
Abstract
Hereditary angioedema (HAE) with C1 esterase inhibitor (C1-INH) deficiency (C1-INH-HAE) is a rare disease characterized by diminished levels or dysfunctional activity of C1-INH, leading to dysregulated plasma kallikrein activity within the kallikrein–kinin pathway. Symptoms manifest as painful, potentially life-threatening swelling of subcutaneous tissues throughout the body and/or submucosal edema in the upper airway or gastrointestinal tract. Attacks recur with unpredictable frequency, intensity, and duration, placing a heavy burden on patients’ daily lives. Despite improved availability of medications for on-demand treatment during attacks and prophylaxis of future attacks, unmet needs remain. Lanadelumab, a fully human monoclonal antibody, may help address some of the limitations of existing prophylactic options (e.g., the need for intravenous administration or frequent dosing). Preclinical studies demonstrate that it is highly potent and specifically inhibits plasma kallikrein, and findings from phase Ia and Ib studies suggest this agent is well tolerated and provides sustained inhibition of plasma kallikrein, allowing for less frequent dosing. The phase III HELP Study (NCT02586805) evaluating the efficacy and safety of lanadelumab in preventing HAE attacks has been completed, and its open-label extension (NCT02741596) is ongoing. Lanadelumab is now approved in the USA and Canada for prophylaxis to prevent attacks of HAE in patients aged ≥ 12 years. This review provides an overview of the discovery and clinical development of lanadelumab, from preclinical through phase Ib studies, characterizing its safety/tolerability, efficacy, and pharmacokinetic and pharmacodynamic profiles. It also highlights how this agent may positively impact clinical care of patients with C1-INH-HAE.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Hereditary angioedema (HAE) with C1 inhibitor deficiency (C1-INH-HAE) is a rare genetic disease characterized by recurring, frequently painful swelling episodes that can disrupt quality of life and may be fatal if treatment is delayed. |
Treatment options for C1-INH-HAE have grown over the last decade, with the publication of several well-designed clinical trials evaluating acute and prophylactic treatment, and subsequent approval of agents with diverse targets within the kallikrein–kinin pathway. In the USA, for instance, seven HAE-specific therapies have been approved by the US FDA since 2008. |
Findings from phase I studies suggest that lanadelumab, a fully human monoclonal antibody targeting plasma kallikrein, is well tolerated and can be dosed less frequently than currently available prophylactic options. This agent was recently approved in the USA and Canada for prophylaxis to prevent attacks of HAE in patients aged ≥ 12 years. |
1 Introduction
Hereditary angioedema (HAE) with C1 esterase inhibitor (C1-INH) deficiency (C1-INH-HAE), an orphan disease affecting approximately 1 per 50,000 people [1, 2], is an autosomal dominant disorder characterized by diminished levels of C1-INH, resulting in reduced functional activity (type I, most common; affecting ~ 85% of cases), or normal or somewhat elevated levels of C1-INH but dysfunctional protein activity (type II, ~ 15% of cases) [3, 4]. Low levels of functional C1-INH lead to dysregulated plasma kallikrein activity within the kallikrein–kinin pathway, resulting in uncontrolled production of bradykinin, a potent vasodilator that causes vascular leakage and angioedema in C1-INH-HAE [5].
Symptoms of C1-INH-HAE manifest clinically as nonpruritic swelling of subcutaneous tissues throughout the body, particularly the extremities, face, neck, or genitalia, and/or submucosal edema in the upper airway or gastrointestinal tract [6,7,8]. Symptoms often begin by 10 years of age and recur with unpredictable frequency, intensity, and duration throughout patients’ lives. Attack frequency and severity among patients is variable; some experience very few attacks and require only on-demand therapy to treat attacks, whereas others have multiple attacks per month or per week, necessitating prophylactic treatment. In some cases, despite receiving prophylactic therapy, patients continue to have “breakthrough” attacks [6, 7, 9, 10]. Attacks are frequently painful and can be debilitating [7]. Symptoms may develop or intensify during puberty or over the course of hormonal changes throughout life, particularly in females, with exposure to estrogens [11, 12]. Although the reasons are unclear, HAE attacks often occur more frequently and with greater severity in women than in men [11, 12]. Left untreated, symptoms can worsen in intensity, spread to other sites, and last up to 5 days [7,8,9]. Although select triggering factors (e.g., physical trauma, emotional distress) and prodromal symptoms (e.g., nausea, erythema marginatum) have been reported, attacks often occur without identifiable triggers or warning signs [7, 13], leading many patients to fear future attacks.
The heavy burden of illness associated with C1-INH-HAE negatively impacts daily quality of life for patients and their families and caregivers [7, 14, 15]. Upper airway edema (affecting approximately 50% of patients at least once in their lifetime) [16, 17] can progress over minutes to hours to upper airway obstruction, which can lead to death from asphyxiation without proper treatment [18]. HAE attacks involving the face and extremities can limit patients’ desire to participate in social functions and disrupt their ability to travel, work, or attend school because of disfigurement [7]. Attacks affecting the abdomen, among the most commonly occurring, are often disabling (associated with severe cramping, nausea, vomiting, diarrhea, and syncope) and can be misdiagnosed as an “acute surgical abdomen” (e.g., appendicitis), leading to inappropriate and unnecessary surgical intervention [7, 8, 17, 19,20,21]. Consequently, many patients are subject to multiple negative experiences that impact their emotional and psychosocial well-being, both during and between attacks [15, 22]. Patients with C1-INH-HAE have shown impairments across multiple health-related quality-of-life measures, regardless of location of attacks, and often experience depression and anxiety [7, 14, 23, 24]. Additionally, C1-INH-HAE is associated with a heavy economic burden stemming from direct and indirect costs, including those related to increased emergency department visits and missed school and work days, respectively, hampering academic and professional progress [25, 26].
