis increases hepatic triglyceride synthesis, leading to overproduction of large triglyceride-rich VLDL particles in patients with T2DM. Furthermore, insulin resistance increases chylomicron particle concentrations by stimulating DNL, increasing microsomal triglyceride transfer protein activity and enhancing intracellular chylomicron stability in the intestine. It also increases FFAs delivery to the enterocytes, impairs insulin signalling and increases intestinal lipid absorption during the postprandial period. Collectively, these effects could increase the enterocytic secretion of apoB-48-containing chylomicrons. Increased competition between chylomicron and VLDL remnants for hepatic receptors also delays the uptake of TRL remnants by this pathway. Insulin resistance also increases apoC-III synthesis, decreases LPL production and downregulates LDL receptor expression, limiting TRL lipolysis and removal [3, 6]. The role of ANGPTL3 and apoC-III in the pathogenesis of HTG is reviewed later.
4 Hypertriglyceridaemia, Atherosclerotic Cardiovascular Disease and Acute Pancreatitis
Several Mendelian randomization studies have consistently demonstrated that genetically elevated remnant cholesterol and apoB-containing TRLs increase the risk of ASCVD events [20,21,22]. An observational analysis from the Copenhagen General Population Study and the Copenhagen City Heart Study has demonstrated that non-fasting plasma triglycerides (≥5 mmol/L) or remnant cholesterol (>2.3 mmol/L) increases risk for aortic stenosis, ischaemic stroke and myocardial infarction by 1.5-fold, 3-fold and 5-fold, respectively [22,23,24]. In a meta-regression analysis of randomized controlled statin and non-statin trials, triglyceride lowering (per 1 mmol/L reduction) is associated with 16% lower risk of major vascular events after adjusting for LDL cholesterol [25]. In a Canadian cohort, approximately 25% of patients with ASCVD had HTG despite controlled LDL cholesterol in the general population [26].
Experimental evidence suggest that small TRL remnants readily infiltrate the subendothelial space, where they are trapped by a connective tissue matrix. These particles are rapidly phagocytosed by arterial wall macrophages, which are then transformed into ‘foam cells’ [27]. Furthermore, TRL remnants impair endothelial function, inhibit fibrinolysis, enhance coagulation and activate monocytes and inflammation. TRL lipolysis also releases toxic products, such as oxidized fatty acids and lysolecithin, that induce endothelial cell inflammation and coagulation [28,29,30,31,32].
Severe HTG is known to increase risk of acute pancreatitis, which accounts for 1–10% of episodes of acute pancreatitis. Mild-to-moderate HTG (non-fasting plasma triglyceride levels ≥ 5 mmol/L) is associated with a 10-fold increase in risk of acute pancreatitis. The mechanism for how HTG contributes to the development of acute pancreatitis is unclear, but may principally involve lipotoxicity and low-grade inflammation [33, 34].
5 Treatment Guidelines for Hypertriglyceridaemia and the REDUCE-IT Trial
TRLs are well recognized as a risk-enhancing factor and target for therapy in recent international lipid guidelines and position statements [4, 8, 35, 36]. The general consensus is to optimize lifestyle and behavioural interventions as a first step, after ruling out secondary causes, followed by statin therapy in those with moderate HTG and elevated 10-year risk of ASCVD. In patients with severe HTG, dietary therapy followed by use of a fibrate is recommended to mitigate risk of acute pancreatitis. Importantly, the use of high pure eicosapentaenoic acid (EPA), as in the REDUCE-IT (Reduction of Cardiovascular Events with Icosapent Ethyl—Intervention Trial) [37], has been incorporated in the most recent American Heart Association/American College of Cardiology (AHA/ACC) guideline for persistent HTG in high-risk patients receiving statin treatment [8]. The use of fibrates, in particular fenofibrate, has been recommended as an add-on to statin therapy for ASCVD prevention in high-risk patients only in the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS) guidelines [35].
REDUCE-IT investigated the effect of icosapent ethyl on ischaemic events in statin-treated patients with elevated triglycerides and cardiovascular disease (CVD) or diabetes [37]. In 8179 patients who were followed for a median of 4.9 years, high-dose icosapent ethyl (4 g/day) significantly reduced the burden of first, subsequent and total ischaemic events [37]. Although icosapent ethyl effectively lowers triglycerides, it also has anti-inflammatory and plaque stabilizing properties, which might have contributed to the benefits seen in the trial. It was also recently suggested that the beneficial effects seen in REDUCE-IT are largely attributable to the negative effect of the placebo corn oil on events [38]. The STRENGTH trial investigated a carboxylic formulation of omega-3 fatty acids on major cardiovascular events in 13,078 statin-treated patients with HTG and high ASCVD risk [39]. However, this study was terminated prematurely owing to futility. A particular complication of high-dose EPA is an attendant risk of atrial fibrillation [40]. An outcome trial is currently underway to investigate the effect of EPA on the incidence of cardiovascular events in patients with CAD (RESPECT-EPA; UMIN000012069).
An international consensus statement testifying to the potential value of pemafibrate, a new selective peroxisome proliferator-activated receptor alpha modulator (SPPARMα), supports the PROMINENT study [41]. The study is currently underway and due for completion in 2022, and is addressing the effect of pemafibrate on cardiovascular outcomes in statin-treated patients with T2DM and mild-to-moderate HTG [42]. Beyond pure EPA and SPPARMα, inhibition of ANGPTL3 and apoC-III could pave the way for more effective and sustained reductions in plasma triglycerides and TRLs and the inclusion of these agents in future clinical practice.
