
Abstract
Background
Sandoz adalimumab SDZ-ADL (GP-2017) is an approved adalimumab biosimilar with similar efficacy and comparable safety and immunogenicity to reference adalimumab (ref-ADL) as confirmed by analytical, pharmacokinetic and confirmatory studies. ADMYRA, a phase III double-blind study, was conducted with an aim to generate efficacy, safety and immunogenicity comparability data in patients with moderate-to-severe rheumatoid arthritis (RA) having inadequate response to disease-modifying anti-rheumatic drugs (DMARDs) including methotrexate (MTX). The study also evaluated an aspect of ‘switching’ reference product to the biosimilar in terms of efficacy, safety and immunogenicity up to Week 48.
Methods
Eligible patients (N = 353) were randomized 1:1 to receive subcutaneous (sc) SDZ-ADL 40 mg (n = 177) or ref-ADL (n = 176) every other week from Week 0 to Week 24. At Week 24, all patients with at least a moderate response by Disease Activity Score-28 including high-sensitivity C-reactive protein (DAS28-CRP) in the SDZ-ADL group continued SDZ-ADL (n = 159), and in the ref-ADL group were switched to SDZ-ADL (n = 166), treated for up to 46 weeks. The primary endpoint was change in DAS28-CRP from baseline at Week 12. Other efficacy endpoints included proportion of patients with European League Against Rheumatism (EULAR) response, EULAR remission, Boolean remission, safety and immunogenicity.
Results
The DAS28-CRP score changes from baseline at Week 12 were similar between SDZ-ADL (− 2.16) and ref-ADL (− 2.18) with a mean difference (95% CI) of 0.02 (− 0.24 to 0.27), which was within the pre-specified equivalence margin of ± 0.6. After switching treatment from ref-ADL to SDZ-ADL, the mean DAS28-CRP change was similar between the SDZ-ADL and ‘ref-ADL/switched SDZ-ADL’ group (− 3.09 vs − 3.05). The proportion of patients with good/moderate EULAR response was 69.2%/29.0% in the SDZ-ADL group and 68.0%/29.6% in the ‘ref-ADL/switched SDZ-ADL’ group. The proportion of patients in EULAR remission was 51.4% and 54.4% and in Boolean remission was 16.8% and 21.6% for SDZ-ADL and ‘ref-ADL/switched SDZ-ADL’ groups, respectively. The secondary endpoints were similar across the treatment groups. The incidence of adverse events (AEs) and injection-site reactions were low and similar between SDZ-ADL and ‘ref-ADL/switched SDZ-ADL’ groups (AEs 70.6% vs 68.8%, injection-site reactions 4.0% vs 6.3%), and most of these patients experienced AEs of mild or moderate severity. Antidrug antibodies were detected in 24.2% and 25.6% of patients treated with SDZ-ADL and ‘ref-ADL/switched SDZ-ADL’, respectively, from baseline to Week 48, of which 72.5% in SDZ-ADL and 79.1% in ‘ref-ADL/switched SDZ-ADL’ groups were neutralizing.
Conclusions
In patients with moderate-to-severe RA who had an inadequate response to DMARDs, SDZ-ADL demonstrated a similar efficacy and a comparable safety and immunogenicity profile to ref-ADL. Efficacy was sustained after switching from ref-ADL to SDZ-ADL with no impact on safety (NCT02744755).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
SDZ-ADL and reference adalimumab (ref-ADL) demonstrated equivalent efficacy (in terms of DAS28-CRP score change) in patients with moderate to severe active rheumatoid arthritis who had an inadequate response to disease-modifying anti-rheumatic drugs. Equivalent efficacy was also shown in terms of DAS28-CRP, ACR20/50/70 response rates, EULAR responses and Boolean remission rates. |
The immunogenicity and safety profile of SDZ-ADL and ref-ADL were similar, and local tolerability of the injections was equally good. Switching from ref-ADL to SDZ-ADL did not have any negative impact on the safety or immunogenicity of adalimumab treatment. |
1 Introduction
Patients with chronic disease require long-term treatment options based on the severity of their disease to avoid complications and to improve their health condition and quality of life. Biological medicinal products have been highly effective in treating patients with moderately and severely chronic diseases. However, biologics are expensive and often not accessible to many patients, and can lead to long-term increased financial burden on patients and on health care systems. Biosimilars are biological medical products that are highly similar to other already approved reference products; they show similar safety and efficacy to the reference product and are high quality alternatives [1, 2] leading to reduction of health care costs. It is noteworthy that the development of a biosimilar follows Health Agencies’ biosimilar guidelines and approval is gained via stringent regulatory approval pathways based on the comprehensive similarity data from analytical, preclinical and clinical stages of development [3,4,5]. Due to lowered health care costs with the availability of biosimilars as a substitute for more costly biologics, patients can be treated with biosimilars at an earlier stage of disease, thus preventing or delaying early disease progression and improving quality of life [6].
SDZ-ADL (Hyrimoz®; Sandoz GmbH., Austria) is a biosimilar of Humira® (adalimumab), a recombinant, fully human anti-tumor necrosis factor (anti-TNF)-α IgG1 monoclonal antibody, which is currently approved in the European Union [7], the United States [8] and other countries for the treatment of rheumatoid arthritis (RA), inflammatory bowel disease and other autoimmune diseases as approved for Humira® [9]. Following the European Medicines Agency (EU) and Food and Drug Administration (USA) guided approach of step-wise totality of evidence [10], the analytical and preclinical similarity between SDZ-ADL and reference adalimumab (ref-ADL; Humira®; AbbVie Inc., United States) has been demonstrated [11] prior to pharmacokinetics and therapeutic equivalence [9, 12]. Similar clinical efficacy of SDZ-ADL and ref-ADL has been demonstrated in a phase III study in patients with moderate-to-severe chronic plaque psoriasis [11, 13,14,15].
