
Abstract
Background
Bevacizumab is an antiangiogenic recombinant humanized monoclonal antibody that inhibits tumor growth. FKB238, a bevacizumab biosimilar, has analytical pharmacokinetic and safety profiles similar to those of bevacizumab.
Objective
This phase III trial (NCT02810457) compared the efficacy and safety of FKB238 with that of bevacizumab in patients with advanced/recurrent non-squamous non-small-cell lung cancer (non-sq-NSCLC).
Methods
This global, multicenter, double-blind, parallel, randomized, comparative clinical trial enrolled and randomized patients with advanced/recurrent non-sq-NSCLC to receive intravenous infusions of either FKB238 15 mg/kg or bevacizumab 15 mg/kg. All patients received intravenous infusions of paclitaxel 200 mg/m2 and carboplatin (area under the curve 6.0) immediately prior to investigational products for 4–6 cycles. FKB238 and bevacizumab were administered on day 1 of each 21-day cycle until objective progressive disease by RECIST version 1.1 or other discontinuation criteria were met. The primary efficacy endpoint was overall response rate (ORR), including complete and partial response and based on blinded independent central review assessment. Other efficacy determinations included progression-free survival (PFS), overall survival (OS), and immunogenicity. Adverse events and severity were reported.
Results
The ORR for the intent-to-treat (ITT) population (N = 731) was 51.6% in the FKB238 arm (N = 364) and 53.7% in the bevacizumab arm (N = 367). The FKB238:bevacizumab ORR ratio (ITT population) was 0.96 (90% confidence interval [CI] 0.86–1.08), and the difference in ORR (per-protocol set) between FKB238 and bevacizumab was − 0.02 (95% CI − 0.09 to 0.06). Both CIs fell within the prespecified equivalence margins. Estimated median PFS was 7.72 and 7.62 months in the FKB238 and bevacizumab arms, respectively (hazard ratio 0.97; 95% CI 0.82–1.16). Treatment-emergent adverse events (TEAEs) were reported for 94.2% and 95.1% of patients in the FKB238 and bevacizumab arms, respectively. Grade 3 or higher TEAEs were reported for 53.6% and 55.5% of patients in the FKB238 and bevacizumab arms, respectively. Serious TEAEs were reported for 25.1% and 26.0% of patients treated with FKB238 and bevacizumab, respectively.
Conclusions
Efficacy equivalence was demonstrated between the two drugs, and safety profiles were similar. There were no meaningful differences in efficacy and safety between FKB238 or bevacizumab in patients with non-sq-NSCLC.
Trial registration number
NCT02810457.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
FKB238 has efficacy and safety profiles within the parameters set by EU and US regulatory authorities for being considered a biosimilar to reference bevacizumab. |
FKB238 has efficacy and safety comparable to that of bevacizumab among patients with non-squamous non-small-cell lung cancer. |
1 Introduction
Lung cancer is the leading cause of cancer-related deaths in the USA, accounting for approximately 23% of all cancer-related deaths and with an overall 5-year survival rate of 21% [1]. Patients with non-small-cell lung cancer (NSCLC) most often present with inoperable, locally advanced or metastatic disease for which no cure is available [2, 3]. Although survival has improved significantly in recent years with the introduction of kinase inhibitors and immune checkpoint inhibitors, the 5-year survival rate is still only about 25% [1, 4, 5]. The standard of care for patients with NSCLC varies with the stage of the disease at diagnosis [6]. Surgery, followed by platinum-based chemotherapy with or without adjuvant radiation therapy are the standard for most patients with stage I to stage IIIA NSCLC [6, 7]. More recently, targeted immunotherapy is becoming the choice of first-line treatment among patients with advanced NSCLC and those that are not amenable to other treatments [6, 8].
Angiogenesis is a complex process mediated by vascular endothelial growth factor (VEGF) which is involved in the growth and metastasis of several cancers, including NSCLC [9, 10]. Angiogenesis inhibitors, including agents that block the activity of VEGF, have been shown to be effective in increasing progression-free survival (PFS) in non-squamous (non-sq)-NSCLC [11]. Bevacizumab (Avastin®) is a recombinant humanized monoclonal antibody that acts as an angiogenesis inhibitor [12]. Bevacizumab binds to soluble VEGF-A, thereby preventing its interaction with its receptors on vascular cells and inhibiting angiogenesis [13]. Bevacizumab is effective for the treatment of several cancers, including non-sq-NSCLC, and is approved by both the US FDA and the European Medicines Agency (EMA) [14,15,16].