Management of C1-INH-HAE focuses on treating symptoms during an HAE attack (with on-demand treatment) and preventing or reducing the frequency and severity of future attacks by short-term prophylaxis (e.g., before scheduled events that may trigger an attack, such as surgeries or dental procedures), and long-term prophylaxis [3, 27,28,29]. A crossover study of patients with C1-INH-HAE showed improved health-related quality of life (as measured by the 36-Item Short Form Survey) in those receiving preventive therapy with intravenous C1-INH every 3–4 days (plus on-demand therapy for HAE attacks, as needed), compared with patients receiving on-demand treatment for HAE attacks only [30]. The decision to use continuous prophylaxis is individualized and based on multiple factors, including the frequency and severity of attacks, the impact of HAE on patients’ daily lives, and patients’ treatment preferences. Additionally, several HAE consensus documents emphasize the importance of continually re-evaluating the appropriateness of long-term prophylaxis, as this treatment modality may or may not be a life-long necessity depending upon fluctuations of disease activity [3, 29].
Treatment options for C1-IHN-HAE have grown over the last decade, with the publication of several well-designed clinical trials evaluating acute and prophylactic treatment, and subsequent approval of agents with diverse targets within the kallikrein–kinin pathway; sites of action of several agents are shown in Fig. 1 [31]. In the USA, for instance, seven HAE-specific therapies have been approved by the US FDA since 2008. Licensing of individual agents varies by region (discussed below).

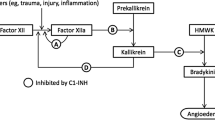
From Zuraw [31], © 2008 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society
Pathways inhibited by C1 inhibitor and newer agents for hereditary angioedema with C1 esterase inhibitor deficiency [31]. Proteolytic activities are indicated with green arrows and point toward the steps they catalyze. Steps inhibited by C1 esterase inhibitor (C1-INH), ecallantide, icatibant, and lanadelumab are shown with red T-bars. In the classic complement pathway, activation of the complement protease C1 is indicated by horizontal bars over the complement names.
Gene therapy for HAE is another emerging area of increasing interest. Encouraging findings have been reported from a preclinical study evaluating the use of a gene transfer vector expressing normal human C1-INH (AAVrh.10hC1EI) in a C1-INH-deficient mouse model mimicking HAE [32].
Based on current labeling in the USA and EU, approved options for on-demand treatment of HAE attacks include the intravenously administered plasma-derived C1-INH concentrates Cinryze® (EU only; Shire Services BVBA, Brussels, Belgium) [33] and Berinert® (USA and EU; CSL Behring, Marburg, Germany) [34, 35]; intravenously administered recombinant C1-INH concentrate Ruconest® (US and EU; Pharming Technologies B.V., the Netherlands) [36, 37]; the subcutaneously administered kallikrein inhibitor ecallantide (Kalbitor® [USA only]; Dyax Corp, Burlington, MA, USA, [now part of Shire Human Genetic Therapies, Inc.] [38]; and the subcutaneously administered bradykinin B2 receptor antagonist icatibant (Firazyr® [USA and EU]; Shire Orphan Therapies, LLC, MA, USA; Shire Pharmaceuticals Ireland Limited, Dublin, Ireland) [39, 40]. Until recently, approved options for long-term prophylaxis included Cinryze (USA and EU; Shire Services BVBA Brussels, Belgium; Viropharma Biologics, Inc. [now part of Shire Human Genetic Therapies, Inc.], Lexington, MA, USA) [33, 41]; the subcutaneously administered C1-INH concentrate Haegarda® (USA only; CSL Behring, Marburg, Germany) [42]; orally administered attenuated androgens such as danazol (USA only; e.g., Danocrine®; Sanofi-Aventis US LLC, Bridgewater, NJ, USA) [43]; and orally administered antifibrinolytic agents such as tranexamic acid (EU only; e.g., Sandoz, Camberley, Surrey, UK) [44, 45].
Although multiple therapeutic options are available, many have limitations. For instance, attenuated androgens have the potential for multiple dose-related adverse events (AEs), including virilization, muscle pain, acne, elevated liver enzymes, altered lipid profiles, menstrual irregularity, depression, and weight gain [28, 46]. These agents are contraindicated in pregnant or breast-feeding women and are not recommended in growing children (e.g., aged < 16 years) [43, 46]. Also, inconvenient administration schedules for prophylactic agents (e.g., twice-weekly dosing) contribute to burden of treatment, and various challenges are associated with the need for frequent use of intravenous treatments, such as loss of venous access or use of venous access ports, which increases the potential for thrombosis and/or infection [47]. Additionally, efficacy of existing treatment options in preventing attacks is incomplete in some patients, placing them at risk for future attacks and requiring the use of higher doses, more frequent administration, and/or rescue therapy with on-demand treatment [28, 48, 49]. Consequently, there remain unmet needs for prophylaxis of HAE attacks.
Lanadelumab (SHP643; previously DX-2930) is a fully human monoclonal antibody (representing a novel structure within the HAE treatment landscape) that is a highly specific inhibitor of plasma kallikrein [50], the enzyme involved in formation of bradykinin [5]. This agent was recently approved in the USA and Canada for prophylaxis to prevent attacks of HAE in patients aged ≥ 12 years, at a recommended dose of 300 mg every 2 weeks (or possibly 300 mg every 4 weeks in well-controlled patients who are attack free for > 6 months) [51, 52], and is designated as an orphan drug by the European Medicines Agency [53]. Lanadelumab has been evaluated in several recent studies. A phase III, randomized, double-blind, placebo-controlled study in patients aged ≥ 12 years with symptomatic C1-INH-HAE (at least one attack per 4 weeks at baseline) evaluated efficacy and safety in prevention of HAE attacks (HELP Study®, NCT02586805). In this study, subcutaneous lanadelumab 150 or 300 mg was administered every 2–4 weeks over a 6-month treatment period [54]. An open-label extension (HELP Study Extension, NCT02741596) is ongoing [55]. As a subcutaneous injection administered once every 2–4 weeks, lanadelumab may offer an advantage over existing preventive treatments, potentially transforming the approach to prophylaxis.
This manuscript provides clinicians with an overview of the discovery and clinical development of lanadelumab, from preclinical through phase Ia and Ib studies, characterizing its safety/tolerability, efficacy, and pharmacokinetic and pharmacodynamic profiles. It also highlights ways in which this agent may positively impact the clinical care of patients with C1-INH-HAE.