The ADMYRA study was conducted not only with an aim to contribute further therapeutic equivalence data in a rheumatology indication but also to support regulatory submissions to obtain health authority approval. The objective of this study was to demonstrate similar efficacy and to compare the safety and immunogenicity of SDZ-ADL and ref-ADL in patients with moderate-to-severe RA with inadequate response to methotrexate (MTX) with or without other disease-modifying anti-rheumatic drugs (DMARDs) over 24 weeks. Noteworthy, the study also evaluated the effect of a single switch from ref-ADL to SDZ-ADL at Week 24 on efficacy, safety and immunogenicity (NCT02744755). Herein, we report the results from the 48-week treatment period of the study, with a focus on a comparison between patients who continued with SDZ-ADL treatment (referred to as the ‘SDZ-ADL’ group) and patients who switched from ref-ADL to SDZ-ADL (referred to as the ‘ref-ADL/switched SDZ-ADL’ group).
2 Methods
2.1 Study Design
ADMYRA was a phase III, randomized, double-blind, parallel-group, 48-week multicenter study conducted in 83 centers in 13 countries from March 2016 to September 2017. Eligible patients were randomized 1:1 to receive either SDZ-ADL 40 mg (50 mg/mL) or ref-ADL (Humira®; AbbVie Inc., USA, AbbVie Ltd,) subcutaneously, every other week, for 24 weeks (Study period 1 [SP1]).
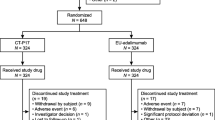
Response was assessed by disease activity score including 28 joint count (DAS28)–C-reactive protein (CRP) score according to European League Against Rheumatism (EULAR) response criteria [16, 17]. At Week 24, patients achieving at least a moderate response in the SDZ-ADL group continued SDZ-ADL (defined as ‘continued SDZ-ADL’ group), and in the ref-ADL group were switched to SDZ-ADL (defined as ‘ref-ADL to SDZ-ADL’ group) for the following period up to Week 46 (Study period 2 [SP2]). During SP2, patients could self-administer SDZ-ADL injections. Patients who did not show at least a moderate EULAR response had their final assessment at Week 24 and did not continue in the study (Fig. 1). End of study visit was performed at Week 48. All patients had to be on a stable dose of MTX at least 4 weeks prior to randomization and continued on the same dose, as well as on a stable dose of folic acid (≥ 5 mg/week) prior to baseline and continued until end of study.

Study design resembled ref-ADL ARMADA trial [25]. Study treatment in Study Period 1: SDZ-ADL or ref-ADL 40 mg/0.8 mL sc injection from Day 1 to W22. Study treatment in Study Period 2: patients with at least moderate EULAR response switched to SDZ-ADL from W24 until W46. In Study Periods 1 and 2, last injections were administered at W22 and W46, respectively; last patient assessments were performed at W24 and W48, respectively. CRP C-reactive protein, DAS disease activity score, EULAR European League Against Rheumatism, Ref-ADL reference adalimumab, sc subcutaneous, SDZ-ADL Sandoz biosimilar adalimumab, W week
The study protocol was reviewed by the Independent Ethics Committee or Institutional Review Board for each center and the study was conducted according to the ethical principles of the Declaration of Helsinki. Informed consent was obtained from each patient and/or legal guardian in writing before entering the study.
2.2 Patients
Patients, aged ≥ 18 years, were included if they had RA according to the American College of Rheumatology (ACR) 1987 or ACR/EULAR 2010 modified criteria for ≥ 6 months before baseline; active disease defined as DAS28-CRP ≥ 3.2 at screening; CRP levels > 5 mg/L or erythrocyte sedimentation rate (ESR) levels above the upper limits of normal. Patients with inadequate clinical response to MTX at a dose of 10–25 mg/week following dose escalation according to local standards were also included. Patients must have had MTX therapy for ≥ 3 months and on a stable dose for at least 4 weeks prior to and at baseline; and on a stable dose of folic acid (≥ 5 mg per week) prior to baseline and during the course of the study. Patients who failed any other DMARD treatment used alone or in combination with MTX were allowed to enter the study after an appropriate wash-out period.
Patients were excluded if they had any prior exposure to adalimumab or other anti-TNFα therapies; prior treatment with a biologic for RA within 6 months or 5 half-lives before baseline; history of hypersensitivity to any recombinant protein drugs or any of the excipients; history of or ongoing inflammatory or autoimmune diseases other than RA; systemic corticosteroids use (> 7.5 mg/day) within 4 weeks prior to baseline; systemic manifestation of RA such as vasculitis, pulmonary fibrosis, peri-, myo-, or endo-carditis etc. (rheumatoid nodules and Sjogren syndrome was not excluded considering these are the most common systemic manifestations of RA and it would be difficult to find such patients); history of tuberculosis or evidence of latent tuberculosis (as assessed by chest X-ray, chest computerized tomography [CT] scan, magnetic resonance imaging [MRI] and/or positive QuantiFERON®-TB Gold test); history of joint surgery within the preceding 2 months before screening; positive for HIV, Hepatitis B or C serology (defined by serological markers, either HBsAg or anti-HBc for Hepatitis B and positive HCVAb or HCV RNA for Hepatitis C); history of persistent chronic infection, recurrent infection or active infections requiring hospitalization or treatment. Patients with functional RA status of class IV according to the ACR 1991 revised criteria [18] at screening and women of child-bearing potential not agreeing to use highly effective contraception methods or nursing or pregnant women were also excluded from the trial.
2.3 Study Endpoints
The primary endpoint was the change from baseline in DAS28-CRP score at Week 12. The key secondary endpoint was the time-weighted averaged change from baseline in DAS28-CRP until Week 24. Other secondary endpoints assessed up to Week 24 and Week 48 included (1) absolute change from baseline in DAS28-CRP scores; (2) proportion of patients achieving EULAR remission (DAS28-CRP < 2.6), or good (improvement of DAS28 from baseline > 1.2) and moderate (improvement of DAS28 from baseline > 0.6 and ≤ 1.2) EULAR response based on DAS28-CRP; (3) proportion of patients achieving remission according to Boolean-based definition (total joint count 28 ≤ 1, swollen joint count 28 ≤ 1, CRP ≤ 1 mg/dL and patient global assessment [PtGA] ≤ 10); (4) proportion of patients achieving an improvement in the ACR20/50/70 response rates and (5) changes in CRP and ESR from baseline.