6 Angiopoietin-Like Protein 3 (ANGPTL3): Molecular and Cardiometabolic Aspects
Angiopoietin-like protein 3 (ANGPTL3) is a 460 amino acid glycoprotein that is secreted predominately by the liver, where its expression is activated by the oxysterol-stimulated liver X receptor [5]. It appears to be active predominately after feeding [43], where it works with ANGPTL8 to inhibit LPL, the enzyme responsible for hydrolysis of circulating triglycerides (Fig. 1). ANGPTL3 also inhibits endothelial lipase (EL) in vitro [44]. Composed of an N-terminal signal peptide, an N-terminal coiled-coil domain, a linker region and a C-terminal fibrinogen-like domain, it shares high sequence homology with both ANGPTL4 and ANGPTL8 [5]. ANGPTL3 undergoes cleavage and glycosylation to give an N-terminal fragment containing the coiled-coil region and a C-terminal fragment containing the fibrinogen-like domain. Intracellular cleavage is mediated mainly via furin (or proprotein convertase subtilisin/kexin type 3, PCSK3) in hepatocytes, while extracellular cleavage is mediated via paired basic amino acid-cleaving enzyme 4 (PACE4 or PCSK6) [45]. The N-terminal coiled-coil domain is involved in binding and inhibiting both LPL and EL, while the C-terminal fibrinogen domain mediates the angiogenic effects [5]. This inhibitory effect appears to be via a mechanism that involves unfolding and dimeric disassociation of the LPL protein, which is enabled by ANGPTL8 [43]. The inhibitory actions of ANGPTL3 on EL are mechanistically unclear, but appear to result in low plasma HDL levels [44]. ANGPTL3 also has secondary and possibly direct effects on glucose metabolism, with bi-allelic loss of function mutations in ANGPTL3 giving rise to increased insulin sensitivity and low plasma glucose levels [46].
Animal experiments utilising ANGPTL3 loss-of-function (LOF) mutations and large-scale genomic studies in humans provide significant evidence for the inhibition of ANGPTL3 as a therapeutic target in ASCVD [5, 47,48,49,50,51,52]. Carriers of LOF mutations in ANGPTL3 have on average a 35% reduction in the risk of CAD events, with smaller studies demonstrating a direct association between plasma ANGPTL3 and imaging evidence of atherosclerosis [50,51,52,53]. Furthermore, the positive association between plasma ANGPTL3 levels and incident CAD events appears to be independent of plasma lipid levels [51], suggesting that inhibiting ANGPTL3 may have a direct effect on atherosclerosis [47].
7 Apolipoprotein C-III (ApoC-III): Molecular and Cardiometabolic Aspects
Apolipoprotein C-III (apoC-III) is a 79 amino acid glycoprotein that is synthesized predominately in the liver, transported freely among plasma lipoproteins, and inhibits both lipolytic activity and hepatic uptake of TRLs (Fig. 1) [3, 6, 54]. Structurally, it contains two amphipathic helices and aromatic tryptophan residues in the carboxyl-terminal, which appear to be important for interaction with TRLs. After synthesis, apoC-III undergoes post-translational modification that results in at least three different glycoforms—unsialylated apoC-IIIo, monosialylated apoC-III1 and disialylated apoC-III2—which pertains to hepatic clearance [55]. The rate of transcription of apoC-III is stimulated by glucose and decreased by insulin, PPARα and farnesoid X receptor [3, 6, 56]. Accordingly, apoC-III expression is upregulated in insulin resistance, with glycaemic control a major determinant of apoC-III secretion [56]. In the circulation, apoC-III is mainly present in TRLs and HDL, and to a lesser extent on LDL. The distribution of apoC-III among lipoproteins is dependent on the metabolic status of the patient, varying with fasting and feeding, and triglyceride levels [6].
ApoC-III has pro-atherogenic properties and is accordingly predictive of cardiovascular mortality [6, 57, 58]. ApoC-III increases the affinity of LDL for artery wall proteoglycans, increasing subendothelial accumulation of lipoprotein, and increases the enrichment of LDL particles with apoC-III [59, 60]. Animal apoC-III knockout models have been shown to have reduced triglyceride levels and increased protection against atherosclerosis [61]. In humans, LOF mutations in APOC3 are associated with reduced plasma triglycerides and reduced risk for ASCVD [57, 58]. Patients with heterozygous APOC3 LOF mutation (R19X) had 35% lower plasma triglyceride levels due to markedly higher fractional clearance rates of VLDL-TG and VLDL-apoB100 but increased production of LDL-apoB [62]. As reviewed later, this supports the effect of apoC-III inhibitors on LDL cholesterol in patients with severe HTG.
8 Pharmacological Approaches to Inhibit ANGPTL3 and ApoC-III
Several promising therapies, using antibodies or nucleic acid inhibition, targeting ANGPTL3 and apoC-III protein or mRNA, respectively, have been developed and trialled [63,64,65,66,67]. Monoclonal antibodies (mAb) work by binding to and inactivating the target protein of interest. These therapies are complex and expensive to produce and can result in the activation of innate immunity and the development of neutralising auto-antibodies [64]. As critically reviewed by others [63,64,65,66,67], antisense oligonucleotides (ASOs) and small interfering RNA (siRNA) therapies work by inhibiting the mRNA transcripts of a selected protein, halting its translation and resulting in the protein mRNA’s degradation [63,64,65,66,67]. ASOs utilize short, single strands of RNA that bind to the specific mRNA, leading to competitive inhibition of translation or to degradation of the resulting complex. By contrast to ASOs, siRNA therapy utilizes double-stranded RNA, which consists of a guide (or anti-sense) strand and a passenger (or sense) strand, and interacts with an endogenous, multi-enzyme complex in the cytoplasm, called the RNA-induced silencing complex (RISC). The passenger strand loads the RNA duplex onto RISC, which contains a key endonuclease called Argonaute 2 (AGO2) that efficiently effectuates mRNA cleavage (see review by Macchi et al. [63]). Inclusion of a triantennary N-acetylgalactosamine (GalNAc3) complex conjugated to the oligonucleotide or siRNA, enhances liver specificity resulting in greater potency at lower doses as well as reduced systemic toxicity (Table 2) [68].
