
Abstract
Mucopolysaccharidosis VII is an extremely rare, autosomal recessive lysosomal storage disorder characterized by a deficiency of β-glucuronidase activity, resulting in partial degradation and accumulation of GAGs in numerous tissues throughout the body, with consequent cellular damage and organ dysfunction. Enzyme replacement therapy (ERT) with intravenous vestronidase alfa (Mepsevii™), a recombinant form of human β-glucuronidase, is the first disease-specific therapy approved for the treatment of mucopolysaccharidosis VII in pediatric and adult patients. In the pivotal, blind start, phase 3 trial, 24 weeks of vestronidase alfa therapy significantly reduced urinary GAG (uGAG) excretion in patients with mucopolysaccharidosis VII. Based on a Multi-Domain Responder Index (MDRI; comprises six clinically important morbidity domains, with prespecified minimally important differences for each domain), most evaluable patients experienced an improvement in ≥ 1 domain during the 24-week primary assessment period (overall positive mean change of 0.5 domains). The clinical benefits of vestronidase alfa were sustained during longer-term treatment, as was the reduction in uGAG excretion. Vestronidase alfa has a manageable tolerability profile, with most adverse reactions of mild to moderate severity. Given the lack of treatment options and the clinical benefits it provides, intravenous vestronidase alfa is an important emerging ERT for patients with mucopolysaccharidosis VII.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Recombinant enzyme intended to replace deficient endogenous lysosomal β-glucuronidase |
Catabolizes accumulated GAGs systemically |
Significantly reduces uGAG excretion from baseline to 24 weeks, with most patients having a clinically meaningful improvement in ≥ 1 MDRI domain |
Maintains clinical benefits during longer-term therapy |
1 Introduction
Mucopolysaccharidosis VII (also known as Sly syndrome) is an extremely rare, autosomal recessive lysosomal storage disorder caused by mutations in the GUSB gene [1, 2]. GUSB encodes β-glucuronidase [3], an enzyme that plays an essential role in the degradation of specific glycosaminoglycans (GAGs) in lysosomes, namely chondroitin sulfate, dermatan sulfate, and heparan sulfate [1, 4]. In patients with mucopolysaccharidosis VII, the enzymatic activity of β-glucuronidase is deficient [2], resulting in partial degradation and accumulation of GAGs in numerous tissues throughout the body, with consequent cellular damage and organ dysfunction [1, 4]. Although the exact prevalence of mucopolysaccharidosis VII is unknown, it is estimated to be < 1 in 1,000,000 individuals [5].
The clinical presentation and onset of mucopolysaccharidosis VII varies greatly, ranging from earlier onset (e.g. at birth) to a later onset (e.g. during childhood) of clinical manifestations [6, 7]. Patients who present in childhood typically exhibit mild skeletal abnormalities, coarse facial features, and corneal clouding; those who present at birth typically have short stature and greater skeletal dysplasia, hepatosplenomegaly, macrocephaly, gingival hypertrophy, hernias, frequent ear infections, and cognitive impairment [7]. Irrespective of age, patients may also have cardiovascular impairments, decreased pulmonary function, and limited mobility [7]. The phenotype of individuals most profoundly impacted is characterized by the presence of non-immune hydrops fetalis (an excessive accumulation of fluid within fetal body cavities and extravascular compartments) [7]. The life expectancy of patients with mucopolysaccharidosis VII is shortened, with the specifics dependent on the nature of clinical manifestations, and ranges from infancy to adulthood [6, 7].
Enzyme replacement therapy (ERT) with vestronidase alfa (Mepsevii™), a recombinant form of human β-glucuronidase, is the first disease-specific treatment for mucopolysaccharidosis VII [8,9,10]. Its development was based on the success of ERT in patients with other types of mucopolysaccharidoses [11,12,13,14,15,16], as well as promising preclinical data of ERT using recombinant mouse β-glucuronidase in murine models of mucopolysaccharidosis VII [17,18,19]. Intravenous vestronidase alfa is approved for the treatment of mucopolysaccharidosis VII in pediatric and adult patients in the USA [8], the EU (non-neurological manifestations only) [9], and Brazil [10]. This article reviews the efficacy and tolerability of vestronidase alfa in this indication, and summarizes its pharmacological properties.
2 Pharmacological Properties of Vestronidase Alfa
Vestronidase alfa, a recombinant homotetramer with the same amino acid sequence as that of human β-glucuronidase, acts as an exogenous source of β-glucuronidase [8, 9]. The mannose-6-phosphate (M6P) residues on its oligosaccharide chains bind to the cation-independent M6P receptor on cell surfaces [8, 20]. Vestronidase alfa is subsequently internalized into cell lysosomes, where it acts to catabolize accumulated GAGs in affected tissues [8, 9].