2 C1-INH-HAE Pathophysiology: Rationale for Plasma Kallikrein Inhibition
Endogenous C1-INH, a member of the serine protease inhibitor (serpin) family [56], inhibits proteases or factors in several key physiologic pathways, including complement, contact system (kallikrein–kinin), and coagulation and fibrinolysis [57]. The contact system is activated upon interaction of blood with negatively charged macromolecules on surfaces, such as heparin, collagen, or fibrin clots in vivo, or dextran sulfate in vitro [58]. Factor XII (FXII) is autoactivated to generate activated FXII (FXIIa), which cleaves prekallikrein, generating plasma kallikrein. In turn, plasma kallikrein activates FXII to generate additional FXIIa and cleaves high-molecular-weight kininogen (HMWK), leading to the release of bradykinin (Fig. 1). Upon release, bradykinin binds to the B2 receptor on endothelial cells and acts as a potent vasodilator. Endogenous C1-INH plays an integral role in regulating this cascade through the inhibition of both FXIIa and plasma kallikrein [5, 59,60,61].
Although the exact biochemical composition of attack triggers in C1-INH-HAE is unknown, dysregulated activation of the kallikrein–kinin pathway has been established as a key underlying defect. Deficient levels of functional C1-INH lead to uncontrolled activation of the kallikrein–kinin cascade, resulting in unopposed activity of plasma kallikrein, excessive proteolysis of HMWK, and, subsequently, pathologic levels of bradykinin. Overproduction of bradykinin results in vascular leakage, with subsequent tissue swelling, characteristic of HAE attacks [5, 59].
The key role of plasma kallikrein in the pathogenesis of C1-INH-HAE provides a strong rationale for targeting this protease in the pharmacologic management of this disease. Usefulness of plasma kallikrein as a drug target has been demonstrated through clinical experience with the subcutaneously administered ecallantide, a selective, short-acting, reversible, 60-amino acid peptide inhibitor of plasma kallikrein approved in the USA for acute treatment of HAE attacks [38, 61,62,63]. The subcutaneously administered lanadelumab, a monoclonal antibody that binds specifically to plasma kallikrein, may provide sustained inhibition of plasma kallikrein to prevent HAE attacks [50], a desirable attribute for prophylactic therapy.
Clinical data from studies evaluating oral plasma kallikrein inhibitors in the pipeline for prevention of HAE attacks have also been published. Whereas a phase III study with avoralstat (BCX4161; BioCryst Pharmaceuticals), a first-generation oral plasma kallikrein inhibitor administered three times daily, did not demonstrate efficacy versus placebo in preventing HAE attacks (NCT02303626) [64], promising findings were shown with the second-generation oral plasma kallikrein inhibitor BCX7353 (BioCryst Pharmaceuticals). In a phase II, randomized, dose-ranging study (NCT02870972), BCX7353 demonstrated a significantly lower rate of HAE attacks versus placebo at doses of ≥ 125 mg. Most AEs were grade 1 (mild); dose-related increases in gastrointestinal AEs were noted [65]. A phase III study evaluating BCX7353 versus placebo in HAE attack prevention is underway (NCT03485911) [66].
3 Lanadelumab Discovery and Initial Characterization: Preclinical Studies
Monoclonal antibodies are beneficial treatment options for multiple diseases (including autoimmune and inflammatory conditions) because of their high potency, long half-life, and good tolerability and have demonstrated improved patient care [67,68,69]. Furthermore, advances in phage display technology have led to generation of fully human monoclonal antibodies [70, 71]. Lanadelumab is a fully human immunoglobulin G1 (IgG1), kappa light chain monoclonal antibody expressed in Chinese hamster ovary cells [50, 72]. Discovered by Dyax Corp. as an antigen-binding fragment using phage display technology, it was selected for further development because of its highly potent (inhibition constant = 125 pM in vitro), specific inhibition of the proteolytic activity of plasma kallikrein [50, 72]. A detailed report of its discovery and characterization is presented by Kenniston et al. [50]; key findings are highlighted below and summarized in Table 1.
Lanadelumab was engineered to bind the active site of plasma kallikrein and block the cleavage of HMWK [50]. The specificity of lanadelumab for the active form of plasma kallikrein, as opposed to the prekallikrein zymogen, was a key candidate selection criterion, as this attribute may help achieve efficacy with lower drug levels while allowing basal plasma kallikrein activity to be maintained. It was reasoned that, by specifically targeting active plasma kallikrein, drug levels would only need to approximate the amount of active enzyme (felt to be lower than the total amount of prekallikrein in the circulation) [73,74,75], and specificity for active plasma kallikrein may enable basal plasma kallikrein activity upon contact system activation.
The high specificity of lanadelumab for plasma kallikrein was demonstrated in preclinical studies; at a concentration of 1 μM it did not inhibit 20 other serine proteases tested, including FXIa, which has significant homology to plasma kallikrein [50]. Furthermore, lanadelumab did not inhibit tissue kallikrein 1 [50], a distinct kininogenase from plasma kallikrein [76]. Structural analysis shows that lanadelumab completely occludes the active site of plasma kallikrein, thus preventing proteolytic activity [50]. In vitro studies demonstrated that lanadelumab bound both free soluble plasma kallikrein and plasma kallikrein bound to HMWK [50]. Additionally, lanadelumab bound plasma kallikrein assembled in vitro on human umbilical vein endothelial cells [50]. The assembly of plasma kallikrein to endothelial cells is mediated via HMWK, which binds the noncatalytic domain of plasma kallikrein and cell surface receptors (e.g., globular complement factor C1q receptor, urokinase plasminogen activator receptor, and cytokeratin 1) [77, 78]. Therefore, it is conceivable that cell surface-bound plasma kallikrein may contribute to attack localization in HAE [78].