Safety endpoints included evaluation of the adverse events (AEs) as well as the local tolerability at injection sites of both medications as assessed by the investigator during the study, including the study period after treatment switch. The proportion of patients with anti-drug antibodies (ADAs) was assessed at baseline and at Weeks 2, 4, 12, 24, 36 and 48 by a validated electrochemiluminescence bridging immunogenicity assay and a validated competitive ligand-binding assay.
2.4 Statistical Analyses
The sample size was based on an expected difference of zero and common standard deviation (SD) of 1.30 for the DAS28-CRP change from baseline at Week 12, an equivalence margin of [− 0.6, 0.6], significance level of 5% and assumed dropout rate of 20%. A sample size of 154 patients per treatment group has 90% power to test the equivalence between SDZ-ADL and ref-ADL, which also provides at least 80% to demonstrate equivalence in terms of key secondary endpoint.
A mixed-model repeated measures (MMRM) analysis was performed for DAS28-CRP change from baseline including treatment, stratification factors (region, body weight category and prior therapy), time, the interaction of time and treatment, all as categorical variables, and baseline DAS28-CRP as a continuous covariate. The primary analysis was performed on the Week 12 per-protocol set (W12 PPS) that consisted of all patients who completed Week 12 and do not have any major protocol deviations regarding the study’s primary objective and have received at least five doses of study drug up to Week 10. Therapeutic equivalence, in terms of change from baseline in DAS28-CRP, could be established if the 95% confidence interval (CI) for the difference in mean changes was contained within the interval (− 0.6 to 0.6).
Time-weighted averaged change from baseline in DAS28-CRP until Week 24 was analyzed using an ANCOVA with treatment group and stratifications as factors, and baseline DAS28-CRP value as continuous covariate. The 95% CI for the mean difference between SDZ-ADL and ref-ADL was compared with the pre-specified equivalence margin of [− 0.6; 0.6]. The key secondary endpoint analysis was performed based on the SP1 PPS, which consisted of all patients in the W12 PPS who completed Week 24 and do not have any major protocol deviations regarding the study’s key secondary objective, and have received at least five doses of study drug from Week 12 to Week 22.
Other secondary endpoints and safety and immunogenicity outcomes were summarized descriptively. The analyses of SP1 and SP2 data were based on SP1 FAS (full analysis set), SP1 SAF (safety analysis set) and SP1 PPS (per-protocol set) and SP2 FAS, SP2 SAF and SP2 PPS, respectively; and the entire study data were analyzed based on SP1 FAS, SP1 SAF and SP2 PPS. SAS 9.4 was used to conduct statistical tests.
3 Results
3.1 Patient Disposition and Baseline Clinical Characteristics (SP1 FAS)
In this 48-week study, 353 patients were randomized to SDZ-ADL (n = 177) or ref-ADL (n = 176) and 331 (SDZ-ADL, n = 163; ref-ADL, n = 168) completed SP1. After 24 weeks, 325 patients (‘continued SDZ-ADL’, n = 159; ‘ref-ADL to SDZ-ADL’, n = 166) entered SP2 and 303 completed SP2 (‘continued SDZ-ADL’, n = 145 and ‘ref-ADL to SDZ-ADL’, n = 158; SP2 FAS). During SP1, 14 (7.9%) of the patients in the SDZ-ADL group and 8 (4.5%) in the ref-ADL group discontinued the study (see electronic supplementary material [ESM], Fig. S1). The main reason for discontinuations in SP1 was withdrawal of consent by patients (8 [4.5%] of the patients in the SDZ-ADL group and 4 (2.3%) of the patients in the ref-ADL group). Of these, worsening of RA and mobility was recorded as the reason for withdrawal in only one patient.
After switching the treatments at Week 24, 14 (8.8%) in the ‘continued SDZ-ADL’ group and 8 (4.8%) in the ‘ref-ADL to SDZ-ADL’ group discontinued the study during SP2.
This numerical imbalance was mainly driven by discontinuation due to AEs, which occurred in 5 (3.1%) patients in the ‘continued SDZ-ADL’ group but did not occur in the ‘ref-ADL to SDZ-ADL’ group. The AEs leading to discontinuations during SP2 were two episodes of dyspepsia, one of severe and another of mild severity, one incidence of encephalopathy of moderate severity, one incidence of worsening of rheumatoid arthritis of mild severity, one incidence of hypersensitivity vasculitis of moderate severity and one incidence of liver function test increased of moderate severity in the ‘continued SDZ-ADL’ group and none in the ‘ref-ADL to SDZ-ADL’ treatment group.
There was no difference in compliance to study treatment between continued patients and switched patients in SP2. Over 85% of patients received all doses in SP2 (138/159 [86.8%] in the ‘continued SDZ-ADL’ group and 146/166 [88%] in the ‘ref-ADL to SDZ-ADL’ group). Of these, approximately 8% of patients in each group who completed the SP2 missed only one dose.
Overall, compliance with study treatment administration was similar in both treatment groups during the entire study; approximately 75% of patients completed the study receiving all treatments (no dose missed). Another 16% of patients completed the study missing in total only one dose.
Patient demographics and baseline characteristics were balanced and comparable between both treatment groups during SP1 and SP2 (SDZ-ADL vs ref-ADL; ‘continued SDZ-ADL’ vs ‘ref-ADL to SDZ-ADL’), except for minor differences in the distribution of male and female patients between the treatment groups in SP1.
There were no clinically relevant differences between treatment groups in the baseline disease characteristics (Table 1).
3.1.1 Prior medication (SP1 FAS)
The use of RA medication by the study participants prior to enrollment was similar in both treatment groups of SP1, with sulfasalazine (SDZ-ADL: 35 [19.8%]; ref-ADL: 32 [18.2%]) and MTX (SDZ-ADL: 36 [20.3%]; ref-ADL: 41 [23.3%]) being two most frequently recorded RA medications taken prior.