In recent years, value-based cancer therapy has been emphasized in American Society of Clinical Oncology (ASCO) and National Comprehensive Cancer Network (NCCN) guidelines [17, 18]. These guidelines focus on providing patients affordable treatments that are effective and well tolerated. Biosimilar agents are biological drugs that have comparable activity to an approved reference biological agent [19,20,21]. Consequently, biosimilars are an important avenue to achieve affordability while retaining the efficacy demonstrated by the reference biologic agent.
A phase I study demonstrated that FKB238 had comparable pharmacokinetics to those of bevacizumab, did not induce antidrug antibodies (ADAs), and was well tolerated [22]. This paper reports on a phase III study (AVANA) to determine whether the efficacy and safety of FKB238 were similar to those of bevacizumab in patients with non-sq-NSCLC (NCT02810457).
2 Methods
2.1 Patients
All patients signed informed consent. Adults with newly diagnosed advanced (stage IV) or recurrent non-sq-NSCLC with at least one measurable lesion were eligible for this study. Other inclusion criteria were an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, life expectancy > 6 months, adequate renal and liver function, and a negative pregnancy test. Important exclusion criteria were a diagnosis of small-cell lung cancer (SCLC) or a combination of SCLC and NSCLC, other cancers within the prior 5 years, unresolved toxicities from prior treatments, tumors invading major blood vessels, previous dosing with a VEGF inhibitor, brain metastases, cardiovascular disease, hepatitis B or C infection, human immunodeficiency virus infection, and major surgery within 28 days of treatment initiation.
The intent-to-treat (ITT) population included all patients randomized to treatment. The per-protocol set (PPS) included all patients randomized to treatment who received at least one dose of investigational product (IP) with no important protocol deviations. The safety population included all patients randomized to treatment who received at least one dose of IP (FKB238 or bevacizumab). All patients in the PPS who were assessed for ADAs at baseline and at least once thereafter were assigned into the ADA-evaluable population.
2.2 Study Design
This was a global, multicenter, double-blind, parallel, randomized, comparative trial (Fig. 1). Patients were randomized 1:1 to receive intravenous infusions of FKB238 or bevacizumab 15 mg/kg (Avastin® approved by the EU) once every 21 (± 3) days until objective progressive disease or other discontinuation criteria were met. Intravenous infusions of paclitaxel 200 mg/m2 and carboplatin (area under the curve 6.0) were administered once every 21 (± 3) days immediately prior to the IP for at least 4 cycles and not more than 6 cycles, as determined by the individual patient’s treatment needs and investigator’s assessment.

Study design. CBDCA carboplatin, DCO discontinuation, NSCLC non-small-cell lung cancer, PS performance status, PTX paclitaxel
2.3 Randomization, Blinding, and Stratification
Patients were randomized and assigned a unique identification number using ClinPhone® RTSM (PAREXEL Informatics). Investigators, site staff, pharmacy staff, patients, contract research organization personnel, and sponsor personnel were all blinded to individual patient treatment assignment throughout the course of the study. Patients were stratified by their epidermal growth factor receptor mutation and anaplastic lymphoma kinase gene status, geographical region, weight loss over the previous 6 months, and disease stage.
2.4 Study Objectives
The primary objective of this study was to demonstrate the efficacy equivalence of FKB238 and bevacizumab in combination with paclitaxel/carboplatin as measured by overall response rate (ORR), assessed as the rate of the best overall response (BOR) of complete response (CR) or partial response (PR) by Response Evaluation Criteria in Solid Tumors (RECIST v1.1) [23], using blinded independent central review (BICR) assessment. The major secondary objectives included comparison of FKB238 with bevacizumab for ORR at week 19, PFS, overall survival (OS), immunogenicity as determined by the presence of ADAs, and safety. Disease progression was defined as an absence of CR, PR, or stable disease, or no evidence of disease.
2.4.1 Immunogenicity Assays
The presence of ADAs and neutralizing antibodies (NAbs) against FKB238 or bevacizumab was detected using validated electrochemiluminescence assays. Serum samples were collected from patients on day 1 of treatment cycles 1, 2, 4, and 6, study treatment discontinuation, and at follow-up visit. Data were normalized against the mean of the negative controls on that assay plate.