Pharmacokinetic analyses in cynomolgus monkeys demonstrated a high bioavailability (66%) and prolonged serum half-life: ~ 12.5 days following a single subcutaneous injection, and ~ 19.3 days following intravenous administration [50]. These values are equivalent to a half-life of ≥ 2 weeks in humans [72]. The pharmacodynamic activity of lanadelumab in cynomolgus monkeys was observed by the inhibition of plasma kallikrein-mediated proteolysis of HMWK at 28 days following subcutaneous dosing [50]. Though not specific to HAE, the carrageenan-induced paw edema model (based on use of carrageenan, a polysaccharide-based irritant, injected into rats or mice to induce an acute swelling of the paw) has been shown to involve various inflammatory mediators, including bradykinin [79,80,81]. In vivo preclinical bioactivity of lanadelumab was observed in a carrageenan-induced paw edema model [50]. Sprague-Dawley rats were pretreated with lanadelumab (1 mg/kg or 30 mg/kg), a control buffer solution, or with indomethacin (as a positive control) before injection of 1% carrageenan (0.1 mL) into the rats’ right rear paws. At each observation time point, paw edema was lower in rats pretreated with lanadelumab (at either dose) than in those receiving the control buffer. However, the reduction in swelling was greater with the higher lanadelumab dose. At 8 h after injection with carrageenan, paw edema in the lanadelumab 30-mg/kg group was 55.6% lower than in the group receiving the control buffer solution (comparable to the 47.2% reduction achieved with indomethacin pretreatment) [50]. This in vivo bioactivity, combined with a demonstration of preclinical safety in toxicity studies conducted in accordance with health authority regulations, supported transitioning lanadelumab into clinical development studies [72].
4 Lanadelumab Clinical Development Program: Phase Ia and Phase Ib Studies
The clinical development program for lanadelumab consists of a phase Ia [72] and a phase Ib [82] study, the previously mentioned phase III trial (NCT02586805) [54], and its long-term open-label extension study (NCT02741596). Key findings from the phase Ia and phase Ib studies are highlighted below and summarized in Table 1.
4.1 Phase Ia Study
A phase Ia randomized, double-blind, single-center trial evaluated the safety/tolerability and pharmacokinetic/pharmacodynamic (PK/PD) profile of lanadelumab versus placebo in healthy individuals aged 18–55 years. In this study, subjects were randomized versus placebo to receive a single dose (0.1, 0.3, 1.0, or 3.0 mg/kg) in ascending cohorts via subcutaneous administration. Within each dosing group of eight subjects, six received lanadelumab and two received placebo [72]. Escalating doses were administered only after approval by the dose-escalation committee, based on safety/tolerability of the previous dose. Key parameters assessed included safety (e.g., treatment-emergent and treatment-related AEs, physical examination, vital signs, electrocardiogram, clinical laboratory assessment), pharmacokinetic parameters, and presence of antidrug antibodies. Additionally, the lanadelumab pharmacodynamic profile was evaluated via two exploratory biomarker assays that monitored plasma kallikrein activity following ex vivo plasma activation using FXIIa—the fluorogenic synthetic peptide substrate activity assay (measuring inhibition of plasma kallikrein), and the Western blot biomarker assay (assessing the presence of cleaved HMWK, to evaluate the ability of lanadelumab to inhibit plasma kallikrein activity toward its endogenous substrate). Each subject was followed for 16 weeks after receiving the single injection of study medication or placebo [72].
A total of 32 subjects were enrolled and randomized (lanadelumab, n = 24; placebo, n = 8), of whom 30 (93.7%) completed the study (neither of the two discontinuations were related to AEs). No treatment-emergent serious AEs or deaths were reported. Aside from one subject with symptoms of an upper respiratory tract infection (deemed not related to treatment), no clinically significant changes in vital signs, physical examination, or electrocardiogram-related findings were reported. Additionally, no lanadelumab-treated subjects developed antidrug antibodies [72]. The most commonly reported treatment-emergent AEs in lanadelumab treated subjects were headache (25% [vs. 25% with placebo]; none severe, all resolved) and upper respiratory tract infection (12.5% [vs. 0% with placebo]). No injection-site reactions were reported, and no clinically significant differences in occurrence of AEs were noted.
Pharmacokinetic results demonstrated a dose-proportional drug exposure; mean lanadelumab peak plasma concentrations (Cmax) for the 0.1-, 0.3-, 1.0-, and 3.0-mg/kg doses were 0.56, 1.37, 5.60, and 14.50 μg/mL, respectively. Additionally, the mean elimination half-life was consistent regardless of dose (20.6, 16.8, 17.6, and 21.2 days for the 0.1-, 0.3-, 1.0-, and 3.0-mg/kg doses, respectively) [72]. Pharmacodynamic findings of the two exploratory biomarker assays demonstrated long-lasting dose-dependent inhibition of plasma kallikrein activity with the 1.0- and 3.0-mg/kg (but not the 0.1- or 0.3-mg/kg) doses; inhibition was detectable > 20 days post-dose. In participants treated with 3.0 mg/kg, a significant decrease (p = 0.001) was noted in cleavage of ex vivo FXIIa-mediated HMWK at day 5, which was sustained at day 28 after dosing (p = 0.003) [72]. The PK/PD findings achieved in this study, along with the favorable safety profile and lack of antidrug antibodies in this trial, supported evaluating the effects of lanadelumab in a phase Ib study in patients with C1-INH-HAE.
4.2 Phase Ib Study
The phase Ib study was a randomized, double-blind, placebo-controlled, multiple ascending dose study in patients with C1-INH-HAE [82]. This trial evaluated the following four dose groups: 30, 100, 300, and 400 mg. Within each dose cohort, patients were randomized to receive two doses (14 days apart) of lanadelumab or placebo in a 2:1 ratio [82]. On-demand treatment was allowed when patients experienced HAE attacks. Eligible patients were aged ≥ 18 years with a documented diagnosis of C1-INH-HAE; age at first onset ≤ 30 years or presence of positive family history; and occurrence of at least two HAE attacks per year, one of which must have occurred within the past 6 months. Patients were excluded if they had received long-term prophylaxis for C1-INH-HAE within the past 90 days or had participated in another investigational trial within the past 90 days. Additionally, those enrolling in the study could not have been treated with C1-INH within 1 week before enrollment or have received monoclonal antibodies (or recombinant protein with an Fc domain) within the previous 5 years [82].