8.1 Monoclonal Antibody-Based Therapies
Evinacumab is a fully human IgG4 mAb that specifically inhibits ANGPTL3 in the circulation [50, 69] inducing ANGPTL3 deficiency and activating both LPL and EL activity [70, 71]. Bioavailability and lipid-lowering potency is greater with intravenous compared with subcutaneous administration and as it is not metabolized by cytochrome P450, evinacumab does not interact with other drugs [72]. However, as with all other mAb therapies, evinacumab is potentially susceptible to the development of an adaptive immune response and autoantibodies could neutralize its therapeutic effects over time [73].
Monoclonal antibodies targeting apoC-III are difficult to produce owing to the relatively high abundance of the lipoprotein and its association with lipoproteins [74]. STT505 is a humanized mAb that targets lipoprotein-bound human apoC-III and promotes its dissociation from lipoproteins. Although only tested in mice expressing human apoC-III, it has been shown to reduce circulating apoC-III levels by 40–60%, predominately due to increased clearance [75].
8.2 Nucleic Acid Based Therapies
8.2.1 Antisense Oligonucleotide (ASO) Therapy
Vupanorsen (AKCEA-ANGPTL3-LRx) is a ligand-conjugated ASO targeted at ANGPTL3 [76]. Mouse models demonstrated significant reductions in plasma triglyceride concentrations that were associated with reduced hepatic triglyceride content, increased insulin sensitivity and reduced atherosclerosis progression [76]. ASO-mediated inhibition of ANGPTL3 has also recently been shown to increase reverse cholesterol transport in mice, as measured by an in vivo macrophage-to-faeces assay utilizing an injection of 3H-cholesterol-labelled macrophage foam cells [77].
Volanesorsen is a 2’-O-methoxyl-modified single-stranded ASO that decreases the mRNA of APOC3 in animal models and can lower plasma triglycerides in humans by > 70% [78,79,80]. Although not yet approved by the US Federal Drug Administration (FDA), volanesorsen has been granted conditional marketing authorization by the European Medicines Agency as an adjunct to diet and triglyceride-lowering therapy for mitigating acute pancreatitis in genetically defined patients with FCS. Olezarsen (formerly AKCEA-APOCIII-LRx) is a next-generation, ligand-conjugated apoC-III ASO that has similar metabolic effects (reductions in triglycerides and apoC-III) to volanesorsen and a better overall safety profile, particularly an absence of significant thrombocytopaenia [81].
8.2.2 Small Interfering RNA (siRNA) Therapy
The most advanced siRNA to date for therapeutic inhibition of ANGPTL3 is the Targeted RNAi Molecule (TRIM™, Arrowhead Pharmaceuticals). In ARO-ANG3, each RNA strand is 2’-methoxy (or 2’-F) and phosphorothioate modified to induce resistance to endonucleases and offset immune activation [63, 82,83,84]. Furthermore, the sense strand is ligand-conjugated to ensure specific delivery to the liver, with dose-dependent, potent and sustained reductions in serum ANGPTL3 and triglycerides.
APOC3 mRNA may also be targeted with a ligand-bound, double-stranded siRNA platform [63, 66, 67]. As reviewed earlier, this approach leverages RISC and can thereby potently and durably lower plasma apoC-III and triglyceride concentrations in humans [85], with no significant adverse effects, particularly thrombocytopaenia.
9 Clinical Trials
The following section reviews the clinical trials targeting ANGPTL3 and apoC-III, focusing on triglyceride-lowering effects in different populations, including those with HTG and FCS. The findings are summarized in Tables 3 and 4.
9.1 ANGPTL3 mAbs
In a phase I, double-blind, single ascending dose study in 83 healthy volunteers with plasma triglyceride levels between 150 and 450 mg/dL, receiving different subcutaneous (SC) or intravenous (IV) doses of evinacumab, resulted in reductions in fasting triglycerides (− 63%) and LDL cholesterol (− 28%) with the highest IV treatment regimen (20 mg/kg body weight) at day 15 [50]. The results of this single ascending dose study were subsequently combined with a multiple ascending dose study in 56 healthy volunteers [69]. Dose-dependent reductions in triglycerides were observed, with a mean reduction of 78% at 8 weeks with the 20-mg/kg IV dose administered every 4 weeks; a significant reduction in plasma LDL-cholesterol concentration (− 35%) was also observed at this dose, with no serious treatment-related adverse events reported [69]. Kinetic data suggest that evinacumab can decrease VLD-apoB-100 production and increase IDL-apoB-100 clearance in patients with homozygous familial hypercholesterolaemia (FH) [70]. Whether a similar effect of evinacumab also applied to patients with HTG remains to be demonstrated.