In clinical trials in patients with mucopolysaccharidosis VII, significant reductions in urinary GAG (uGAG) excretion occurred following multiple doses of vestronidase alfa (Sect. 3). According to population pharmacokinetic (PPK) analyses, the relationship between uGAG excretion (both urinary chondroitin sulfate and urinary dermatan sulfate) and vestronidase alfa serum exposure is described by an inhibitory maximal effect model [21]. Treatment with vestronidase alfa 4 mg/kg once every other week [recommended dosage (Sect. 5)] was associated with greater pharmacodynamic responses and pharmacokinetic exposure than dosages of 1 or 2 mg/kg once every other week. A flat exposure-response relationship was seen with vestronidase alfa serum exposures above a cut-off value that approximately corresponds to the area under the concentration-time curve at 40 μg·h/mL. This cut-off exposure level is exceeded with a 4 mg/kg dose, indicating that uGAG reduction had likely reached a plateau, with any increase in dose > 4 mg/kg not predicted to improve treatment outcomes [21].
Vestronidase alfa exhibited approximately proportional serum exposure within a 1−4 mg/kg dose range in patients with mucopolysaccharidosis VII [8]. With repeated dosing, its pharmacokinetics were time-independent [9]. In patients receiving vestronidase alfa 4 mg/kg every other week, the mean maximal serum concentration was 20 μg/mL [9, 20]. Drug concentrations in patients aged < 5 years were similar to those in older children (aged ≥ 5 years) and adults [8]. Vestronidase alfa has a mean total volume of distribution of 0.26 L/kg [8, 9] or 0.19 L/kg (based on PPK analyses) [21], indicating that the distribution of the drug into the extravascular space is limited [21].
Vestronidase alfa is eliminated by proteolytic degradation into amino acids and small peptides [8, 9]. The mean total clearance of vestronidase alfa is ≈ 0.079 L/h/kg [8, 9] with an inter- and intra-subject variability of 59 and 13% [8]. Vestronidase alfa has a short mean elimination half-life (t½) of 2.6 h in patients with mucopolysaccharidosis VII [8, 9], and an extensive intracellular t½ of ≈ 40 d in mucopolysaccharidosis VII fibroblasts in vitro (supporting the recommended dosing frequency; Sect. 5) [20]. No excretion studies of vestronidase alfa have been conducted in humans; the drug is not expected to be eliminated through fecal or renal excretion [8, 9].
In PPK analyses, the only covariate that significantly altered the pharmacokinetics of vestronidase alfa was baseline bodyweight [21]. Estimates of allometric baseline bodyweight scalars for clearances and volumes were 0.59 and 0.48, supporting an infusion rate of vestronidase alfa based on bodyweight (Sect. 5) [21].
3 Therapeutic Efficacy of Vestronidase Alfa
The efficacy of intravenous vestronidase alfa (≈ 4 h infusion) in 23 treatment-naive pediatric and/or adult patients (aged 5 months to 25 years at enrollment; 19 were aged < 18 years) with confirmed mucopolysaccharidosis VII was investigated in an open-label, dose-exploration phase 1/2 trial [22] and its ongoing long-term extension (Sect. 3.3), an open-label phase 2 trial [23] (Sect. 3.3), and a pivotal, randomized, placebo-controlled, blind start, multicenter, phase 3 trial [24] (Sect. 3.1) and its ongoing open-label extension [25] (Sect. 3.2). The dosage of vestronidase alfa in the phase 3 trial was based on the optimal regimen in the dose-exploration phase 1/2 trial [26] (Sect. 3.3).
Eligible patients had uGAG excretion levels that were a minimum of twofold (phase 1/2 [22] and phase 2 [23]) or threefold (phase 3 [24]) above the mean normal for age at screening. In the phase 3 trial, other key inclusion criteria were no prior vestronidase alfa treatment and the presence of clinical signs of lysosomal storage disease (i.e. ≥ 1 of the following: joint limitations, airway obstruction/pulmonary problems, enlarged liver and spleen, and/or limitation of mobility while still ambulatory) [26, 27]. Two patients in each trial had non-immune hydrops fetalis [26]. Key exclusion criteria included a previous successful bone marrow transplant (BMT) or any degree of detectable chimerism in donor cells and/or a concurrent disease or condition that would interfere with the participation of the study or affect the safety of the patient [22, 23, 27].