2.5 Statistical Methods
2.5.1 Sample Size
The sample size was determined as requiring approximately 730 randomized patients (365 patients in each treatment arm) to meet both FDA and EMA requirements, assuming a 10% dropout rate and 35% response rate in each treatment arm determined by two one-sided tests. The sample size had 80% power to show that the 90% confidence interval (CI) for the ORR risk ratio comparing FKB238 with bevacizumab was entirely within the margin of 0.73 and 1.38, as agreed with the FDA, and the 95% CI of ORR risk difference between the two treatment arms was completely within the ± 0.1221 equivalence margin as agreed with the EMA.
2.5.2 Analyses
Primary analyses of the FKB238:bevacizumab ORR ratio on the ITT population and difference in respective ORRs on the PPS population were performed as required by the FDA and EMA, respectively. Analyses of ORR at week 19 were also performed as a secondary endpoint.
For time-to-event secondary endpoints, PFS was defined as the time from randomization to first documentation of disease progression or death due to any reason, and OS was defined as the time in months from date of randomization to death due to any cause. PFS and OS were analyzed using the Kaplan–Meier method. The hazard ratio (HR) was estimated using the Cox regression model, including treatment arm and the baseline covariates of the randomization stratification factors, ECOG performance status at baseline, sex, smoking history, and age. Patients without a reported event of death at the end of the study were censored for OS at the last date when they were known to be alive.
The safety population included all patients randomized to treatment and who received at least one dose of IP. Adverse events (AE) were coded using Medical Dictionary for Regulatory Activities (MedDRA, v 21.1) [24], and severity was reported according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE, v 4.0) [25]. AEs were presented as number and percentage of patients experiencing treatment-emergent AEs (TEAEs).
3 Results
3.1 Patients
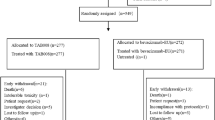
A total of 731 patients with advanced or recurrent non-sq-NSCLC were randomized 1:1 to either the FKB238 arm (n = 364) or the bevacizumab arm (n = 367). Among these, 728 patients received at least one dose of FKB238 (n = 362) or one dose of bevacizumab (n = 366). The disposition of the patients is provided in Fig. 2. By the end of the study, 36 (9.9%) and 49 (13.4%) patients withdrew consent in the FKB238 and bevacizumab arms, respectively. Patient demographics and disease characteristics at baseline were similar between the two treatment arms (Table 1).

Patient disposition. DCO discontinuation, IP investigational product, ITT intent to treat
3.2 Efficacy
3.2.1 Overall Response Rate (ORR)
Based on BICR assessments for the ITT population, four (1.1%) patients experienced CR and 184 (50.5%) patients experienced PR for an ORR of 51.6% (95% CI 46.38–56.89) in the FKB238 arm (n = 364), and two (0.5%) patients experienced CR and 195 (53.1%) experienced PR for an ORR of 53.7% (95% CI 48.43–58.87) in the bevacizumab arm (n = 367). The ratio of FKB238:bevacizumab ORRs was 0.96 (90% CI 0.86–1.08), which fell entirely within the 0.73 and 1.38 margins, indicating equivalence in efficacy between the two products per the FDA requirement (Table 2). Based on the PPS, four (1.1%) patients experienced CR and 178 (50.6%) experienced PR for an ORR of 51.7% (95% CI 46.35–57.03) in the FKB238 arm (n = 352), and one (0.3%) patient experienced CR and 188 (53.1%) experienced PR for an ORR of 53.4% (95% CI 48.04–58.68) in the bevacizumab arm (n = 354). The risk difference between the two treatment arms was − 0.02 (95% CI − 0.09 to 0.06), which was entirely within the ± 0.1221 equivalence margin, indicating equivalence in efficacy between the two products per the EMA requirement (Table 2).