The key objectives for the study were to assess lanadelumab safety (including AEs, vital signs, physical examination, clinical laboratory measurements, and electrocardiogram), PK/PD parameters, and antidrug antibody plasma levels. The pharmacodynamics of lanadelumab were evaluated via the same two exploratory assays as used in the phase Ia study described above: the Western blot assay (evaluating presence of cleaved HMWK) and the fluorogenic assay (assessing inhibition of plasma kallikrein). However, in this study, the Western blot assay was performed using citrated plasma with and without FXIIa to activate the plasma kallikrein–kinin cascade. The frequency of attacks and the use of on-demand therapy in the two higher-dose treatment groups (i.e., 300 or 400 mg vs. placebo), in individuals who also had at least two attacks within the 3 months before enrollment, were additional endpoints. These groups for therapeutic efficacy measurement were prespecified before study enrollment. Based on the time to Cmax, determined from the phase Ia study, the number of attacks per week was evaluated from days 8 to 50 [82].
Altogether, 37 patients were randomized (62% female; mean age 39.9 years; 100% White) with a similar mean number of attacks in the past 3 and 12 months. Safety was assessed in patients receiving at least one dose of lanadelumab (n = 24) or placebo (n = 13). At least one AE occurred in 58% and 77% of patients receiving lanadelumab and placebo, respectively. The most commonly reported treatment-related AEs in the lanadelumab versus placebo groups were injection-site pain (25 vs. 23%, respectively) and headache (8 vs. 15%, respectively). Two patients receiving lanadelumab had severe treatment-related AEs, including injection-site pain in a patient receiving the 30-mg dose and worsening headache and prolonged night sweats in a patient receiving 400 mg. No AE-related deaths or discontinuations occurred during the study, no serious AEs emerged with lanadelumab treatment, and no clinically meaningful abnormalities or changes in laboratory parameters, physical examination, vital signs, and electrocardiogram evaluations were reported. The antidrug antibodies identified in two patients in this study were non-neutralizing and did not impact the pharmacokinetic or pharmacodynamic profiles of lanadelumab [82].
The pharmacokinetic profile was evaluated in 22 patients who received at least one dose of lanadelumab (and whose blood samples were sufficient), whereas the pharmacodynamic profile was evaluated in 35 patients (22 patients from the lanadelumab group and 13 patients who received at least one dose of placebo). Mean maximum plasma concentrations were dose proportional: 3895, 7890, 27,460, and 45,322 ng/mL for the 30-, 100-, 300-, and 400-mg doses, respectively. Half-life was 13.8–15.0 days across all lanadelumab dose groups. Detectable drug concentrations were observed across all dose cohorts through day 120 [82].
The Western blot assay showed significant decreases compared with predose levels (p < 0.05) in cleaved HMWK levels in plasma not activated with FXII from patients who received lanadelumab 300- and 400-mg doses on days 8 and 22; levels approached those in healthy volunteers, demonstrating the potential for lanadelumab to decrease activation of the contact system in patients with C1-INH-HAE. In contrast, pharmacodynamic bioactivity of lanadelumab in the 30 and 100 mg dose groups was minimal. Additionally, lanadelumab dose dependently suppressed cleavage of HMWK by ex vivo-mediated activation by FXII; statistical significance was reached for all lanadelumab dosing groups at day 8, for the 300 and 400 mg dose groups at day 22, and for the 400 mg dose group at day 50. The fluorogenic assay demonstrated dose-dependent inhibition of plasma kallikrein with the 100-, 300-, and 400-mg doses: peak inhibition values were ~ 30%, 60%, and 70%, respectively, on the second administration of lanadelumab. In contrast, minimal pharmacodynamic inhibition was observed with the 30-mg and placebo doses [82]. The observation that plasma levels of cleaved HMWK, though reduced, were detectable in patients who received a sufficient dose of lanadelumab to prevent HAE attacks suggests that basal plasma kallikrein activity was maintained.
Findings from the prespecified primary efficacy analysis (from day 8 to 50) in patients in the higher dose lanadelumab groups demonstrated a significantly lower weekly attack rate with lanadelumab versus placebo for both the 300- (p < 0.001) and 400-mg doses (p = 0.005). The percentage of patients who were attack free was 100% for lanadelumab 300 mg, 82% for lanadelumab 400 mg, and 27% in the placebo group. Overall, patients in the combined 300- and 400-mg groups had 91% fewer attacks than patients receiving placebo. Baseline severity of HAE attacks did not influence treatment response, and reduction in HAE attacks correlated with drug exposure [82]. Although not included in the primary efficacy analysis, the incidence of attacks was decreased in the 30- and 100-mg lanadelumab dose groups versus placebo [82].
Overall, this phase Ib study demonstrated that two doses of lanadelumab, separated by 14 days, were well tolerated up to a dose of 400 mg. No new safety concerns were found or safety-related study discontinuations reported. The linear, dose-dependent pharmacokinetic profile and long half-life were consistent with findings in healthy volunteers in the phase Ia study [72, 82]. Pharmacodynamic findings demonstrated in vivo biologic activity, and treatment at the higher doses significantly reduced attack rates versus placebo. Collectively, these findings demonstrated the potential for lanadelumab to inhibit degradation of HMWK and to provide long-term prevention of attacks, supporting its continued evaluation in larger trials.
5 Summary
Despite improved availability of prophylactic and on-demand medications for the management of C1-INH-HAE, many unmet needs remain. Some patients with C1-INH-HAE continue to fear future attacks, which are frequently painful, disrupt quality of life, and may be fatal [14, 15, 83, 84]. The goal is to develop a therapy that is well tolerated and decreases the frequency (or, ideally, eliminates the occurrence) of attacks, thus reducing patients’ daily burden of disease and disability [3, 85]. The fully human monoclonal antibody lanadelumab may help address some of the limitations of existing prophylactic options (i.e., intravenous administration and the need for frequent dosing schedules). The results from preclinical studies demonstrate that it is highly potent and specifically inhibits plasma kallikrein [50], whereas findings from phase Ia and Ib studies suggest lanadelumab is well tolerated, and has a PK/PD profile that provides sustained inhibition of plasma kallikrein, allowing for less frequent dosing [72, 82]. Notably, the safety of long-term inhibition of plasma kallikrein is unknown and will remain a continuing focus as clinical experience with agents targeting plasma kallikrein continues to be gained. Larger studies of longer duration are ongoing in the phase III clinical program. The 26-week primary study has been completed (its findings have led to the recent approval of lanadelumab for prophylactic use in the USA and Canada), whereas the open-label extension study is underway.