3.1 Pivotal Trial
Four male and eight female patients (aged 8−25 years; median age 14 years) were randomized in a blinded manner to vestronidase alfa 4 mg/kg every other week for 48 weeks (Group A) or placebo with crossover to vestronidase alfa 4 mg/kg at prespecified timepoints (weeks 8, 16 or 24 in Groups B, C and D, respectively) for a minimum of 24 weeks treatment (Fig. 1) [24].

Adapted from Harmatz et al. [24], with permission. PL placebo, VES intravenous vestronidase alfa 4 mg/kg every other week
Study design of the pivotal, blind start, phase 3 trial.
The primary endpoint in all countries except the USA was the percentage change from baseline in uGAG dermatan sulfate excretion (assessed by liquid chromatography-mass spectrometry/mass spectrometry) after 24 weeks of vestronidase alfa [26]. In the USA, efficacy was based on the totality of the clinical data on a per participant basis, with no primary endpoint declared [26]. Other major efficacy outcomes were assessments across a spectrum of clinical morbidities commonly observed in mucopolysaccharidosis diseases, which were assessed using the Multi-Domain Responder Index (MDRI) comprising six clinical domains: 6-minute walk test (6MWT), predicted forced vital capacity (FVC%pred), shoulder flexion, visual acuity, and Bruininks–Oseretsky test (BOT-2) of fine and gross motor proficiency [24]. Each domain was measured against prespecified minimally important difference (MID) thresholds (Table 1), permitting translation of numerous clinical measures into a combined endpoint without hindering the results by non-assessable data [24]. In post hoc analyses, a fatigue domain was included in the MDRI (MDRI-fatigue), with fatigue assessed using the Pediatric Quality of Life Inventory™-Multidimensional Fatigue Scale [24]. Due to the extreme rarity of mucopolysaccharidosis VII, the study population had a high degree of heterogeneity with varying physical and/or cognitive clinical manifestations. At baseline, three patients could not walk, nine were not able to perform pulmonary function tests, five could not perform the BOT-2 gross motor test, and five could not perform visual acuity tests [28].
At 24 weeks, there was a significant reduction from baseline in uGAG dermatan sulfate excretion with vestronidase alfa treatment, with uGAG dermatan sulfate levels reduced by 64.8% (Table 1) [24]. There was also a significant reduction in uGAG chondroitin sulfate excretion (Table 1). These reductions occurred rapidly (i.e. within 2 weeks of treatment initiation) and were sustained until the end of treatment [24]. All patients achieved ≥ 50% reduction from baseline in uGAG dermatan sulfate excretion (i.e. were responders) on at least one visit during the initial 24 weeks of vestronidase alfa therapy [9].
There was a positive overall mean change in the MDRI of 0.5 domains after 24 weeks’ vestronidase alfa therapy, although this difference was not statistically significant (p = 0.0527; Table 1). MDRI-fatigue assessments showed a significant overall positive improvement of 0.8 domains (Table 1) [24]. Most patients (83%; 10 of 12 patients) achieved prespecified MID criteria in one or more MDRI domains during the 24-week primary assessment period. Although the overall LSM change in the 6MWT distance of + 20.8 m did not reach the prespecified MID of 23 m, three patients did exceed MID criteria, with clinically meaningful improvements in 6MWT distance (increases of 65–83 m) and ≥ 10% change from baseline in 6MWT distance (Table 1). Most patients could not perform pulmonary function testing and thus, no conclusions could be drawn; one assessable patient had an improvement in lung function (Table 1). There were minimal changes in shoulder flexion during the 24-week assessment period (Table 1), as all patients had minimal restriction in shoulder flexion at baseline (mean for tighter shoulder 139° vs. normal flexion range of ≈ 160°). Uncorrected visual acuity improved by ≈ 1 line in both the left and right eyes (Table 1), with four of seven assessable patients improving by ≥ 2 lines on the Snellen eye chart in one or both eyes. There were minimal changes in BOT-2 fine motor and gross motor domains (Table 1) [24].
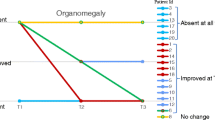
According to heat map assessments categorized into three domains (mobility, fatigue, and fine motor and self-care), after 8 weeks of treatment, four patients improved in the mobility domain, five improved in the fatigue domain, and three improved in the fine motor and self-care domain (with two showing improvements in ≥ 2 domains) [28]. Liver and spleen volume assessments (n = 7) showed mostly normal or below normal liver and spleen volumes at baseline and on average, were unchanged with vestronidase alfa therapy [8].