3.2.2 ORR at Week 19
In the ITT population, the ORR at week 19 was 47.8% (174 patients) and 51.0% (187 patients) in the FKB238 and bevacizumab arms, respectively (Fig. 3), with a FKB238:bevacizumab ratio in ORR between the two treatment arms of 0.94 (90% CI 0.83–1.06). In the PPS, the ORR at week 19 was 47.7% (168 patients) and 50.8% (180 patients) in the FKB238 and bevacizumab arms, respectively (Fig. 3), with a risk difference between the two treatment arms of − 0.03 (95% CI − 0.10 to 0.04). The analyses of ORR at week 19 showed similar response rates and supported the efficacy equivalence between the two products.

Comparison of overall response rate (ORR) at week 19 between FKB238 and bevacizumab based on the intent-to-treat (ITT) population and per-protocol set (PPS). ORR is defined as the rate of the best overall response of complete response or partial response by RECIST v1.1 using blinded independent central review assessment. The black bar indicates the ORR at week 19 for the FKB238 arm, and the white bar indicates the ORR at week 19 for the bevacizumab arm. ORR-ITT represents the ORR at week 19 for the ITT population, and ORR-PPS represents the ORR for the PPS
3.2.3 Progression-Free Survival
Based on the assessments for the ITT population, 246 (67.6%) patients progressed or died in the FKB238 arm compared with 255 (69.5%) patients in the bevacizumab arm. Disease progression by RECIST occurred in 173 (47.5%) and 196 (53.4%) patients in the FKB238 and bevacizumab arms, respectively, with the corresponding deaths in the absence of RECIST progression occurring in 20.1 and 16.1% of patients. The estimated HR for FKB238:bevacizumb comparison based on Cox regression model was 0.97 (95% CI 0.82–1.16). Based on a Kaplan–Meier analysis of this population, the estimated median PFS was 7.72 (95% CI 7.46–7.98) months in the FKB238 arm and 7.62 (95% CI 6.90–7.82) months in the bevacizumab arm (Fig. 4). These analyses showed similar results for the two treatment arms.

Kaplan–Meier estimates for progression-free survival based on the intent-to-treat population. The numbers of patients at risk in each treatment arm are shown below the survival curves
3.2.4 Overall Survival
Based on a Kaplan–Meier analysis, for the ITT population, the estimated median OS was 14.13 months (95% CI 12.52–16.56) in the FKB238 arm and 16.95 months (95% CI 14.65–19.02) in the bevacizumab arm (Fig. 5). The estimated HR for FKB238:bevacizumb comparison based on Cox regression model was 1.18 (95% CI 0.96–1.45). The 95% CI contained the value 1.0, and the estimated HR was not different from 1.

Kaplan–Meier estimates for overall survival based on the intent-to-treat population. The numbers of patients at risk for each treatment arm are shown below the survival curves
3.3 Immunogenicity
In the FKB238 and bevacizumab treatment arms, 305 patients were ADA evaluable (all patients in the PPS who had at least one ADA assessment before and after baseline data collection). In both treatment arms, 3.0% (nine patients) tested positive for ADAs at any visit, whereas the incidence of treatment-emergent ADAs was 2.3% (seven patients) in each treatment arm. Only one patient in each treatment arm tested positive for NAbs at any visit.
3.4 Safety
Overall, in the safety population, TEAEs were experienced by 341 (94.2%) patients in the FKB238 arm and 348 (95.1%) patients in the bevacizumab arm (Table 3). The incidence of TEAEs causally related to IP was lower in the FKB238 (148 [40.9%] patients) arm than in the bevacizumab arm (174 [47.5%] patients). CTCAE grade 3 or higher TEAEs were reported for 194 (53.6%) and 203 (55.5%) patients in the FKB238 and bevacizumab arms, respectively. Treatment-emergent serious AEs (TESAEs) related to IPs were reported for 26 (7.2%) and 21 (5.7%) patients in the FKB238 and bevacizumab arms, respectively. Treatment interruptions were reported for eight (2.2%) patients in the FKB238 arm and 12 (3.3%) patients in the bevacizumab arm. Interruptions due to AEs occurred in five (1.4%) patients in the FKB238 arm compared with eight (2.2%) patients in the bevacizumab arm. TEAEs leading to discontinuation of IPs and associated with these drugs were reported for 14 (3.9%) and 18 (4.9%) patients in the FKB238 and bevacizumab arms, respectively. TEAEs leading to death occurred in 8.3 and 6.3% of patients in the FKB238 and bevacizumab arms, respectively.