9.2 ANGPTL3 Nucleic Acid Therapies
In a phase I dose-finding study in 44 healthy volunteers, vupanorsen resulted in dose-dependent lowering of plasma triglyceride concentrations, with mean reductions after 6 weeks of 60 mg SC weekly being 50%, associated with a decrease in LDL cholesterol (− 33%) and apoB (− 22%) concentrations; no serious adverse events were reported [76]. In a placebo-controlled phase II study in patients with fasting triglycerides > 150 mg/dL, T2DM and hepatic steatosis [86], significant mean reductions in triglycerides of 36%, 53% and 47% at 6 months were observed with 40 mg every 4 weeks (Q4W), 80 mg Q4W and 20 mg weekly, respectively, of SC vupanorsen. However, there was no improvement in indices of insulin resistance, glycaemia and hepatic steatosis. Despite these findings, in a press release on 31 January 2022, Pfizer announced the discontinuation of the clinical development programme for vupanorsen. This was based on two findings from the TRANSLATE-TIMI 70 study: first, the magnitude of lowering triglycerides and non-HDL cholesterol was considered sub-optimal; second, there was an increase in quantity of liver fat with higher dose of the investigational product [87]. In a phase I study of healthy individuals with fasting triglycerides >100 mg/dL, repeated doses of ARO-ANG3 given SC at day 1 and day 29 achieved a mean decrease of 66% in serum triglycerides with the 300-mg SC dose after 16 weeks [83, 84]. There were no serious adverse events and no clinically significant adverse changes in platelet count and liver function tests.
9.3 ApoC-III Nucleic Acid Therapies
The APPROACH study was a phase III, double-blind randomized trial to evaluate the safety and efficacy of volanesorsen in patients with FCS and plasma triglycerides >500 mg/dL [79]. At 3 months there was an 84% decrease in plasma apoC-III levels in patients receiving the ASO compared with placebo, which was accompanied by a 77% decrease in triglycerides and a 136% increase in LDL-cholesterol concentrations. A significant portion of the volanesorsen group experienced injection-site reactions, which were not observed in the placebo group, and 45% of the volanesorsen group also experienced severe thrombocytopenia [79].
The COMPASS study was a multi-centre, placebo-controlled phase III trial of the efficacy and safety of subcutaneously administered volanesorsen in patients with predominantly MCS and plasma triglycerides > 500 mg/dL [80]. Given the findings from APPROACH, a protocol change was made in COMPASS after 13 weeks of treatment that required the dose of volanesorsen to be decreased, which helped offset thrombocytopenia. After 3 months, volanesorsen markedly lowered plasma apoC-III (− 76%) and triglyceride (− 71%) concentrations, but increased plasma LDL cholesterol (+ 96%), which were sustained over 6 months of treatment. This was associated with a reduction in acute pancreatitis. Mild injection-site reactions and mild thrombocytopenia were again more frequently seen in patients receiving volanesorsen [80].
Table 4 also summarizes other key studies utilizing apoC-III inhibitors in subjects with HTG. The findings from the COMPASS trial augment the findings of these trials with volanesorsen [81, 88,89,90] and demonstrate that this intervention is efficacious across a wide range of plasma triglyceride concentrations and particularly in patients with MCS. Furthermore, the efficacy of treatment appears to be independent of the type of genetic variants impairing the activity of LPL, supporting the notion that volanesorsen can operate via LPL-independent mechanisms [90], including by inhibition of TRL production. Another important finding was that volanesorsen was equally efficacious in lowering triglycerides in patients both taking and not taking lipid-lowering treatments, including fibrates, statins and fish oils [88]. A potential limitation of these studies, however, is the increase in LDL-cholesterol concentration with volanesorsen treatment (see Table 3), although this can be offset with use of statin therapy. Whether such increase in plasma LDL cholesterol is clinically relevant remains to be determined. In contrast to findings in the APPROACH and COMPASS trials, volanesorsen or olezarsen ASO treatment did not significantly alter plasma LDL-cholesterol concentration in patients with mild-to-moderate HTG [81, 88]. This suggests that the effect of apoC-III inhibition on LDL cholesterol is dependent on fasting triglyceride or TRL concentrations. There is currently an ongoing clinical trial (BALANCE study) which investigates the effect of olezarsen in patients with FCS (ClinicalTrials.gov identifier: NCT04568434) and is due for completion in mid-2023.
In a phase I study to evaluate the effect of ARO-APOC3 (50 mg SC Q4W), significant reductions at 16 weeks were observed in apoC-III (98% and 96%), triglycerides (85% and 88%) and non-HDL cholesterol (45% and 59%) in patients with HTG (≥300 mg/dL) or chylomicronaemia (>800 mg/dL), respectively [85]. There were no serious adverse events reported. More recently, a phase II, dose-ranging study investigating the effect of olezarsen in patients with fasting triglycerides 200–500 mg/dL and were at high risk for or had established ASCVD was conducted. Treatment with olezarsen resulted in reductions in triglycerides of 23% for 10 mg Q4W, 56% for 15 mg Q2W and 60% for 10 mg weekly and 50 mg Q4W. Significant reductions were also seen for apoC-III, VLDL, non-HDL cholesterol and apoB [91].
10 Implications and Conclusions
Significant evidence supports the causal role of elevated TRLs, specifically remnant particles, in the development of ASCVD in both primary and secondary prevention [1,2,3,4, 8]. International guidelines have highlighted increased plasma TRL and apoB concentrations as risk-enhancing factors for the prevention of ASCVD [8, 35, 36]. Elevated TRLs are also related to the increased risk of metabolic-associated fatty liver disease, aortic stenosis and acute pancreatitis [3, 22, 33, 34]. Despite best standard of care, including optimal LDL cholesterol concentrations, a significant proportion of patients with ASCVD remain at increased residual risk due to increased plasma concentrations of TRLs [25, 26]. The therapeutic use of high-dose pure EPA is supported by clinical trial evidence for benefit in secondary prevention of ASCVD [37]. However, the value of this trial has been challenged, with evidence suggesting that benefits are not mediated by reduction in TRLs and that the positive findings may relate to acceleration in risk of ASCVD in the group treated with ‘placebo’ oil [38]. New therapeutic targets, such as ANGPTL3 and apoC-III [5, 6], are therefore welcomed for addressing gaps in the management of HTG in secondary prevention, as well as for addressing the management of severe HTG as a risk factor for acute pancreatitis [8]; a particular advantage of these agents is the durability of effects that may address poor adherence to orally administered therapies.