3.2 Open-Label Extension
All patients in the pivotal trial subsequently enrolled in an open-label extension trial, with interim results available for nine patients [29]. Continuous treatment with intravenous vestronidase alfa 4 mg/kg every other week for ≤ 120 weeks resulted in sustained reductions in uGAG excretion, with some patients (not all patients were assessable for each endpoint) showing sustained benefits or improvements in the 6MWT, total fatigue score, and visual acuity assessments. The impact of long-term therapy on fine and gross motor proficiency was variable, with some patients showing continued improvements in fine and gross motor skills, and others showing decreased manual dexterity and/or balance. One patient discontinued treatment after receiving only one vestronidase alfa infusion (due to non-compliance); their uGAG levels slowly reverted to baseline levels, although they continued to show improvements in manual dexterity and fine motor precision skills [29].
3.3 Other Studies
In a 36-week, dose-exploration (vestronidase alfa 1, 2 or 4 mg/kg every other week) phase 1/2 trial (n = 3; aged 5−25 years [9]), the mean percent reduction from baseline (averaged from measurements at weeks 14, 22, 30 and 38) in uGAG dermatan sulfate excretion was 61.8% with a 4 mg/kg dose compared with 42.2 and 52.4% with 1 and 2 mg/kg doses [26]. Thus, the optimal dosage of vestronidase alfa was 4 mg/kg once every other week, which is consistent with PPK analysis evidence (Sect. 2). In the ongoing extension phase, all patients showed some clinical improvement during ≤ 120 weeks’ vestronidase alfa therapy [9, 30]. One patient showed a 21% improvement over baseline in pulmonary function testing (FVC%pred) and a 105 m improvement in the 6MWT at 120 weeks and two patients with hepatosplenomegaly at baseline had a reduction in liver volume of 24 and 53% and spleen volume of 28 and 47% at 36 weeks [9].
In the open-label phase 2 trial, five of eight enrolled patients (aged 1.7−5 years) completed 48 weeks of treatment with intravenous vestronidase alfa 4 mg/kg every other week [31, 32]. One of these patients had been previously treated with vestronidase alfa through an emergency Investigational New Drug (IND) approval [20]. A 64% reduction from baseline in mean uGAG excretion was observed at week 4 and maintained through week 48. [31, 32]. At 48 weeks, there were numerical increases in mean standing height (92 vs. 86 cm at baseline) and mean growth velocity post-treatment (6.84 vs. 5.06 cm/year within two years of pre-treatment), with splenomegaly and hepatomegaly (n = 2 and 3) resolved in one and two patients. Clinical Global Impression (CGI) scores improved or remained unchanged, with two patients with reliable functional development evaluation data experiencing an improvement in CGI score by week 48. The number of patients capable of achieving all motor function evaluation milestones increased from none to two [31, 32].
Emergency IND applications for compassionate use of intravenous vestronidase alfa were approved for two patients with mucopolysaccharidosis VII [20]. An infant (aged 5 months at the start of treatment) received vestronidase alfa 2 mg/kg every other week between weeks 1−18 and 4 mg/kg every other week from week 18 onwards [33, 34], and a child (aged 12 years) received vestronidase alfa 2 mg/kg every other week for 24 weeks [35]. Both patients had rapid reductions in uGAG excretion levels from baseline (−74 and −92% after 4 and 48 weeks in the infant patient [33]; −59 and −75% after 2 and 6 weeks, with stabilized reductions thereafter up to week 24 in the other patient [35]). Both patients also had other improvements, including in pulmonary function (e.g. decreased frequency of hypoxic episodes [33] or proportion of time per day off a ventilator without an increase in end-tidal carbon dioxide levels [35]) and developmental progress [e.g. improved visual tracking and sound recognition after 48 weeks (infant) [33]].
Another pediatric patient with mucopolysaccharidosis VII who required continuous ventilatory support received vestronidase alfa through expanded access; after 164 weeks, this patient had a reduced need for mechanical ventilation (9 h daily off ventilator support) [8, 34].
A global (EU, USA and Latin America), prospective, longitudinal Disease Monitoring Program (DMP; NCT03604835) is currently recruiting patients with mucopolysaccharidosis VII (target enrolment of 35 patients) [36]. The aims of the DMP are to characterize mucopolysaccharidosis VII disease presentation and progression over time in patients treated and not treated with vestronidase alfa, assess the long-term effectiveness and safety of vestronidase (including hypersensitivity reactions and immunogenicity), and assess the longitudinal change in the biomarker(s), clinical assessments, patient/caregiver-reported outcome measures, and other possible predictors of mucopolysaccharidosis VII disease progression and mortality [36].