4 Discussion
The cost of biologic pharmaceuticals is very high relative to small-molecule drugs and is rising rapidly [26, 27]. The introduction of biosimilars into the market is expected to decrease the cost of biologic medicines. In its Biosimilar Action Plan, the FDA encouraged the development of biosimilars to increase innovation and competition among biologics and potentially reduce the cost of these drugs [28]. Both ASCO and NCCN have also adopted guidelines that encourage cost-effective treatment of cancer [17, 18]. Thus, there is a strong rationale for the development of FKB238 as a new bevacizumab biosimilar [29].
The requirements for a biologic agent to be designated a biosimilar are very rigorous [19,20,21]. These involve the totality of evidence approach, which include detailed analytical studies of various characteristics (such as primary amino acid sequence, pharmacokinetics, efficacy, safety, and immunogenicity) using state-of-the-art technologies to compare the new agent with the reference biologic. At least seven other biosimilars of bevacizumab are approved in different parts of the world, although only two (Mvasi™ and Zirabev™) have been approved by the EMA and the FDA [30]. The indications for which these drugs have been approved include metastatic colorectal cancer; advanced, recurrent, or metastatic non-sq-NSCLC; recurrent glioblastoma in adults; metastatic renal cell carcinoma; persistent, recurrent, or metastatic cervical cancer; and ophthalmological indications. An earlier study demonstrated that the pharmacokinetics, safety profile, and immunogenicity of FKB238 were similar to those of bevacizumab in healthy volunteers [22]. The goal of our study was to demonstrate biologic equivalence between FKB238 and bevacizumab with respect to efficacy, safety, and immunogenicity in patients with non-sq-NSCLC in accordance with guidance from the FDA and EMA.
Bevacizumab, in combination with paclitaxel and carboplatin, has proven efficacy and safety as first-line therapy in NSCLC and other indications; this trial recruited only patients with non-sq-NSCLC with other indications accessed in accordance with the principles of extrapolation [14, 29]. For the ITT population in our study, our observed ORR for FKB238 (51.7%) was very similar to that for bevacizumab (53.7%), with the ratio staying entirely within the limits for equivalence set by the FDA. Similarly, for the PPS, the ORR for FKB238 (51.7%) was similar to that observed for bevacizumab (53.4%), with the risk difference staying entirely within the limits for equivalence set by the EMA. Other efficacy endpoints, ORR at week 19, PFS, and OS, were also similar between the two drugs. Although the OS was 14.1 months for FKB238 compared with 17.0 months for bevacizumab, the estimated HR was not significantly different from 1, and the 95% CI contained the value 1. This phenomenon might be more of a reflection of the advanced stage of disease at presentation, of the patients included in this study, and of the impact of missing survival status for some patients than the efficacy of the drug, since this study was not powered to detect differences [31]. Also as previously noted, more patients in the bevacizumab arm withdrew consent, had a worse AE profile, had worse prognostic factors, and were consequently at a higher risk of information loss on OS than patients in the FKB238 arm. In our study, the safety profile of FKB238 was similar to that of bevacizumab and comparable to historic data for bevacizumab [14]. Furthermore, the incidence of ADAs was low and similar for both FKB238 and bevacizumab.
One limitation of this study is the inclusion of patients with very advanced disease who may not respond as well as patients with less advanced disease. Testing both FKB238 and bevacizumab in patients with less advanced disease may have yielded results demonstrating higher efficacy and survival. However, since the original study that demonstrated the efficacy of bevacizumab in patients with NSCLC was conducted in patients with advanced disease, it was incumbent that this study with a goal to demonstrate biosimilarity between FKB238 and bevacizumab was also conducted in a similar patient population [14]. Another potential issue is the choice of ORR instead of OS as the primary endpoint, since survival-based endpoints are preferred when trying to demonstrate clinical benefit for anticancer therapies. However, they are not suitable for demonstrating biosimilarity [19]. ORR, on the other hand, is a direct measure of the efficacy of treatment, is approved by the FDA and the EMA for comparing the antitumor activity of the putative biosimilar with that of the reference biologic, and has previously been used successfully for this purpose [19, 20, 32, 33].