The recent clinical trial evidence that we have reviewed suggest that ANGPTL3 and apoC-III inhibitors may be equally effective in lowering markedly elevated plasma triglyceride concentrations (see Tables 3 and 4), with possibly greater efficacy with the latter agent. However, ANGPTL3 inhibitors may have the advantage of greater lowering of plasma LDL concentrations and this may be particularly relevant to patients with mixed atherogenic dyslipidaemia. Both agents need to be tested in a wide range of lipid disorders characterized by impairment in the metabolism of TRLs. Relevant trials are currently underway. Figure 2 compares the putative application for ANGPTL3 and apoC-III inhibitors for treating lipid disorders ranging from severe HTG through mixed atherogenic dyslipidaemia to severe FH [92]. Such applications are contingent on use of best background standard of care, especially lifestyle measures and proven pharmacotherapies including statins, ezetimibe, fibrates and PCSK9 inhibitors [8].

Angiopoietin-like protein 3 (ANGPTL3) and apolipoprotein C-III (apoC-III) inhibition may be equally effective in correcting hypertriglyceridaemia in chylomicronaemia syndromes, metabolic-associated fatty liver disease, insulin resistance/diabetes and mixed hyperlipidaemia, but ANGPTL3 inhibition may more effectively lower low-density lipoprotein (LDL) cholesterol and have extended applications in mixed atherogenic dyslipidaemia and more severe forms of familial hypercholesterolaemia
Confirmation of the safety of long-term inhibition of ANGPTL3 and apoC-III with RNA-based therapies is evidently essential. Notwithstanding the small numbers of studies, current evidence confirms safety and good tolerability following the utilization of GalNAc3 conjugated nucleic acids [81, 83,84,85]. The costs of these potent agents and their effectiveness needs to be clearly demonstrated in different clinical contexts. Nucleic acid is likely to be more effective than mAb-based therapies for inhibiting both ANGPTL3 and apoC-III. Whether there are synergistic effects of ANGPTL3 and apoC-III inhibition in correcting severe HTG remains to be demonstrated. siRNA may in turn also be less expensive than ASO, owing to the former intrinsic mode of action via RISC and hence the lower frequency of subcutaneous drug administration. In aggregate, whilst it may be premature to speculate, given safety, tolerability and cost effectiveness remain to be confirmed, siRNA inhibition of ANGPTL3 may offer a futuristic panacea for targeting HTG in a wide range of dyslipidaemic settings, including residual elevation in LDL cholesterol in FH [92].
References
Laufs U, Parhofer KG, Ginsberg HN, Hegele RA. Clinical review on triglycerides. Eur Heart J. 2020;41:99–109.
Nordestgaard BG. Triglyceride-rich lipoproteins and atherosclerotic cardiovascular disease: new insights from epidemiology, genetics, and biology. Circ Res. 2016;118:547–63.
Packard CJ, Boren J, Taskinen MR. Causes and consequences of hypertriglyceridemia. Front Endocrinol (Lausanne). 2020;11:252.
Ginsberg HN, Packard CJ, Chapman MJ, Borén J, Aguilar-Salinas CA, Averna M, et al. Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur Heart J. 2021;42:4791–806.
Kersten S. Angiopoietin-like 3 in lipoprotein metabolism. Nat Rev Endocrinol. 2017;13:731–9.
Boren J, Packard CJ, Taskinen MR. The roles of apoC-III on the metabolism of triglyceride-rich lipoproteins in humans. Front Endocrinol (Lausanne). 2020;11:474.
Cooper AD. Hepatic uptake of chylomicron remnants. J Lipid Res. 1997;38:2173–92.
Virani SS, Morris PB, Agarwala A, Ballantyne CM, Birtcher KK, Kris-Etherton PM, et al. 2021 ACC expert consensus decision pathway on the management of ASCVD risk reduction in patients With persistent hypertriglyceridemia: a report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol. 2021;78:960–93.
Goldberg RB, Chait A. A comprehensive update on the chylomicronemia syndrome. Front Endocrinol. 2020;11:593931.
Gill PK, Dron JS, Hegele RA. Genetics of hypertriglyceridemia and atherosclerosis. Curr Opin Cardiol. 2021;36:264–71.
Johansen CT, Kathiresan S, Hegele RA. Genetic determinants of plasma triglycerides. J Lipid Res. 2011;52:189–206.
Watts GF, Ooi EM, Chan DC. Demystifying the management of hypertriglyceridaemia. Nat Rev Cardiol. 2013;10:648–61.
Mahley RW, Huang Y, Rall SC. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J Lipid Res. 1999;40:1933–49.
Trinder M, Vikulova D, Pimstone S, Mancini GBJ, Brunham LR. Polygenic architecture and cardiovascular risk of familial combined hyperlipidemia. Atherosclerosis. 2022;340:35–43.
Brahm HRA. Hypertriglyceridemia. Nutrients. 2013;5:981–1001.
Aguilar-Salinas CA, Hugh P, Barrett R, Pulai J, Zhu XL, Schonfeld G. A familial combined hyperlipidemic kindred with impaired apolipoprotein B catabolism. Kinetics of apolipoprotein B during placebo and pravastatin therapy. Arterioscler Thromb Vasc Biol. 1997;17:72–82.