4 Tolerability of Vestronidase Alfa
Intravenous vestronidase alfa had a manageable tolerability profile in the four clinical trials discussed in Sect. 3, with the following discussion focusing on a pooled analysis of these trials (duration ≤ 132 weeks as of the data lock point) [26]. Most adverse reactions were mild to moderate in severity [9]. No patient withdrew from a study or discontinued treatment with vestronidase alfa as a result of an adverse event (AE) or serious AE (SAE), and no deaths were reported [26]. No clinically significant changes in vital signs were observed in any patient exposed to vestronidase alfa [26].
In the pooled analysis (n = 23, where patients from the phase 3 and open-label extension trials were only counted once), treatment-emergent AEs were reported in 96% of patients, the most common of which were vomiting (48%), cough (48%), upper respiratory tract infection (44%), infusion site extravasation (35%), and diarrhea, rash, and pyrexia (all 30%) [26]. Two patients had anaphylactic reactions following treatment with vestronidase alfa, with subsequent infusions tolerated without recurrence [8, 10]. Anaphylaxis can be life-threatening and it is recommended that vestronidase alfa is administered in the presence of appropriate medical support [8,9,10].
Eight patients experienced SAEs across all trials [26]. Four of which were considered to be related to vestronidase alfa treatment including: a febrile convulsion occurring in a patient within 3 days of a diphtheria, tetanus, and pertussis (DTP) vaccination, which subsequently resolved (and was assessed as possibly related to treatment due to temporal association with the infusion); an anaphylactoid reaction in a patient after inadvertent bolus infusion of vestronidase alfa, which resolved and the infusion was completed in the same day without recurrence of symptoms; and urticaria and bronchospasm experienced by a patient during infusion of the drug, which resolved with medical treatment [26].
Seventy percent of patients (16 of 23) developed anti-recombinant human β-glucuronidase antibodies, and on at least one occasion, nine of these patients further developed neutralizing antibodies [9]. The high proportion of patients who developed anti-recombinant human β-glucuronidase antibodies was consistent with the proportion of patients in clinical trials who developed antibodies to ERTs for other mucopolysaccharidosis [12, 14, 15]. No definitive correlation has been reported between antibody titer and the development of neutralizing antibodies, and the presence of anti-drug antibodies does not appear to affect reductions in uGAG excretion [9].
5 Dosage and Administration of Vestronidase Alfa
Intravenous vestronidase alfa is indicated in pediatric and adult patients for the treatment of mucopolysaccharidosis VII in the USA [8] and Brazil [10] or non-neurological manifestations of mucopolysaccharidosis VII in the EU [9]. Its effects on neurological manifestations originating in the CNS have not yet been established [8, 10]. Vestronidase alfa is not expected to cross the blood-brain barrier, and is therefore not likely to impact neurological manifestations of the disorder [9].
The recommended dosage of vestronidase alfa is 4 mg/kg of bodyweight administered by intravenous infusion every other week [8,9,10]. The infusion should be administered over ≈ 4 h. To minimize the risk of hypersensitivity reactions, administration of a non-sedating antihistamine with or without an antipyretic medicinal product is recommended 30–60 min before initiating infusion of vestronidase alfa [8,9,10]. Administration of vestronidase alfa may be slowed, temporarily interrupted or discontinued if hypersensitivity reactions occur [9, 10] (e.g. a severe systemic reaction [8]). In the EU, it is recommended that treatment with vestronidase alfa is periodically evaluated, and discontinuation of vestronidase alfa should be considered in patients where clear benefits (including stabilization of disease manifestations) are not observed [9]. Local prescribing information should be consulted for the recommended infusion rate by patient bodyweight guidelines, and additional warnings and precautions.
6 Current Status of Vestronidase Alfa in Mucopolysaccharidosis VII
Despite a relatively comprehensive understanding of the underlying pathophysiology of mucopolysaccharidosis VII, the development of effective therapeutic options for the disease has, until recently, been slow. This largely reflects the rarity and heterogeneous clinical presentations of the disease, which has hindered the use of traditional study design approaches [24]. Prior to the approval of vestronidase alfa, there were no disease-specific treatments for mucopolysaccharidosis VII, and disease management predominantly focused on symptomatic and supportive care (e.g. physiotherapy and hydrotherapy) [37,38,39]. BMTs have been performed in a small number of patients with mucopolysaccharidosis VII, some of whom experienced marked clinical improvements, especially in motor function, although effects on neurological manifestations were equivocal [7, 38,39,40]. There are considerable limitations with BMT and its efficacy is variable in this population [7, 38,39,40].