3.2 Efficacy
3.2.1 Primary Endpoint
3.2.1.1 DAS28-CRP Scores From Baseline at Week 12 (W12 PPS)
The least square (LS) mean (standard error [SE]) change in DAS28-CRP from baseline at Week 12 was − 2.16 (0.114) for SDZ-ADL and − 2.18 (0.110) for ref-ADL (Fig. 2A). The LS means difference (95% CI) was 0.02 (− 0.24 to 0.27), which was within the pre-specified equivalence margin of ± 0.6, demonstrating therapeutic equivalence between SDZ-ADL and ref-ADL. Equivalence was further confirmed with the analysis on FAS population, with a DAS28-CRP response of − 2.02 (0.10) in the SDZ-ADL group and − 2.15 (0.09) in the ref-ADL group (LS mean difference 0.13 [− 0.10 to 0.37]).

a Mean changes from baseline in DAS28-CRP scores over 12 weeks (W12 PPS). A mixed-model repeated measures analysis was performed for DAS28-CRP change from baseline including treatment, stratification factors, time (visits), the interaction between time (visits) and treatment, all as categorical variables, and baseline DAS28-CRP value as a continuous variable. *One patient had more than two joint assessments missing and therefore DAS28-CRP was not calculated. b Mean change in DAS28-CRP scores over 48 weeks (SP2 PPS). The boxed values represent actual mean values for comparison. At BL, actual mean DAS28-CRP for SDZ-ADL and ref-ADL/switched SDZ-ADL were 5.74 and 5.73, respectively. BL baseline, CI confidence interval, CRP C-reactive protein, DAS disease activity score, LS least squares, N number of patients per treatment group, PPS per-protocol set, ref-ADL reference adalimumab, SD standard deviation, SDZ-ADL Sandoz biosimilar adalimumab, SE standard error, SP2 PPS study period 2 per protocol set: all patients in the SP1 PPS who completed the full study and do not have any major protocol deviations during the entire study
3.2.2 Key Secondary Endpoint
3.2.2.1 Time-Averaged Change From Baseline in DAS28-CRP Score until Week 24 (SP1 PPS)
The time-weighted average change from baseline in DAS28-CRP until Week 24 was similar between SDZ-ADL and ref-ADL (SDZ-ADL − 1.85 [0.10]; ref-ADL − 1.93 [0.09]). The LS means difference between the treatment groups was 0.08 (95% CI − 0.11 to 0.27). The 95% CI were completely contained within the pre-specified equivalence margin of [− 0.6, 0.6], demonstrating therapeutic equivalence between SDZ-ADL and ref-ADL until Week 24. Similar responses were observed with the analysis on the FAS (SP1 FAS) population (LS means difference between the treatment groups was 0.11 [95% CI − 0.06 to 0.28]).
3.2.3 Other Secondary Endpoints
Until Week 24, mean change in DAS28 CRP, ACR20/50/70 responses, EULAR responses and mean proportion of patients achieving remission according to EULAR and Boolean definition was similar between both SDZ-ADL and ref-ADL treatment groups [19, 20]. The analysis on the FAS population further confirmed the results (see ESM, Tables S2–S5).
3.2.3.1 Treatment Outcomes, Until Week 48 Including Single Switch
DAS28-CRP Responses (SP2 PPS) Efficacy was maintained throughout the study in both treatment groups, including the single switch until Week 48; similar mean (SD) DAS28-CRP change from baseline was observed between the SDZ-ADL (− 3.09 [1.09]) and ‘ref-ADL/switched SDZ-ADL’ (− 3.05 [1.27]) groups. The mean DAS28-CRP change from baseline to Week 48 including a switch of treatment was similar between the SDZ-ADL and ‘ref-ADL/switched SDZ-ADL’ groups (− 2.74 vs − 2.92) in the FAS population (SP1 FAS). The absolute mean DAS28-CRP scores over 48 weeks was similar between the SDZ-ADL and ‘ref-ADL/switched SDZ-ADL’ groups (Fig. 2b).
DAS28-ESR Responses (SP2 PPS) The DAS28-ESR change from baseline up to Week 48 showed no clinically meaningful differences in terms of LS means and the 95% CI of the LS means difference between the two treatment groups (SDZ-ADL [− 3.25], ‘ref-ADL/switched SDZ-ADL’ [− 3.19]; LS mean difference −0.06 (95% CI [− 0.37 to 0.25]).
EULAR Responses (SP2 PPS) After a single switch, a similar proportion of patients achieved EULAR ‘good’ or ‘moderate’ responses at Week 48 based on DAS28-CRP between SDZ-ADL and ‘ref-ADL/switched SDZ-ADL’ groups: EULAR good/moderate responses, 69.2%/29.0% versus 68.0%/29.6% (Fig. 3a).

Proportion of patients achieving EULAR response and proportion of patients with EULAR and Boolean remission over time (SP2 PPS). a EULAR good/moderate responses up to Week 48. b EULAR and Boolean remission up to Week 48. Dotted lines indicate the treatment switch from ref-ADL group to SDZ-ADL group. EULAR European League Against Rheumatism, ref-ADL reference adalimumab, SDZ-ADL Sandoz biosimilar adalimumab, SP2 PPS study period 2 per protocol set, Wk week
At Week 48, the proportion of patients with good/moderate EULAR response was 63.4%/28.0% in the SDZ-ADL group and 61.7/32.7 in the ‘ref-ADL/switched SDZ-ADL’ group in the FAS population (SP1 FAS).
Remission (EULAR and Boolean) (SP2 PPS) After switching, a similar proportion of patients achieved EULAR remission in the SDZ-ADL (51.4%) and ‘ref-ADL/switched SDZ-ADL’ (54.4%) groups (Fig. 3b). Similarly, the remission statuses were comparable according to the Boolean definition in both treatment groups (SDZ-ADL [16.8%], ‘ref-ADL/switched SDZ-ADL’ [21.6%]).
At Week 48, the proportion of patients in EULAR remission was 46.0% and 49.7% and in Boolean remission was 17.4% and 19.6% for SDZ-ADL and ‘ref-ADL/switched SDZ-ADL’ groups, respectively, in the FAS population (SP1 FAS).
ACR Responses (SP2 PPS) After the switch, ACR20/50/70 responses were increased to 86.1%, 66.7% and 45.4% in the SDZ-ADL and 88.1%, 64.3% and 43.7% in the ‘ref-ADL/switched SDZ-ADL’ groups, respectively (Fig. 4), and the proportion of patients achieving ACR responses was comparable in both treatment groups over 48 weeks. At Week 24 and after switch, the individual components of the ACR criteria were comparable between SDZ-ADL and ‘ref-ADL/switched SDZ-ADL’ groups (see ESM Table S1).