Cruz-Bautista I, Huerta-Chagoya A, Moreno-Macías H, Rodríguez-Guillén R, Ordóñez-Sánchez ML, Segura-Kato Y, et al. Familial hypertriglyceridemia: an entity with distinguishable features from other causes of hypertriglyceridemia. Lipids Health Dis. 2021;20:14.
Kesaniemi YA, Grundy SM. Dual defect in metabolism of very-low-density lipoprotein triglycerides. Patients with type 5 hyperlipoproteinemia. JAMA. 1984;251:2542–7.
Vergès B. Pathophysiology of diabetic dyslipidaemia: where are we? Diabetologia. 2015;58:886–99.
Sarwar N, Danesh J, Eiriksdottir G, Sigurdsson G, Wareham N, Bingham S, et al. Triglycerides and the risk of coronary heart disease:10 158 incident cases among 262 525 participants in 29 Western Prospective Studies. Circulation. 2007;115:450–8.
Varbo A, Benn M, Tybjærg-Hansen A, Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61:427–36.
Kaltoft M, Langsted A, Nordestgaard BG. Triglycerides and remnant cholesterol associated with risk of aortic valve stenosis: Mendelian randomization in the Copenhagen General Population Study. Eur Heart J. 2020;41:2288–99.
Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384:626–35.
Varbo A, Nordestgaard BG. Remnant cholesterol and risk of ischemic stroke in 112,512 individuals from the general population. Ann Neurol. 2019;85:550–9.
Marston NA, Giugliano RP, Im K, Silverman MG, O’Donoghue ML, Wiviott SD, et al. Association between triglyceride lowering and reduction of cardiovascular risk across multiple lipid-lowering therapeutic classes: A systematic review and meta-regression analysis of randomized controlled trials. Circulation. 2019;140:1308–17.
Lawler PR, Kotrri G, Koh M, Goodman SG, Farkouh ME, Lee DS, et al. Real-world risk of cardiovascular outcomes associated with hypertriglyceridaemia among individuals with atherosclerotic cardiovascular disease and potential eligibility for emerging therapies. Eur Heart J. 2020;41:86–94.
Goldstein JL, Ho YK, Brown MS, Innerarity TL, Mahley RW. Cholesteryl ester accumulation in macrophages resulting from receptor-mediated uptake and degradation of hypercholesterolemic canine beta-very low density lipoproteins. J Biol Chem. 1980;255:1839–48.
Zheng XY, Liu L. Remnant-like lipoprotein particles impair endothelial function: direct and indirect effects on nitric oxide synthase. J Lipid Res. 2007;48:1673–80.
Kohler HP, Grant PJ. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. 2000;342:1792–801.
Moyer MP, Tracy RP, Tracy PB, van Veer C, Sparks CE, Mann KG. Plasma lipoproteins support prothrombinase and other procoagulant enzymatic complexes. Arterioscler Thromb Vasc Biol. 1998;18:458–65.
Alipour A, van Oostrom AJ, Izraeljan A, Verseyden C, Collins JM, Frayn KN, et al. Leukocyte activation by triglyceride-rich lipoproteins. Arterioscler Thromb Vasc Biol. 2008;28:792–7.
Wang L, Gill R, Pedersen TL, Higgins LJ, Newman JW, Rutledge JC. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J Lipid Res. 2009;50:204–13.
Pedersen SB, Langsted A, Nordestgaard BG. Nonfasting mild-to-moderate hypertriglyceridemia and risk of acute pancreatitis. JAMA Intern Med. 2016;176:1834–42.
Hansen SEJ, Madsen CM, Varbo A, Nordestgaard BG. Low-grade inflammation in the association between mild-to-moderate hypertriglyceridemia and risk of acute pancreatitis: a study of more than 115000 individuals from the general population. Clin Chem. 2019;65:321–32.
Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111–88.
Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the american college of cardiology/american heart association task force on clinical practice guidelines. Circulation. 2019;139:e1082–143.
Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. 2019;380:11–22.
Olshansky B, Chung MK, Budoff MJ, Philip S, Jiao L, Doyle RT Jr, et al. Mineral oil: safety and use as placebo in REDUCE-IT and other clinical studies. Eur Heart J Suppl. 2020;22:J34–48.
Nicholls SJ, Lincoff AM, Garcia M, Bash D, Ballantyne CM, Barter PJ, et al. Effect of high-dose omega-3 fatty acids vs corn oil on major adverse cardiovascular events in patients at high cardiovascular risk: the strength randomized clinical trial. JAMA. 2020;324:2268–80.
Gencer B, Djousse L, Al-Ramady OT, Cook NR, Manson JE, Albert CM. Effect of long-term marine ɷ-3 fatty acids supplementation on the risk of atrial fibrillation in randomized controlled trials of cardiovascular outcomes: a systematic review and meta-analysis. Circulation. 2021;144:1981–90.
Fruchart JC, Santos RD, Aguilar-Salinas C, Aikawa M, Al Rasadi K, Amarenco P, et al. The selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha) paradigm: conceptual framework and therapeutic potential : A consensus statement from the International Atherosclerosis Society (IAS) and the Residual Risk Reduction Initiative (R3i) Foundation. Cardiovasc Diabetol. 2019;18:71.
Pradhan AD, Paynter NP, Everett BM, Glynn RJ, Amarenco P, Elam M, et al. Rationale and design of the pemafibrate to reduce cardiovascular outcomes by reducing triglycerides in patients with diabetes (PROMINENT) study. Am Heart J. 2018;206:80–93.
Chen YQ, Pottanat TG, Siegel RW, Ehsani M, Qian YW, Zhen EY, et al. Angiopoietin-like protein 8 differentially regulates ANGPTL3 and ANGPTL4 during postprandial partitioning of fatty acids. J Lipid Res. 2020;61:1203–20.