5 Conclusion
Our data show that FKB238 is similar to bevacizumab in efficacy, safety, and immunogenicity in patients with advanced non-sq-NSCLC. The phase I study demonstrated that FKB238 was similar to bevacizumab in healthy human volunteers with respect to pharmacokinetics, immunogenicity, and safety profiles. Taken together, the totality of data show that FKB238 meets the criteria for biosimilarity set by the FDA and EMA.
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. https://doi.org/10.3322/caac.21654.
Pilkington G, Boland A, Brown T, Oyee J, Bagust A, Dickson R. A systematic review of the clinical effectiveness of first-line chemotherapy for adult patients with locally advanced or metastatic non-small cell lung cancer. Thorax. 2015;70(4):359–67. https://doi.org/10.1136/thoraxjnl-2014-205914.
Crino L, Weder W, van Meerbeeck J, Felip E, Group EGW. Early stage and locally advanced (non-metastatic) non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v103–15. https://doi.org/10.1093/annonc/mdq207.
Assi HI, Kamphorst AO, Moukalled NM, Ramalingam SS. Immune checkpoint inhibitors in advanced non–small cell lung cancer. Cancer. 2018;124(2):248–61. https://doi.org/10.1002/cncr.31105.
Kang J, Zhang C, Zhong W-Z. Neoadjuvant immunotherapy for non-small cell lung cancer: State of the art. Cancer Commun. 2021;41(4):287–302. https://doi.org/10.1002/cac2.12153.
Mascaux C, Tomasini P, Greillier L, Barlesi F. Personalised medicine for nonsmall cell lung cancer. Eur Respir Rev. 2017;26(146): 170066. https://doi.org/10.1183/16000617.0066-2017.
Hoang T, Schiller JH. Advanced NSCLC: from cytotoxic systemic chemotherapy to molecularly targeted therapy. Expert Rev Anticancer Ther. 2002;2(4):393–401. https://doi.org/10.1586/14737140.2.4.393.
Ettinger DS, Aisner DL, Wood DE, Akerley W, Bauman J, Chang JY, et al. NCCN guidelines insights: non-small cell lung cancer, version 5.2018. 2018;16(7):807–821. https://doi.org/10.6004/jnccn.2018.0062.
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–6. https://doi.org/10.1056/NEJM197111182852108.
Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358(19):2039–49. https://doi.org/10.1056/NEJMra0706596.
Manzo A, Montanino A, Carillio G, Costanzo R, Sandomenico C, Normanno N, et al. Angiogenesis inhibitors in NSCLC. Int J Mol Sci. 2017;18(10):2021. https://doi.org/10.3390/ijms18102021.
Genentech. AVASTIN® (bevacizumab). South San Francisco, CA, 2019.
Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis. 2012;15(2):171–85. https://doi.org/10.1007/s10456-011-9249-6.
Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. New Engl J Med. 2006;355(24):2542–50. https://doi.org/10.1056/NEJMoa061884.
United States Food and Drug Administration. Avastin (bevacizumab) Information. In: Services DoHaH, editor. United States Food and Drug Administration; 2018.
European Medicines Agency. Avastin (bevacizumab). London: European Medicines Agency; 2017.
American Society of Clinical Oncolgy. Advocacy priorities. American Society of Clinical Oncology, Alexandria, VA, USA. 2020. https://www.asco.org/advocacy/advocacy-agenda-initiatives/advocacy-priorities. Accessed 22 Apr 2020.
National Comprehensive Cancer Network. Non-small cell lung cancer: NCCN Evidence Blocks™. In: NCCN clinical practice guidelines in oncology (NCCN Guidelines®). NCCN.org. 2020. https://www.nccn.org/professionals/physician_gls/pdf/nscl_blocks.pdf. Accessed 12 Apr 2020.
European Medicines Agency. Guideline on similar biological medicinal products. London: European Medicines Agency; 2014.
United States Food and Drug Administration. Scientific Considerations in demonstrating biosimilarity to a reference product: guidance for industry. In: Services DoHaH, editor. Silver Spring: United States Food and Drug Administration; 2015.
World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs). World Health Organization. 2010. https://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf.
Kaito H, van den Berg F, Niewiarowski A, Boyce M, editors. A randomized, double-blind, single dose comparison of pharmacokinetics and safety of FKB238 with bevacizumab. In: American Society of Clinical Oncology 2017 Annual Meeting; 2017; Chicago, IL, USA: J Clin Oncol.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–47. https://doi.org/10.1016/j.ejca.2008.10.026.