ACR20/50/70 responses over 48 weeks (SP2 PPS). Dotted lines indicate the treatment switch from ref-ADL group to SDZ-ADL group. ACR American College of Rheumatology, ref-ADL reference adalimumab, SDZ Sandoz biosimilar adalimumab, SP2 PPS study period 2 per protocol set, Wk week
At Week 48, the proportions of patients achieving ACR20/50/70 responses were 75.4%, 56.9% and 36.5% in the SDZ-ADL groups and 84.8%, 61.8% and 40.0% in the ‘ref-ADL/switched SDZ-ADL’ groups, respectively, in the FAS population (SP1 FAS).
CRP and ESR (SP2 PPS) At baseline, CRP and ESR levels were similar between SDZ-ADL and ref-ADL treatment groups. No clinically meaningful difference in CRP levels was observed between the two treatment groups, up to Week 48 (Fig. 5). However, at Week 24, with SP1 PPS the absolute change from baseline in the CRP levels in SDZ-ADL-treated patients was lower compared with that in the ‘ref-ADL/switched SDZ-ADL’-treated patients (− 2.51 vs − 5.30).

Mean change in CRP and ESR levels over 48 weeks (SP2 PPS). a CRP, b ESR. Dotted lines indicate the treatment switch from ref-ADL group to SDZ-ADL group. CRP C-reactive protein, ESR erythrocyte sedimentation rate, ref-ADL reference adalimumab, SDZ-ADL Sandoz biosimilar adalimumab, SP2 PPS study period 2 per protocol set
In the FAS population (SP1 FAS), the change from baseline in CRP was − 3.4 versus − 5.6 and ESR was − 16.7 versus − 18.1 in the SDZ-ADL and ‘ref-ADL/switched SDZ-ADL’ groups, respectively.
3.3 Safety (SP1 SAF; SP2 SAF)
The duration of exposure to study drug (median [min, max]) during the entire study was similar between SDZ-ADL (323 days [1, 353]) and ‘ref-ADL/switched SDZ-ADL’ (323 days [1, 345]) groups. Over 48 weeks, the incidence of treatment-emergent AEs (TEAEs) was comparable in patients with at least one TEAE in the SDZ-ADL (n = 125, 70.6%) and ‘ref-ADL/switched SDZ-ADL’ (n = 121, 68.8%) groups (Table 2).
Viral upper respiratory tract infection was reported with highest incidence in both SDZ-ADL and ‘ref-ADL/switched SDZ-ADL’ groups (16.4% vs 10.8%) followed by upper respiratory tract infection (7.9% vs 5.1%).
The incidence of TEAEs was similar between SDZ-ADL and ref-ADL groups up to Week 24 [20]. Up to Week 24 (SP1), the incidence of severe AEs was identical in both treatment groups (SDZ-ADL 3/177, 1.7% and ref-ADL 3/176, 1.7%), and most patients experienced AEs of mild or moderate severity. The incidence of TEAEs during the switch was also similar between ‘continued SDZ-ADL’ (36.5%, n = 58) and ‘ref-ADL to SDZ-ADL’ (32.5%, n = 54) groups (Table 3).
No deaths were reported throughout the study. The incidence of SAEs during the entire study duration were low and similar in both treatment groups (SDZ-ADL 7 [4.0%]; ‘ref-ADL/switched SDZ-ADL’ 10 [5.7%]; Table 4). Up to Week 24, the incidence of SAEs was similar in both treatment groups (SDZ-ADL 5 [2.8%] and ref-ADL 4 [2.3%]). The proportion of patients with at least one SAE during the switch was low and comparable between ‘continued SDZ-ADL’ (4 [2.5%]) and ‘ref-ADL to SDZ-ADL’ (6 [3.6%]) groups.
No SAEs with a suspected causal relationship to study drug were reported until Week 24. After switching, two patients in the ‘ref-ADL to SDZ-ADL’ group were reported with SAEs (bronchitis 1 [0.6%]; pneumonia 1 [0.6%]) suspected to be related to study drug (SDZ-ADL) by the investigator.
During SP1, two (1.1%) patients in both SDZ-ADL and ref-ADL groups were reported with AEs leading to study treatment discontinuation. In the SDZ-ADL group, one (0.6%) patient discontinued study treatment due to severe bacterial pneumonia and one (0.6%) patient discontinued because of pregnancy. In the ref-ADL group, one (0.6%) patient discontinued study treatment due to localized mild infection and one (0.6%) patient because of a severe benign brain neoplasm and nervous system disorders. During SP2, seven (4.4%) patients in the ‘continued SDZ-ADL’ (rheumatoid arthritis, 3 [1.9%] and hypersensitivity vasculitis, dyspepsia, liver function test increased, encephalopathy, 1 [0.6%] patient each) and one (0.6%) patient in the ‘ref-ADL to SDZ-ADL’ group (utricaria,1 [0.6%]) discontinued study treatment due to TEAEs. None of the RA events (3 [1.9%]) were suspected to be related to SDZ-ADL.
The overall incidence of TEAEs of special interest was comparable between SDZ-ADL (27 [15.3%]) and ‘ref-ADL/switched SDZ-ADL’ (26 [14.8%]) groups over 48 weeks. Up to Week 24, the incidence of TEAEs of special interest was similar in the SDZ-ADL group (19 [10.7%]) and ref-ADL group (18 [10.2%]). After switch, the incidence of TEAEs was higher in the ‘continued SDZ-ADL’ group (14 [8.8%]), compared with the ‘ref-ADL to SDZ-ADL’ (8 [4.8%]) group.
3.3.1 Injection-Site Reactions (ISRs)
The proportion of patients with ISRs before the switch was lower in the SDZ-ADL group (7 [4.0%]) compared with the ref-ADL group (11 [6.3%]). All except two ISRs (injection-site discoloration and injection pruritus) reported in the ref-ADL group were suspected to be related to study drug. Most importantly, after switching, the proportion of patients with ISRs was highly similar and very low in both treatment groups (‘continued SDZ-ADL’ 1 [0.6%]; ‘ref-ADL to SDZ-ADL’ 2 [1.2%]). All ISRs during the switching period were suspected to be study drug related. All ISRs were of mild or moderate severity and none led to study drug discontinuation.