Shimamura M, Matsuda M, Yasumo H, Okazaki M, Fujimoto K, Kono K, et al. Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler Thromb Vasc Biol. 2007;27:366–72.
Liu J, Afroza H, Rader DJ, Jin W. Angiopoietin-like protein 3 inhibits lipoprotein lipase activity through enhancing its cleavage by proprotein convertases. J Biol Chem. 2010;285:27561–70.
Robciuc MR, Maranghi M, Lahikainen A, Rader D, Bensadoun A, Öörni K, et al. Angptl3 deficiency is associated with increased insulin sensitivity, lipoprotein lipase activity, and decreased serum free fatty acids. Arterioscler Thromb Vasc Biol. 2013;33:1706–13.
Lupo MG, Ferri N. Angiopoietin-Like 3 (ANGPTL3) and atherosclerosis: Lipid and mon-lipid related effects. J Cardiovasc Dev Dis. 2018;5:2.
Arca M, D’Erasmo L, Minicocci I. Familial combined hypolipidemia: angiopoietin-like protein-3 deficiency. Curr Opin Lipidol. 2020;31:41–8.
Ruhanen H, Haridas PAN, Jauhiainen M, Olkkonen VM. Angiopoietin-like protein 3, an emerging cardiometabolic therapy target with systemic and cell-autonomous functions. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865:158791.
Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C, Schurmann C, Gottesman O, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med. 2017;377:211–21.
Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, et al. ANGPTL3 deficiency and protection against coronary artery disease. J Am Coll Cardiol. 2017;69:2054–63.
Romeo S, Yin W, Kozlitina J, Pennacchio LA, Boerwinkle E, Hobbs HH, et al. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. 2009;119:70–9.
Hatsuda S, Shoji T, Shinohara K, Kimoto E, Mori K, Fukumoto S, et al. Association between plasma angiopoietin-like protein 3 and arterial wall thickness in healthy subjects. J Vasc Res. 2007;44:61–6.
Meyers NL, Larsson M, Vorrsjo E, Olivecrona G, Small DM. Aromatic residues in the C terminus of apolipoprotein C-III mediate lipid binding and LPL inhibition. J Lipid Res. 2017;58:840–52.
Rodríguez M, Rehues P, Iranzo V, Mora J, Balsells C, Guardiola M, Ribalta J. Distribution of seven ApoC-III glycoforms in plasma, VLDL, IDL, LDL and HDL of healthy subjects. J Proteomics. 2022;251:104398.
Adiels M, Taskinen MR, Björnson E, Andersson L, Matikainen N, Söderlund S, et al. Role of apolipoprotein C-III overproduction in diabetic dyslipidaemia. Diabetes Obes Metab. 2019;21:1861–70.
Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41.
Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31.
Olin-Lewis K, Krauss RM, La Belle M, Blanche PJ, Barrett PH, Wight TN, et al. ApoC-III content of apoB-containing lipoproteins is associated with binding to the vascular proteoglycan biglycan. J Lipid Res. 2002;43:1969–77.
Hiukka A, Stahlman M, Pettersson C, Levin M, Adiels M, Teneberg S, et al. ApoCIII-enriched LDL in type 2 diabetes displays altered lipid composition, increased susceptibility for sphingomyelinase, and increased binding to biglycan. Diabetes. 2009;58:2018–26.
Yan H, Niimi M, Matsuhisa F, Zhou H, Kitajima S, Chen Y, et al. Apolipoprotein CIII deficiency protects against atherosclerosis in knockout rabbits. Arterioscler Thromb Vasc Biol. 2020;40:2095–107.
Reyes-Soffer G, Sztalryd C, Horenstein RB, Holleran S, Matveyenko A, Thomas T, et al. Effects of APOC3 heterozygous deficiency on plasma lipid and lipoprotein metabolism. Arterioscler Thromb Vasc Biol. 2019;39:63–72.
Macchi C, Sirtori CR, Corsini A, Santos RD, Watts GF, Ruscica M. A new dawn for managing dyslipidemias: The era of rna-based therapies. Pharmacol Res. 2019;150:104413.
Norata GD, Tsimikas S, Pirillo A, Catapano AL. Apolipoprotein C-III: From pathophysiology to pharmacology. Trends Pharmacol Sci. 2015;36:675–87.
Jia X, Liu J, Mehta A, Ballantyne CM, Virani SS. Lipid-lowering biotechnological drugs: from monoclonal antibodies to antisense therapies-a clinical perspective. Cardiovasc Drugs Ther. 2021;35:1269–79.
Tsimikas S. RNA-targeted therapeutics for lipid disorders. Curr Opin Lipidol. 2018;29:459–66.
Nordestgaard BG, Nicholls SJ, Langsted A, Ray KK, Tybjærg-Hansen A. Advances in lipid-lowering therapy through gene-silencing technologies. Nat Rev Cardiol. 2018;15:261–72.
Wang Y, Yu RZ, Henry S, Geary RS. Pharmacokinetics and clinical pharmacology considerations of GalNAc3-conjugated antisense oligonucleotides. Expert Opin Drug Metab Toxicol. 2019;15:475–85.
Ahmad Z, Banerjee P, Hamon S, Chan KC, Bouzelmat A, Sasiela WJ, et al. Inhibition of angiopoietin-like protein 3 with a monoclonal antibody reduces triglycerides in hypertriglyceridemia. Circulation. 2019;140:470–86.
Adam RC, Mintah IJ, Alexa-Braun CA, Shihanian LM, Lee JS, Banerjee P, et al. Angiopoietin-like protein 3 governs LDL-cholesterol levels through endothelial lipase-dependent VLDL clearance. J Lipid Res. 2020;61:1271–86.