ERT with vestronidase alfa is the first disease-specific therapy approved for patients with mucopolysaccharidosis VII [8,9,10]. In the pivotal phase 3 trial in pediatric and adult patients (which utilized a novel blind start design), treatment with vestronidase alfa for 24 weeks significantly reduced uGAG excretion levels compared with baseline (Sect. 3.1), with results from phase 1 and 2 trials supporting these data (Sect. 3.3). Most assessable patients treated with vestronidase alfa experienced an improvement in ≥ 1 domain of the MDRI over 24-week primary assessment period in the pivotal trial (Sect. 3.1; Table 1). Although the clinical relevance of these effects remains to be established, most patients showed a positive trend towards improvement, or at least stabilization, of some disease symptoms, which are considered beneficial for the patient [26]. These clinical benefits were sustained or further improved during longer-term treatment (≤ 120 weeks) in an ongoing open-label extension study, as was the reduction in uGAG excretion (Sect. 3.2). There is limited data on the use of vestronidase alfa for ≥ 120 weeks [26] and further clinical experience is essential to fully elucidate the long-term efficacy of the drug, along with its efficacy on various clinical manifestations such as pulmonary function for which available data were too limited to draw conclusions in the pivotal trial (Sect. 3.1).
Vestronidase alfa had a manageable tolerability profile in clinical trials, with most adverse reactions being mild to moderate in severity (Sect. 4). Given the global rarity of mucopolysaccharidosis VII, the safety data for vestronidase alfa was considered to be adequate [26, 34].
Despite the clinical benefits of vestronidase alfa therapy in patients with mucopolysaccharidosis VII, it is also associated with several limitations. Firstly, like other ERTs, vestronidase alfa therapy is limited by the need for life-long intravenous treatment [41], which may be associated with infusion-related reactions. Secondly, the effects of vestronidase alfa on neurological manifestations originating in the CNS have not yet been determined; however, vestronidase alfa is not expected to cross the blood brain barrier (Sect. 5). A further limitation of vestronidase alfa therapy is the financial burden (e.g. the long-term cumulative cost); the manufacturer has provided a treatment access scheme to assist with financial support for vestronidase alfa therapy when needed [42].
There are currently no global treatment guidelines available for mucopolysaccharidosis VII [43]; however, vestronidase alfa is indicated for patients of all ages (Sect. 5). It is argued that ERT should be initiated as early as possible following a diagnosis of mucopolysaccharidosis, including before the onset of clinical disease, in order to obtain better long-term treatment outcomes [44]. The mucopolysaccharidosis VII DMP, which is currently recruiting patients, will provide a prospective, comprehensive, standardized dataset, alongside long-term efficacy and safety data for vestronidase alfa therapy, and is likely to lead to a greater understanding of the disease [36] (Sect. 3.3).
Given the rarity of the disease, there are no available data pertaining to the use of vestronidase alfa in specific populations (i.e. pregnant or breastfeeding women, and patients with hepatic and/or renal impairment) or its effect on fertility in humans [9, 26]. In preclinical studies, treatment with vestronidase alfa during organogenesis stages in pregnant animals resulted in no adverse developmental outcomes at doses ≤ 20 mg/kg [9]. There was also no impact of vestronidase alfa on fertility in males and female animals [10].
In conclusion, given the lack of treatment options and the clinical benefits it provides, vestronidase alfa is an important emerging ERT for pediatric and adult patients with mucopolysaccharidosis VII.
Data Selection Vestronidase Alfa: 100 records
Duplicates removed | 7 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; not randomized trial) | 19 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 30 |
Cited efficacy/tolerability articles | 22 |
Cited articles not efficacy/tolerability | 22 |
Search Strategy: EMBASE, MEDLINE and PubMed from 1946 to present. Clinical trial registries/databases and websites were also searched for relevant data. Key words were vestronidase alfa, Mepsevii, mucopolysaccaridosis type 7, Sly syndrome, recombinant beta-glucuronidase, enzyme replacement therapy. Records were limited to those in English language. Searches last updated 25 February 2019 | |
Change history
16 April 2019
The article Vestronidase Alfa: A Review in Mucopolysaccharidosis VII, written by Emma H. McCafferty and Lesley J. Scott, was originally published Online First without open access.
References
Tomatsu S, Montaño AM, Dung VC, et al. Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly syndrome). Hum Mutat. 2009;30(4):511–9.
Sly WS, Quinton BA, McAlister WH, et al. Beta glucuronidase deficiency: report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis. J Pediatr. 1973;82(2):249–57.