References
Davis-Lorton M. An update on the diagnosis and management of hereditary angioedema with abnormal c1 inhibitor. J Drugs Dermatol. 2015;14(2):151–7.
Lumry WR. Hereditary angioedema: the economics of treatment of an orphan disease. Front Med (Lausanne). 2018;5:22.
Zuraw BL, Banerji A, Bernstein JA, Busse PJ, Christiansen SC, Davis-Lorton M, et al. US Hereditary Angioedema Association Medical Advisory Board 2013 recommendations for the management of hereditary angioedema due to C1 inhibitor deficiency. J Allergy Clin Immunol Pract. 2013;1(5):458–67.
Gower RG, Busse PJ, Aygoren-Pursun E, Barakat AJ, Caballero T, Davis-Lorton M, et al. Hereditary angioedema caused by C1-esterase inhibitor deficiency: a literature-based analysis and clinical commentary on prophylaxis treatment strategies. World Allergy Organ J. 2011;4(2 Suppl):S9–21.
Kaplan AP, Joseph K. Pathogenesis of hereditary angioedema: the role of the bradykinin-forming cascade. Immunol Allergy Clin N Am. 2017;37(3):513–25.
Johnston DT. Diagnosis and management of hereditary angioedema. J Am Osteopath Assoc. 2011;111(1):28–36.
Banerji A. The burden of illness in patients with hereditary angioedema. Ann Allergy Asthma Immunol. 2013;111(5):329–36.
Lane CJ, Grant JA, Dougherty D 3rd. Life with hereditary angioedema: then and now. Postgrad Med. 2012;124(1):144–51.
Ghazi A, Grant JA. Hereditary angioedema: epidemiology, management, and role of icatibant. Biologics. 2013;7:103–13.
Farkas H, Martinez-Saguer I, Bork K, Bowen T, Craig T, Frank M, et al. International consensus on the diagnosis and management of pediatric patients with hereditary angioedema with C1-inhibitor deficiency. Allergy. 2016;72(2):300–13.
Bouillet L, Gompel A. Hereditary angioedema in women: specific challenges. Immunol Allergy Clin N Am. 2013;33(4):505–11.
Banerji A, Riedl M. Managing the female patient with hereditary angioedema. Womens Health (Lond). 2016;12(3):351–61.
Caballero T, Maurer M, Longhurst HJ, Aberer W, Bouillet L, Fabien V. Triggers and prodromal symptoms of angioedema attacks in patients with hereditary angioedema. J Investig Allergol Clin Immunol. 2016;26(6):383–6.
Caballero T, Prior N. Burden of illness and quality-of-life measures in angioedema conditions. Immunol Allergy Clin N Am. 2017;37(3):597–616.
Longhurst H, Bygum A. The humanistic, societal, and pharmaco-economic burden of angioedema. Clin Rev Allergy Immunol. 2016;51(2):230–9.
Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006;119(3):267–74.
Gill P, Betschel SD. The clinical evaluation of angioedema. Immunol Allergy Clin N Am. 2017;37(3):449–66.
Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol. 2012;130(3):692–7.
Jalaj S, Scolapio JS. Gastrointestinal manifestations, diagnosis, and management of hereditary angioedema. J Clin Gastroenterol. 2013;47(10):817–23.
Nzeako UC. Diagnosis and management of angioedema with abdominal involvement: a gastroenterology perspective. World J Gastroenterol. 2010;16(39):4913–21.
Zanichelli A, Longhurst HJ, Maurer M, Bouillet L, Aberer W, Fabien V, et al. Misdiagnosis trends in patients with hereditary angioedema from the real-world clinical setting. Ann Allergy Asthma Immunol. 2016;117(4):394–8.
Aygoren-Pursun E, Bygum A, Beusterien K, Hautamaki E, Sisic Z, Wait S, et al. Socioeconomic burden of hereditary angioedema: results from the hereditary angioedema burden of illness study in Europe. Orphanet J Rare Dis. 2014;04(9):99.
Fouche AS, Saunders EF, Craig T. Depression and anxiety in patients with hereditary angioedema. Ann Allergy Asthma Immunol. 2014;112(4):371–5.
Lumry WR, Castaldo AJ, Vernon MK, Blaustein MB, Wilson DA, Horn PT. The humanistic burden of hereditary angioedema: impact on health-related quality of life, productivity, and depression. Allergy Asthma Proc. 2010;31(5):407–14.
Wilson DA, Bork K, Shea EP, Rentz AM, Blaustein MB, Pullman WE. Economic costs associated with acute attacks and long-term management of hereditary angioedema. Ann Allergy Asthma Immunol. 2010;104(4):314–20.
Lumry WR. Pharmacoeconomics of orphan disease treatment with a focus on hereditary angioedema. Immunol Allergy Clin N Am. 2017;37(3):617–28.
Cicardi M, Bork K, Caballero T, Craig T, Li HH, Longhurst H, et al. Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy. 2012;67(2):147–57.
Longhurst H, Zinser E. Prophylactic therapy for hereditary angioedema. Immunol Allergy Clin N Am. 2017;37(3):557–70.
Maurer M, Magerl M, Ansotegui I, Aygoren Pursun E, Betschel S, Bork K, et al. The international WAO/EAACI guideline for the management of hereditary angioedema—the 2017 revision and update. Allergy. 2018;73(8):1575–96.
Lumry WR, Miller DP, Newcomer S, Fitts D, Dayno J. Quality of life in patients with hereditary angioedema receiving therapy for routine prevention of attacks. Allergy Asthma Proc. 2014;35(5):371–6.
Zuraw BL. Clinical practice. Hereditary angioedema. New Engl J Med. 2008;359(10):1027–36.
Qiu T, Chiuchiolo MJ, Whaley AS, Russo AR, Sondhi D, Kaminsky SM, et al. Gene therapy for C1 esterase inhibitor deficiency in a murine model of hereditary angioedema. Allergy. 2018. https://doi.org/10.1111/all.13582 (Epub ahead of print).