Wang Y, Gusarova V, Banfi S, Gromada J, Cohen JC, Hobbs HH. Inactivation of ANGPTL3 reduces hepatic VLDL-triglyceride secretion. J Lipid Res. 2015;56:1296–307.
Ruscica M, Zimetti F, Adorni MP, Sirtori CR, Lupo MG, Ferri N. Pharmacological aspects of ANGPTL3 and ANGPTL4 inhibitors: New therapeutic approaches for the treatment of atherogenic dyslipidemia. Pharmacol Res. 2020;153:104653.
Katzmann JL, Packard CJ, Chapman MJ, Katzmann I, Laufs U. Targeting RNA with antisense oligonucleotides and small interfering RNA: JACC State-of-the-Art Review. J Am Coll Cardiol. 2020;76:563–79.
Valanti EK, Dalakoura-Karagkouni K, Siasos G, Kardassis D, Eliopoulos AG, Sanoudou D. Advances in biological therapies for dyslipidemias and atherosclerosis. Metabolism. 2021;116:154461.
Khetarpal SA, Zeng X, Millar JS, Vitali C, Somasundara AVH, Zanoni P, et al. A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat Med. 2017;23:1086–94.
Graham MJ, Lee RG, Brandt TA, Tai LJ, Fu W, Peralta R, et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N Engl J Med. 2017;377:222–32.
Bell TA, Liu M, Donner AJ, Lee RG, Mullick AE, Crooke RM. Antisense oligonucleotide-mediated inhibition of angiopoietin-like protein 3 increases reverse cholesterol transport in mice. J Lipid Res. 2021;62:100101.
Graham MJ, Lee RG, Bell TA 3rd, Fu W, Mullick AE, Alexander VJ, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112:1479–90.
Witztum JL, Gaudet D, Freedman SD, Alexander VJ, Digenio A, Williams KR, et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N Engl J Med. 2019;381:531–42.
Gouni-Berthold I, Alexander VJ, Yang Q, Hurh E, Steinhagen-Thiessen E, Moriarty PM, et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021;9:264–75.
Alexander VJ, Xia S, Hurh E, Hughes SG, O’Dea L, Geary RS, et al. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur Heart J. 2019;40:2785–96.
Butler AA, Graham JL, Stanhope KL, Wong S, King S, Bremer AA, et al. Role of angiopoietin-like protein 3 in sugar-induced dyslipidemia in rhesus macaques: suppression by fish oil or RNAi. J Lipid Res. 2020;61:376–86.
Watts GF, Scott R, Gladding P, Sullivan D, Baker J, Clifton P, et al. RNA interference targeting hepatic angiopoietin-like protein 3 results in prolonged reductions in plasma triglycerides and LDL-C in human subjects. Circulation. 2019;140:E987–8.
Watts GF, Scott R, Gladding P, Sullivan D, Baker J, Clifton P, et al. RNAi inhibition of angiopoietin-like protein 3 (ANGPTL3) with ARO-ANG3 mimics the lipid and lipoprotein profile of combined hypolidemia. Eur Heart J. 2020;41:3331.
Clifton PS, Baker J, Schwabe C, Thackwray S, Scott R, Hamilton J, et al. Pharmacodynamic effect of ARO-APOC3, an investigational hepatocyte-targeted RNA interference therapeutic targeting apolipoprotein C3 in patients with hypertriglyceridemia and multifactorial chylomicronemia. Circulation. 2020;142:12594.
Gaudet D, Karwatowska-Prokopczuk E, Baum SJ, Hurh E, Kingsbury J, Bartlett VJ, et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. Eur Heart J. 2020;41:3936–45.
Pfizer and Ionis Announce Discontinuation of Vupanorsen Clinical Development Program. https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-ionis-announce-discontinuation-vupanorsen
Digenio A, Dunbar RL, Alexander VJ, Hompesch M, Morrow L, Lee RG, et al. Antisense-mediated lowering of plasma apolipoprotein C-III by volanesorsen improves dyslipidemia and insulin sensitivity in type 2 diabetes. Diabetes Care. 2016;39:1408–15.
D’Erasmo L, Gallo A, Di Costanzo A, Bruckert E, Arca M. Evaluation of efficacy and safety of antisense inhibition of apolipoprotein C-III with volanesorsen in patients with severe hypertriglyceridemia. Expert Opin Pharmacother. 2020;21:1675–84.
Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. 2014;371:2200–6.
Tardif J, Karwatowska-Prokopczuk E, St. Amour E, Ballantyne CM, Shapiro MD, Moriarty PM, et al. Apolipoprotein C-III reduction in subjects with moderate triglyceridaemia and at high cardiovascular risk. Eur Heart J. 2022. https://doi.org/10.10193/eurheartj/ehab820.
Watts GF, Raal FJ, Chan DC. Transcriptomic therapy for dyslipidemias utilizing nucleic acids targeted at ANGPTL3. Future Cardiol. 2021. https://doi.org/10.2217/fca-2021-0096.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Ethics Approval
Not applicable.
Consent to Participate/Publish
Not applicable.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable
Author Contributions
All authors contributed to the drafting and editing of the review, as well as subsequent revisions.
Conflicts of Interest
NCW: none. DCC: none. GFW: reports funding or honoraria from Amgen, Pfizer, Esperion, Arrowhead, Regeneron, Sanofi, AstraZeneca, Novartis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ward, N.C., Chan, D.C. & Watts, G.F. A Tale of Two New Targets for Hypertriglyceridaemia: Which Choice of Therapy?. BioDrugs 36, 121–135 (2022). https://doi.org/10.1007/s40259-022-00520-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-022-00520-2