Miller RD, Hoffmann JW, Powell PP, et al. Cloning and characterization of the human beta-glucuronidase gene. Genomics. 1990;7(2):280–3.
Naz H, Islam A, Waheed A, et al. Human beta-glucuronidase: structure, function, and application in enzyme replacement therapy. Rejuvenation Res. 2013;16(5):352–63.
Orphanet. Mucopolysaccharidosis type 7. 2019. http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=584. Accessed 31 Jan 2019.
National Institutes of Health. Mucopolysaccharidosis type VII. 2018. https://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-vii#. Accessed 30 Nov 2018.
Montano AM, Lock-Hock N, Steiner RD, et al. Clinical course of Sly syndrome (mucopolysaccharidosis type VII). J Med Genet. 2016;53(6):403–18.
Ultragenyx Pharmaceutical Inc. Mepsevii™ (vestronidase alfa-vjbk): US prescribing information. 2017. http://www.fda.gov/. Accessed 21 Nov 2018.
European Medicines Agency. Vestronidase alfa (Mepsevii): summary of product characteristics. 2018. http://www.ema.europa.eu/. Accessed 2018.
UNO Healthcare Comércio de Medicamentos Ltda. Mepsevii™: Brazilian prescribing information. São Paulo: UNO Healthcare Comércio de Medicamentos Ltda; 2018.
Kakkis ED, Muenzer J, Tiller GE, et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med. 2001;344(3):182–8.
Wraith JE, Clarke LA, Beck M, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human α-l-iduronidase (laronidase). J Pediatr. 2004;144(5):581–8.
Muenzer J, Wraith JE, Beck M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med. 2006;8(8):465–73.
Harmatz P, Giugliani R, Schwartz I, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J Pediatr. 2006;148(4):533.e6–539.e6.
Hendriksz CJ, Burton B, Fleming TR, et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis. 2014;37(6):979–90.
Hendriksz CJ, Giugliani R, Harmatz P, et al. Multi-domain impact of elosulfase alfa in Morquio A syndrome in the pivotal phase III trial. Mol Genet Metab. 2015;114(2):178–85.
O’Connor LH, Erway LC, Vogler CA, et al. Enzyme replacement therapy for murine mucopolysaccharidosis type VII leads to improvements in behavior and auditory function. J Clin Invest. 1998;101(7):1394–400.
Vogler C, Sands MS, Levy B, et al. Enzyme replacement with recombinant β-glucuronidase in murine mucopolysaccharidosis type VII: impact of therapy during the first six weeks of life on subsequent lysosomal storage, growth, and survival. Pediatr Res. 1996;39:1050–4.
Sands MS, Vogler C, Kyle JW, et al. Enzyme replacement therapy for murine mucopolysaccharidosis type VII. J Clin Invest. 1994;93(6):2324–31.
US Food and Drug Administration. Vestronidase alfa (Mepsevii™): clinical pharmacology and biopharmaceuticals review. 2017. http://www.fda.gov/. Accessed 21 Nov 2018.
Qi Y, McKeever K, Taylor J, et al. Pharmacokinetic and pharmacodynamic modeling to optimize the dose of vestronidase alfa, an enzyme replacement therapy for treatment of patients with mucopolysaccharidosis type VII: results from three trials. Clin Pharmacokinet. 2018. https://doi.org/10.1007/s40262-018-0721-y.
US National Institutes of Health. ClinicalTrials.gov identifier NCT01856218. 2018. http://clinicaltrials.gov/. Accessed 27 Nov 2018.
US National Institutes of Health. ClinicalTrials.gov identifier NCT02418455. 2018. http://clinicaltrials.gov/. Accessed 27 Nov 2018.
Harmatz P, Whitley CB, Wang RY, et al. A novel blind start study design to investigate vestronidase alfa for mucopolysaccharidosis VII, an ultra-rare genetic disease. Mol Genet Metab. 2018;123(4):488–94.
US National Institutes of Health. ClinicalTrials.gov identifier NCT02432144. 2018. http://clinicaltrials.gov/. Accessed 27 Nov 2018.
European Medicines Agency. Vestronidase alfa (Mepsevii): assessment report. 2018. http://www.ema.europa.eu/. Accessed 26 Nov 2018.
US National Institutes of Health. ClinicalTrials.gov identifier NCT02230566. 2018. http://clinicaltrials.gov/. Accessed 27 Nov 2018.
Haller C, Song W, Cimms T, et al. Individual heat map assessments demonstrate ERT treatment response in highly heterogeneous MPS VII study population [abstract no. 595 plus poster]. J Inborn Errors Metab Screen. 2017;5:268–9.