European Medicines Agency. Cinryze 500 IU powder and solvent for solution for injection [summary of product characteristics]. 2016. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001207/WC500108895.pdf. Accessed 6 Dec 2017.
European Medicines Agency. Berinert 500 IU powder and solvent for solution for injection/infusion [summary of product characteristics]. 2016. https://www.medicines.org.uk/emc/medicine/21650. Accessed 6 Dec 2017.
Behring CSL. Berinert [package insert]. Kanakee, IL. 2016.
Pharming Technologies B.V. Ruconest [package insert]. Leiden, the Netherlands. 2015.
European Medicines Agency. Ruconest 2100 U powder for solution for injection (summary of product characteristics). 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001223/WC500098542.pdf. Accessed 6 Dec 2017.
Dyax Corp. Kalbitor [package insert]. Burlington, MA. 2015.
Shire. Firazyr [package insert]. Lexington, MA. 2015.
European Medicines Agency. Firazyr 30 mg solution for injection in pre-filled syringe [summary of product characteristics]. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000899/WC500022966.pdf. Accessed 6 Dec 2017.
Shire. Cinryze [package insert]. Lexington, MA. 2018.
CSL Behring. Haegarda [package insert]. Kanakee, IL. 2017.
Sanofi-Aventis. Danocrine [package insert]. Bridgewater, NJ. 2011. https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/017557s033s039s040s041s042lbl.pdf. Accessed 6 Dec 2017.
Greve J, Strassen U, Gorczyza M, Dominas N, Frahm UM, Muhlberg H, et al. Prophylaxis in hereditary angioedema (HAE) with C1 inhibitor deficiency. J Dtsch Dermatol Ges. 2016;14(3):266–75.
European Medicines Agency. Tranexamic acid 500 mg tablets [summary of product characteristics]. 2013. https://www.medicines.org.uk/EMC/medicine/24325/SPC/Tranexamic+Acid+500mg+Tablets/. Accessed 6 Dec 2017.
Riedl MA. Critical appraisal of androgen use in hereditary angioedema: a systematic review. Ann Allergy Asthma Immunol. 2015;114(4):281–288.e7.
Chen M, Riedl MA. Emerging therapies in hereditary angioedema. Immunol Allergy Clin N Am. 2017;37(3):585–95.
Banerji A, Busse P, Christiansen SC, Li H, Lumry W, Davis-Lorton M, et al. Current state of hereditary angioedema management: a patient survey. Allergy Asthma Proc. 2015;36(3):213–7.
Dominas N, Hoffmann TK, Bas M, Greve J. Improving patient outcomes in hereditary angioedema: reducing attack frequency using routine prevention with C1 inhibitor concentrate. BMJ Case Rep. 2014;2014:1–3.
Kenniston JA, Faucette RR, Martik D, Comeau SR, Lindberg AP, Kopacz KJ, et al. Inhibition of plasma kallikrein by a highly specific active site blocking antibody. J Biol Chem. 2014;289(34):23596–608.
Shire. Takhzyro [package insert]. Lexington, MA. 2018.
TakhzyroTM lanadelumab injection Canadian product monograph. https://www.shirecanada.com/-/media/shire/shireglobal/shirecanada/pdffiles/product%20information/takhzyro-pm-en.pdf. Accessed 1 Oct 2018.
Reichert JM. Antibodies to watch in 2017. mAbs. 2017;9(2):167–81.
Banerji A, Riedl M, Bernstein J, Cicardi M, Longhurst H, Zuraw B, et al. OR034 lanadelumab for prevention of attacks in hereditary angioedema: results from the phase 3 HELP study. Ann Allergy Asthma Immunol. 2017;119(5):S5.
Riedl MA, Bernstein JA, Craig T, Banerji A, Magerl M, Cicardi M, et al. An open-label study to evaluate the long-term safety and efficacy of lanadelumab for prevention of attacks in hereditary angioedema: design of the HELP study extension. Clin Transl Allergy. 2017;7:36.
Zuraw BL. The pathophysiology of hereditary angioedema. World Allergy Organ J. 2010;3(9 Suppl):S25–8.
Walford HH, Zuraw BL. Current update on cellular and molecular mechanisms of hereditary angioedema. Ann Allergy Asthma Immunol. 2014;112(5):413–8.
Hofman ZL, Relan A, Zeerleder S, Drouet C, Zuraw B, Hack CE. Angioedema attacks in patients with hereditary angioedema: local manifestations of a systemic activation process. J Allergy Clin Immunol. 2016;138(2):359–66.
Davis AE 3rd. The pathophysiology of hereditary angioedema. Clin Immunol. 2005;114(1):3–9.
Tse K, Zuraw BL. Recognizing and managing hereditary angioedema. Cleve Clin J Med. 2013;80(5):297–308.
Levy JH, O’Donnell PS. The therapeutic potential of a kallikrein inhibitor for treating hereditary angioedema. Expert Opin Investig Drugs. 2006;15(9):1077–90.
Cicardi M, Levy RJ, McNeil DL, Li HH, Sheffer AL, Campion M, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363(6):523–31.
Levy RJ, Lumry WR, McNeil DL, Li HH, Campion M, Horn PT, et al. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol. 2010;104(6):523–9.
Riedl MA, Aygoren-Pursun E, Baker J, Farkas H, Anderson J, Bernstein JA, et al. Evaluation of avoralstat, an oral kallikrein inhibitor, in a phase 3 hereditary angioedema prophylaxis trial: the OPuS-2 study. Allergy. 2018;73(9):1871–80.
Aygoren-Pursun E, Bygum A, Grivcheva-Panovska V, Magerl M, Graff J, Steiner UC, et al. Oral plasma kallikrein inhibitor for prophylaxis in hereditary angioedema. N Engl J Med. 2018;379(4):352–62.
Efficacy and safety study of BCX7353 as an oral treatment for the prevention of attacks in HAE (APeX-2) NCT03485911. https://clinicaltrials.gov/ct2/show/NCT03485911. Accessed 13 Sep 2018.
Lai Y, Wang R, Chen X, Tang D, Hu Y, Cai J, et al. Emerging trends and new developments in monoclonal antibodies: a scientometric analysis (1980–2016). Hum Vaccin Immunother. 2017;13(6):1–10.