Medical Dictonary for Regulatory Activities (MedDRA) v 21.1. McLean, VA, USA. 2018. https://www.meddra.org/how-to-use/support-documentation/english. Accessed 20 May 2020.
National Cancer Institute. Common terminology criteria for adverse events (CTCAE) v4.0. Bethesda: National Institutes of Health; 2010.
Blackstone EA, Joseph PF. The economics of biosimilars. Am Health Drug Benefits. 2013;6(8):469–78.
Stiff KM, Cline A, Feldman SR. Tracking the price of existing biologics when drugs enter the market. Expert Rev Pharmacoecon Outcomes Res. 2019;19(4):375–7. https://doi.org/10.1080/14737167.2019.1630274.
United States Food and Drug Administration. Biosimilars action plan: balancing innovation and competition. In: Services DoHaH, editor. Silver Spring: United States Food and Drug Administration.
Melosky B, Reardon DA, Nixon AB, Subramanian J, Bair AH, Jacobs I. Bevacizumab biosimilars: scientific justification for extrapolation of indications. Future Oncol. 2018;14(24):2507–20. https://doi.org/10.2217/fon-2018-0051.
Biosimilars of Bevacizumab. Generics and biosimilars initiative (GaBi), Mol, Belgium. 2019. http://gabionline.net/Biosimilars/General/Biosimilars-of-bevacizumab. Accessed 18 Dec 2020.
Wang T, Nelson RA, Bogardus A, Grannis FW Jr. Five-year lung cancer survival. Cancer. 2010;116(6):1518–25. https://doi.org/10.1002/cncr.24871.
Reinmuth N, Bryl M, Bondarenko I, Syrigos K, Vladimirov V, Zereu M, et al. PF-06439535 (a Bevacizumab biosimilar) compared with reference bevacizumab (Avastin(®)), both plus paclitaxel and carboplatin, as first-line treatment for advanced non-squamous non-small-cell lung cancer: a randomized, double-blind study. BioDrugs. 2019;33(5):555–70. https://doi.org/10.1007/s40259-019-00363-4.
Thatcher N, Goldschmidt JH, Thomas M, Schenker M, Pan Z, Paz-Ares Rodriguez L, et al. Efficacy and safety of the biosimilar ABP 215 compared with bevacizumab in patients with advanced nonsquamous non-small cell lung cancer (MAPLE): a randomized, double-blind, phase III study. Clin Cancer Res. 2019;25(7):2088. https://doi.org/10.1158/1078-0432.CCR-18-2702.
Acknowledgements
The authors thank the patients who participated in the trial and all the investigators who contributed to this study. Medical writing assistance was provided by Mukund Nori, PhD, MBA, CMPP of rareLife solutions and funded by AstraZeneca.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Funding
This study was funded by Centus Biotherapeutics.
Conflict of interest
Doctors Syrigos, Abert, Andric, Bondarenko, Dvorkin, Galic, Galiulin, Kuchava, Sriuranpong, Trukhin, and Zhavrid received research support from Centus Biotherapeutics for the conduct of the AVANA trial. Dr. Fu is an employee of AstraZeneca. Mr. Kassalow and Doctors Bashir and Jones are consultants to Centus Biotherapeutics.
Ethics approval
This study was approved by the individual institutional review boards of the study sites and performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments.
Consent
All patients gave informed consent to participate in the trial.
Availability of Data and Material
Data are posted on http://www.ClinicalTrials.gov and can be accessed at: https://www.clinicaltrials.gov/ct2/show/results/NCT02810457?term=NCT02810457&draw=2&rank=1.
Author contributions
The manuscript was developed under the primary guidance of Drs. Syrigos, Bashir, Fu, and Kassalow. All authors reviewed the manuscript at each stage and provided significant direction on the interpretation of the data and its significance.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Syrigos, K., Abert, I., Andric, Z. et al. Efficacy and Safety of Bevacizumab Biosimilar FKB238 Versus Originator Bevacizumab: Results from AVANA, a Phase III Trial in Patients with Non-Squamous Non-Small-Cell Lung Cancer (non-sq-NSCLC). BioDrugs 35, 417–428 (2021). https://doi.org/10.1007/s40259-021-00489-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-021-00489-4