Martins EM, Wang RYW, Francisco Da Silva Franco JF, et al. Sustained efficacy and safety of long-term vestronidase alfa (rhGUS) enzyme replacement therapy in patients with MPS VII [abstract no. P-372 plus poster]. J Inherit Metab Dis. 2018;41(Suppl. 1):S188.
Coker M, Gonzales-Meneses Lopez A, Song W, et al. Long-term outcomes with rhGUS in a phase I/II clinical trial in MPS VII [abstract no. O-044]. J Inherit Metab Dis. 2016;39(Suppl. 1):S48–9.
Gonzalez-Meneses Lopez AGL, Beuno MB, Lau HL, et al. Vestronidase alfa stabilizes or improves disease manifestations in subjects with MPS VII [abstract no. P-371]. J Inherit Metab Dis. 2018;41(Suppl. 1):S187–8.
Ali QA, Lopez AG-M, Viskochil DV, et al. The enzyme replacement therapy vestronidase alfa stabilizes or improves disease manifestations in subjects with MPS VII <5-years old [abstract]. In: Canadian Symposium on Lysosomal Diseases. 2018.
Lau HA, Parmar S, Kazachkov M, et al. Enzyme replacement therapy with investigational rhGUS in an infant with non-immune hydrops fetalis and mucopolysaccharidosis type VII [abstract no. 173 plus poster]. Mol Genet Metab. 2016;117(2):S71.
US Food and Drug Administration. UX003/rhGUS: clinical review. 2017. http://www.fda.gov/. Accessed 21 Nov 2018.
Fox JE, Volpe L, Bullaro J, et al. First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol Genet Metab. 2015;114(2):203–8.
Marsden D, Hostutler R, Cimms T, et al. The MPS VII Disease Monitoring Program is a novel longitudinal, cohort program with a rigor beyond a traditional registry [abstract no. 229 plus poster]. In: WORLD Symposium. 2019.
Society for Mucopolysaccharide Diseases. Guide to understanding mucopolysaccharidosis VII (MPS VII). 2015. http://www.mpssociety.org.uk/. Accessed 4 Dec 2018.
US Food and Drug Administration. Vestronidase alfa (Mepsevii™): risk assessment and risk mitigation review. 2017. http://www.fda.gov/. Accessed 21 Nov 2018.
Sisinni L, Pineda M, Coll MJ, et al. Haematopoietic stem cell transplantation for mucopolysaccharidosis type VII: a case report. Pediatr Transplant. 2018;22(7):e13278.
Yamada Y, Kato K, Sukegawa K, et al. Treatment of MPS VII (Sly disease) by allogeneic BMT in a female with homozygous A619V mutation. Bone Marrow Transplant. 1998;21:629–34.
Ginocchio VM, Brunetti-Pierri N. Recent progress in gene therapies for mucopolysaccharidoses. Expert Opin Orphan Drugs. 2018;6(10):611–23.
Ultracare™. Ultracare: assistance in gaining access to Mepsevii (vestronidase alfa-vjbk). 2019. http://ultracaresupport.com/mepsevii/. Accessed 31 Jan 2019.
Stapleton M, Hoshina H, Sawamoto K, et al. Critical review of current MPS guidelines and management. Mol Genet Metab. 2018. https://doi.org/10.1016/j.ymgme.2018.07.001.
Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63–72.
Acknowledgements
During the peer review process, the manufacturer of vestronidase alfa was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
Emma McCafferty and Lesley Scott are salaried employees of Adis/Springer, are responsible for the article content and declare no relevant conflicts of interest.
Additional information
The manuscript was reviewed by:N. Brunetti-Pierri, Department of Translational Medicine, Federico II University, Naples, Italy; R. Giugliani, Medical Genetics Service, UFRGS, Hospital de Clínicas de Porto Alegre, Rua Ramiro Barcelos, 2350, Porto Alegre, RS 90035-903, Brazil; A.M. Montaño, Department of Pediatrics, and Department of Biochemistry and Molecular Biology, Doisy Research Center, School of Medicine St. Louis University, St. Louis, MO, USA; S. Tomatsu, Nemours/Alfred I DuPont Hospital for Children, Thomas Jefferson University, Wilmington, DE, USA.
The original version of this article was revised due to a retrospective Open Access request.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
About this article

Cite this article
McCafferty, E.H., Scott, L.J. Vestronidase Alfa: A Review in Mucopolysaccharidosis VII. BioDrugs 33, 233–240 (2019). https://doi.org/10.1007/s40259-019-00344-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-019-00344-7