Mitchell PD, El-Gammal AI, O’Byrne PM. Emerging monoclonal antibodies as targeted innovative therapeutic approaches to asthma. Clin Pharmacol Ther. 2016;99(1):38–48.
Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–58.
Chan CE, Lim AP, MacAry PA, Hanson BJ. The role of phage display in therapeutic antibody discovery. Int Immunol. 2014;26(12):649–57.
Nixon AE, Sexton DJ, Ladner RC. Drugs derived from phage display: from candidate identification to clinical practice. mAbs. 2014;6(1):73–85.
Chyung Y, Vince B, Iarrobino R, Sexton D, Kenniston J, Faucette R, et al. A phase 1 study investigating DX-2930 in healthy subjects. Ann Allergy Asthma Immunol. 2014;113(4):460–6.e2.
Schapira M, Silver LD, Scott CF, Schmaier AH, Prograis LJ Jr, Curd JG, et al. Prekallikrein activation and high-molecular-weight kininogen consumption in hereditary angioedema. N Engl J Med. 1983;308(18):1050–3.
Kaufman N, Page JD, Pixley RA, Schein R, Schmaier AH, Colman RW. Alpha 2-macroglobulin-kallikrein complexes detect contact system activation in hereditary angioedema and human sepsis. Blood. 1991;77(12):2660–7.
Silverberg M, Reddigari SR, Kaplan AP. The contact system and its disorders. In: Handin RI, Lux SE, Stossel RP, editors. Blood: principles and practice of hematology; J.B. Lippincott Company, Phila, copyright 1995. pp. 1127–50.
Waeckel L, Potier L, Richer C, Roussel R, Bouby N, Alhenc-Gelas F. Pathophysiology of genetic deficiency in tissue kallikrein activity in mouse and man. Thromb Haemost. 2013;110(3):476–83.
Isordia-Salas I, Pixley RA, Sainz IM, Martinez-Murillo C, Colman RW. The role of plasma high molecular weight kininogen in experimental intestinal and systemic inflammation. Arch Med Res. 2004;35(5):369–77.
Sexton DJ, Kenniston JA, Faucette RR, Nixon AE, TenHoor C, Chyung Y, et al. Comparison of plasma kallikrein inhibition by the endogenous C1-inhibitor versus DX-2930, a monoclonal antibody inhibitor. Blood. 2013;122:1066.
Wirth KJ, Alpermann HG, Satoh R, Inazu M. The bradykinin antagonist Hoe 140 inhibits carrageenan- and thermically induced paw oedema in rats. Agents Actions Suppl. 1992;38(Pt 3):428–31.
Morris CJ. Carrageenan-induced paw edema in the rat and mouse. In: Winyard PG, Willoughby DA, editors. Inflammation protocols. Totowa: Humana Press; 2003. p. 115–21.
Whiteley PE, Dalrymple SA. Models of inflammation: carrageenan-induced paw edema in the rat. Curr Protoc Pharmacol. 2001. https://doi.org/10.1002/0471141755.ph0504s00.
Banerji A, Busse P, Shennak M, Lumry W, Davis-Lorton M, Wedner HJ, et al. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N Engl J Med. 2017;376(8):717–28.
Bygum A, Aygoren-Pursun E, Beusterien K, Hautamaki E, Sisic Z, Wait S, et al. Burden of illness in hereditary angioedema: a conceptual model. Acta Derm Venereol. 2015;95(6):706–10.
Caballero T, Aygoren-Pursun E, Bygum A, Beusterien K, Hautamaki E, Sisic Z, et al. The humanistic burden of hereditary angioedema: results from the Burden of Illness Study in Europe. Allergy Asthma Proc. 2014;35(1):47–53.
Craig T, Aygoren-Pursun E, Bork K, Bowen T, Boysen H, Farkas H, et al. WAO guideline for the management of hereditary angioedema. World Allergy Organ J. 2012;5(12):182–99.
Acknowledgements
The authors thank Yi Wang, Senior Director, Clinical Pharmacology and Pharmacokinetics, Shire, for providing content direction on the PK/PD findings reported in this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Under the direction of the authors, Sophia Shumyatsky, PharmD, of Excel Scientific Solutions, provided writing assistance for this manuscript. Editorial assistance in formatting, proofreading, copy editing, and fact checking also was provided by Excel Scientific Solutions. Shire Human Genetic Therapies provided funding to Excel Scientific Solutions for support in writing and editing this manuscript. The data within the published studies reviewed were interpreted by the authors independently.
Conflict of interest
P.J. Busse has received research support and served on advisory boards for Shire, CSL Behring, and BioCryst. H. Farkas has received research support from Shire; is on the advisory boards for CSL Behring, Shire, BioCryst, and Sobi; has received consultancy fees from CSL Behring, Shire, BioCryst, and Sobi; and has received lecture fees from CSL Behring, Pharming Group, and Sobi. A. Banerji has received research funding from Shire; and is on advisory boards for Alnylam, BioCryst, CSL Behring, and Shire. W. Lumry has received consultant fees from Adverum, BioCryst, CSL Behring, Pharming, and Shire; research grants from BioCryst, CSL Behring, Pharming, and Shire; payments for lectures from CSL Behring, Pharming, and Shire; and is a member of the Medical Advisory Board of the US Hereditary Angioedema Association. H. J. Longhurst has received research grant support and/or speaker/consultancy fees from BioCryst, CSL Behring, Shire, and Sobi. D. J. Sexton is a full-time employee of Shire. M. A. Riedl has received research grants from Shire, CSL Behring, BioCryst, and Pharming; consultant fees from Shire, CSL Behring, BioCryst, Pharming, Adverum, Ionis, and Kalvista; payments for lectures from Shire, Pharming, and CSL Behring, and is a member of the Medical Advisory board of the US Hereditary Angioedema Association.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Busse, P.J., Farkas, H., Banerji, A. et al. Lanadelumab for the Prophylactic Treatment of Hereditary Angioedema with C1 Inhibitor Deficiency: A Review of Preclinical and Phase I Studies. BioDrugs 33, 33–43 (2019). https://doi.org/10.1007/s40259-018-0325-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-018-0325-y