In this study, incidence of injection-site pain was collected as a part of injection-site reactions. Overall, injection-site pain was reported in two (1.1%) of the patients in the SDZ-ADL group and one (0.6%) of the patients in the ‘ref-ADL/switched SDZ-ADL’ group, which was similar to that observed before switching (24 weeks). Importantly, in SP2, no patients reported injection-site pain when switching from ref-ADL to SDZ-ADL in either of the groups.
3.3.2 Immunogenicity
The incidence of ADAs was similar in the SDZ-ADL (40 [24.2%]) and ‘ref-ADL/switched SDZ-ADL’ (43 [25.6%]) groups over 48 weeks and the incidence of neutralizing ADAs was also reported with no meaningful differences between the two treatment groups (SDZ-ADL: 29 [72.5%] vs ‘ref-ADL/switched SDZ-ADL’: 34 [79.1%]). After the single switch at Week 24, 36 (24.0%) and 42 (26.3%) patients were positive for ADAs in ‘continued SDZ-ADL’ and ‘ref-ADL to SDZ-ADL’ groups, respectively, and 26 (72.2%) versus 34 (81.0%) of these were neutralizing.
4 Discussion
This study demonstrated similar efficacy of the adalimumab biosimilar, SDZ-ADL, and ref-ADL in patients with moderate-to-severe RA. Efficacy was sustained after switching study participants from ref-ADL to SDZ-ADL and was comparable between SDZ-ADL and ‘ref-ADL to SDZ-ADL’ treatment groups. This 48-week data on efficacy and safety in patients with RA supports the biosimilarity established between SDZ-ADL and ref-ADL and further contributes to the totality of evidence established from previous studies [13, 21].
A DAS28-CRP score was used for primary endpoint evaluation in this study. DAS28-CRP change from baseline, a continuous endpoint, is considered as a better measure to follow up the disease course and therefore a sensitive efficacy endpoint [22], compared to a binary or dichotomous endpoint like ACR20, which is associated with the limitation that a small change in disease activity may have a substantial impact on the allocated disease outcome category [23]. Additionally, a dichotomous endpoint like ACR20 does not necessarily reflect treatment targets of remission or low disease activity. As their clinical relevance may not be immediately clear, improvement of response outcomes are in general not considered as primary endpoints [24].
Differences were observed in SDZ-ADL and ref-ADL treated patients in EULAR remission based on DAS28-CRP and ACR20/50/70 response rates, which could be attributed to the differences in the CRP. Mean CRP levels (absolute changes from baseline) in SDZ-ADL-treated patients at Week 24 were half that in the ref-ADL-treated patients. This observation of CRP levels was consistent with the higher frequency of upper respiratory tract (indicative of viral exposure) infection in the SDZ-ADL-treated patients as compared to the ref-ADL-treated patients.
Overall, EULAR and ACR20/50/70 response rates observed in this study are consistent or better than those reported earlier [25]. Also, the proportion of patients achieving EULAR/ACR Boolean remission criteria throughout the study was similar between SDZ-ADL and ref-ADL treatment groups. Based on the totality of generated data, also in more sensitive variables such as DAS28-CRP, these differences in the EULAR remission rate and ACR20/50/70 are not indicative of true differences between the treatment groups and were not considered clinically meaningful.
Regulatory guidelines mandate rigorous evaluation and confirmation of biosimilarity between the proposed biosimilar and the approved reference products based on the totality of evidence comprising biochemical, nonclinical and clinical data [26, 27].
Although physicians support biosimilars, they may be less confident in extrapolating indications and switching patients from a biologic to a biosimilar [28] due to lack of adequate data supporting each approved indication or the switching of treatment, or even inadequate guidelines. There are concerns for AEs and potentially increased immunogenicity with adalimumab, which has higher intrinsic immunogenicity than other anti-TNFs [29].
In recent years, numerous data became available from several studies that reported no difference in outcomes between reference and patients switched to the biosimilar. On the other hand, studies have shown that patients switching from reference biologic medicinal product to biosimilars experience worsened feeling or symptoms due to patients’ negative perception of biosimilars (i.e. nocebo effect) [30, 31]. However, it has been reported that a single and multiple switches from a reference biologic medicinal product to a biosimilar is safe and effective [32,33,34,35].
Real-world evidence from studies assessing the differences or similarities between a reference biologic medicinal product and the biosimilar would be advantageous to address these concerns. As the manufacturing processes are not exactly identical but similar for the two products, switching is still a concern among physicians as various factors such as impact on drug exposure, safety and efficacy need to be considered. Such data could, in addition, aid physicians in prescribing biosimilars, and in addressing fears of negative consequences upon switching to a biosimilar [36, 37].
The switch from reference biologic medicinal product to a biosimilar was also evaluated in this study and results are consistent with the findings from the study in patients with moderate-to-severe chronic plaque-type psoriasis, which also showed that switching between ref-ADL and SDZ-ADL does not have an impact on efficacy, safety, or immunogenicity of ref-ADL [13]. The safety profile of SDZ-ADL was consistent with the known safety profile of ref-ADL [38, 39]. Over 48 weeks, incidence of TEAEs was comparable between the two treatment groups. Switching of patients from ref-ADL to SDZ-ADL at Week 24 did not reveal any new safety concerns by the end of the study at Week 48. Antibodies directed against the biological medicine (ADAs) could neutralize the medicine’s activity and reduce its efficacy. Thus, potential immunogenicity always needs to be evaluated for all biological medicines [3]. In this study, immunogenicity in terms of ADAs and neutralizing antibodies was similar for patients in the SDZ-ADL and ref-ADL groups before and after the switch. ISRs including injection-site pain, erythema, itching, hemorrhage or swelling have been reported as one of the most common adverse drug reactions across indications. Patient discomfort due to injection-site pain, in particular, may lead to treatment non-compliance in case of chronic diseases such as RA [12]. In this study, no increased incidence of injection-site pain was noted in patients switching to SDZ-ADL. These results support data on switching from reference product to a biosimilar and efficacy, safety and immunogenicity were similar between patients who switched or continued treatment.
4.1 Study Limitations
Clinicians are concerned about switching patients on stable treatment with the reference medicine to a biosimilar. The ADMYRA study evaluates efficacy, safety and immunogenicity after treatment switch from the reference product (ref-ADL) to the biosimilar (SDZ-ADL). The study was not designed to assess the effect of treatment switching, but rather the primary focus of the study was to assess therapeutic equivalence in patients with rheumatoid arthritis. Nonetheless, the descriptive analysis of switching data does indicate that the treatment effect was sustained and no impact on safety and immunogenicity was observed.
5 Conclusions
In patients with moderate-to-severe RA who had an inadequate response to DMARDs, switching to SDZ-ADL demonstrated a similar efficacy and a comparable safety and immunogenicity profile from the head-to-head comparison to ref-ADL. Efficacy was sustained after switching from ref-ADL to SDZ-ADL with no impact on safety and immunogenicity.
References
Braun J, Kudrin A. Switching to biosimilar infliximab (CT-P13): evidence of clinical safety, effectiveness and impact on public health. Biologicals. 2016;44(4):257–66.
Smolen JS, Goncalves J, Quinn M, Benedetti F, Lee JY. Era of biosimilars in rheumatology: reshaping the healthcare environment. RMD Open. 2019;5(1):e000900.
EuropeanMedicinesAgency. Biosimilars in the EU: Information guide for healthcare professionals. 2019. https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf. Accessed 07 May 2019.
EuropeanMedicinesAgency. Guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues 2012. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical_en.pdf. Accessed 07 May 2019.
FDA. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. 2015.
Kuek A, Hazleman BL, Ostor AJ. Immune-mediated inflammatory diseases (IMIDs) and biologic therapy: a medical revolution. Postgrad Med J. 2007;83(978):251–60.
EuropeanMedicinesAgency. Summary Of Product Characteristics. Hyrimoz. https://www.ema.europa.eu/en/documents/product-information/hyrimoz-epar-product-information_en.pdf. Accessed 07 May 2019.
FDA. Prescribing information. Hyrimoz. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761071.pdf. Accessed 07 May 2019.
FDA. Highlights of prescribing information. Humira. https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/125057s0110lbl.pdf. Accessed 07 May 2019.
Markus R, Liu J, Ramchandani M, Landa D, Born T, Kaur P. Developing the totality of evidence for biosimilars: regulatory considerations and building confidence for the healthcare community. BioDrugs. 2017;31(3):175–87.
da Silva A, Fritsch C, Kronthaler U, Hofmann H-P, Hainzl O, Seidl A. Non-clinical pharmacological characterization and in-vivo efficacy of the proposed adalimumab biosimilar GP2017 compared to the originator adalimumab: 617. Am J Gastroenterol. 2016;111:S284.
EuropeanMedicinesAgency. Summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/humira-epar-product-information_en.pdf. Accessed 07 May 2019.
Blauvelt A, Lacour JP, Fowler JF Jr, Weinberg JM, Gospodinov D, Schuck E, et al. Phase III randomized study of the proposed adalimumab biosimilar GP2017 in psoriasis: impact of multiple switches. Br J Dermatol. 2018;179(3):623–31.
Jauch-Lembach J, Skerjanec A, Haliduola H, Hass N, von Richter O, Fuhr R, et al. Randomized, double-blind, single-dose, three-arm parallel trial to determine the pharmacokinetics and safety of GP2017, EU- and US-adalimumab in healthy male subjects [abstract 2443]. Arthritis Rheumatol. 2017;69(S10).
Blauvelt A, Lacour J, Fowler J, Schuck E, Jauch-Lembach J, Balfour A, et al. Long-term efficacy, safety and immunogenicity results from a randomized, double-blind, phase III confirmatory efficacy and safety study comparing GP2017, a proposed biosimilar, with reference adalimumab [abstract 2440]. Arthritis Rheumatol. 2017;69(S10).
Fransen J, van Riel PL. The Disease Activity Score and the EULAR response criteria. Rheum Dis Clin North Am. 2009;35(4):745–57, vii–viii.
van Gestel AM, Prevoo ML, van ‘t Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism Criteria. Arthritis Rheum. 1996;39(1):34–40.
Hochberg MC, Chang RW, Dwosh I, Lindsey S, Pincus T, Wolfe F. The American College of Rheumatology 1991 revised criteria for the classification of global functional status in rheumatoid arthritis. Arthritis Rheum. 1992;35(5):498–502.
Wiland P, Jeka S, Dokoupilova E, Limon JMM, Jauch-Lembach J, Thakur A, et al. Efficacy, safety, and immunogenicity results of the switch from reference adalimumab (ref-ADL) to sandoz biosimilar adalimumab (GP2017, SDZ-ADL) from ADMYRA phase 3 study in patients with moderate-to-severe rheumatoid arthritis (RA). Ann Rheum Dis. 2019;78:706–7.
Wiland P, Jeka S, Dokoupilová E, Miranda Limón JM, Jauch-Lembach J, Thakur A, et al. A randomized, double-blind, parallel-group, multicenter study to compare the efficacy, safety and immunogenicity of a proposed adalimumab biosimilar (GP2017) with reference adalimumab in patients with moderate-to-severe active rheumatoid arthritis [abstract 1936]. Arthritis Rheumatol. 2018;70(S10).
Schiestl M, Roesli C. Development and analytical characterization of GP2017: a proposed adalimumab biosimilar. Am J Gastroenterol. 2016;111:S289-S.
van Riel PL. The development of the disease activity score (DAS) and the disease activity score using 28 joint counts (DAS28). Clin Exp Rheumatol. 2014;32(5 Suppl 85):S-65-74.
van Riel PL, van Gestel AM. Area under the curve for the American College of Rheumatology improvement criteria: a valid addition to existing criteria in rheumatoid arthritis? Arthritis Rheum. 2001;44(7):1719–21.
EuropeanMedicinesAgency. Guideline on clinical investigation of medicinal products for the treatment of rheumatoid arthritis. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-treatment-rheumatoid-arthritis_en.pdf. Accessed 07 May 2019.
Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48(1):35–45.
Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Narayanan G, et al. Biosimilars-why terminology matters. Nat Biotechnol. 2011;29(8):690–3.
EuropeanMedicinesAgency. Guideline on similar biological medicinal products. WC500180219. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf. Accessed 07 May 2019.
Hemmington A, Dalbeth N, Jarrett P, Fraser AG, Broom R, Browett P, et al. Medical specialists’ attitudes to prescribing biosimilars. Pharmacoepidemiol Drug Saf. 2017;26(5):570–7.
Moots RJ, Xavier RM, Mok CC, Rahman MU, Tsai WC, Al-Maini MH, et al. The impact of anti-drug antibodies on drug concentrations and clinical outcomes in rheumatoid arthritis patients treated with adalimumab, etanercept, or infliximab: results from a multinational, real-world clinical practice, non-interventional study. PLoS One. 2017;12(4):e0175207.
Petersen GL, Finnerup NB, Colloca L, Amanzio M, Price DD, Jensen TS, et al. The magnitude of nocebo effects in pain: a meta-analysis. Pain. 2014;155(8):1426–34.
Rezk MF, Pieper B. Treatment outcomes with biosimilars: be aware of the nocebo effect. Rheumatol Ther. 2017;4(2):209–18.
Cohen SB, Alten R, Kameda H, Hala T, Radominski SC, Rehman MI, et al. A randomized controlled trial comparing PF-06438179/GP1111 (an infliximab biosimilar) and infliximab reference product for treatment of moderate to severe active rheumatoid arthritis despite methotrexate therapy. Arthritis Res Ther. 2018;20(1):155.
Kay J, Schoels MM, Dorner T, Emery P, Kvien TK, Smolen JS, et al. Consensus-based recommendations for the use of biosimilars to treat rheumatological diseases. Ann Rheum Dis. 2018;77(2):165–74.
Weinblatt ME, Baranauskaite A, Dokoupilova E, Zielinska A, Jaworski J, Racewicz A, et al. Switching from reference adalimumab to SB5 (adalimumab biosimilar) in patients with rheumatoid arthritis: fifty-two-week phase III randomized study results. Arthritis Rheumatol. 2018;70(6):832–40.
Gerdes S, Thaci D, Griffiths CEM, Arenberger P, Poetzl J, Wuerth G, et al. Multiple switches between GP2015, an etanercept biosimilar, with originator product do not impact efficacy, safety and immunogenicity in patients with chronic plaque-type psoriasis: 30-week results from the phase 3, confirmatory EGALITY study. J Eur Acad Dermatol Venereol. 2018;32(3):420–7.
Moots R, Azevedo V, Coindreau JL, Dorner T, Mahgoub E, Mysler E, et al. Switching between reference biologics and biosimilars for the treatment of rheumatology, gastroenterology, and dermatology inflammatory conditions: considerations for the clinician. Curr Rheumatol Rep. 2017;19(6):37.
Wiland P, Batko B, Brzosko M, Kucharz EJ, Samborski W, Swierkot J, et al. Biosimilar switching—current state of knowledge. Reumatologia. 2018;56(4):234–42.
Burmester GR, Panaccione R, Gordon KB, McIlraith MJ, Lacerda AP. Adalimumab: long-term safety in 23 458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn’s disease. Ann Rheum Dis. 2013;72(4):517–24.
Sator P. Safety and tolerability of adalimumab for the treatment of psoriasis: a review summarizing 15 years of real-life experience. Ther Adv Chronic Dis. 2018;9(8):147–58.
Acknowledgements
The authors thank all investigators (Clinicaltrials.gov: NCT02744755) and participating patients who contributed to the successful conduct of this study, and Deepak Pakalapati (Product Lifecycle Services-NBS, Novartis Healthcare Pvt. Ltd., Hyderabad, India) for medical writing and editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Hexal AG, a Sandoz company.
Conflict of interest
Piotr Wiland: None declared. Sławomir Jeka: None declared. Eva Dokoupilová: Research grants from AbbVie, Eli Lilly, GSK, Novartis, Pfizer, UCB, Sanofi-Aventis and Hexal AG. Jan Brandt-Jürgens: None declared. Juan Manuel Miranda Limón: None declared. Miguel Cantalejo Moreira: None declared. Raul Veiga Cabello: None declared. Julia Jauch-Lembach: Employee of Sandoz. Anjali Thakur: Employee of Sandoz. Halimuniyazi Haliduola: Employee of Sandoz. Ines Brueckmann: Employee of Sandoz. Norman B. Gaylis: None declared.
Ethical approval
The study was conducted in accordance with the ethical principles derived from the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practices and in compliance with local regulatory requirements. The study protocols were approved by the Independent Ethics Committee or Institutional Review Board for each center.
Informed consent
All patients provided written informed consent before entering the study.
Consent for publication
All patients provided their consent for their data to be published.
Availability of data and material
All data generated or analyzed are included in this article and the supplementary information files. Anonymized datasets and related documents such as the statistical analysis plan, protocol and amendments can be shared upon reasonable request through a data sharing agreement.
Code availability
Not applicable.
Author contributions
Julia Jauch-Lembach and Halimuniyazi Haliduola contributed to the design of the study; Piotr Wiland, Sławomir Jeka, Eva Dokoupilová, Jan Brandt-Jürgens, Juan Manuel Miranda Limón, Miguel Cantalejo Moreira, Raul Veiga Cabello and Norman B. Gaylis contributed to the conduct of the clinical study; Halimuniyazi Haliduola, Julia Jauch-Lembach, Anjali Thakur and Ines Brueckmann contributed to the analysis and interpretation of the results. All authors discussed the results and contributed to the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Wiland, P., Jeka, S., Dokoupilová, E. et al. Switching to Biosimilar SDZ-ADL in Patients with Moderate-to-Severe Active Rheumatoid Arthritis: 48-Week Efficacy, Safety and Immunogenicity Results From the Phase III, Randomized, Double-Blind ADMYRA Study. BioDrugs 34, 809–823 (2020). https://doi.org/10.1007/s40259-020-00447-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-020-00447-6