
Abstract
Background
Ixekizumab (IXE) is an interleukin (IL)-17A antagonist approved for the treatment of adults with moderate-to-severe psoriasis.
Objective
The objective of this study was to determine if the immune response to tetanus and pneumococcal vaccines in healthy subjects administered IXE was noninferior to control.
Methods
In a randomized, open-label, parallel-group study, adult subjects received vaccinations alone (N = 42, control) or in combination with 160 mg IXE subcutaneously 2 weeks prior to vaccination and 80 mg IXE on the day of vaccination (N = 41, IXE). Response to tetanus vaccination was defined as anti-tetanus antibodies ≥ 1.0 IU and a ≥ 1.5-fold increase if baseline was ≤ 1.0 IU or a ≥ 2.5-fold increase if baseline was > 1.0 IU. Response to pneumococcal vaccination was defined as a ≥ 2-fold increase from baseline in anti-pneumococcal antibodies against > 50% of the 23 serotypes. The primary outcomes were the percentages of patients with a response to the tetanus and pneumococcal vaccines 4 weeks after vaccination. A noninferiority analysis of IXE to control using a 40% margin was evaluated for the primary outcomes. Safety and pharmacokinetics were also assessed.
Results
IXE (38 completers) was noninferior to control (41 completers) based on the difference in the proportion of responders to tetanus [1.4%; 90% confidence interval (CI) − 16.6 to 19.2] and pneumococcal (− 0.8%; 90% CI − 12.9 to 11.0) vaccines. Twenty subjects (14 IXE, six control) reported 43 mild treatment-emergent adverse events.
Conclusion
IXE does not suppress the humoral immune response to non-live vaccines and was well tolerated in healthy subjects.
ClinicalTrial.gov identifier: NCT02543918.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Ixekizumab (IXE), a high-affinity monoclonal antibody that selectively targets interleukin-17A, approved for the treatment of adult moderate-to-severe psoriasis, does not suppress the humoral immune response to non-live vaccines, as assessed by administration of tetanus and pneumococcal vaccines in healthy subjects. |
IXE was well tolerated in healthy subjects. |
1 Introduction
Ixekizumab (IXE) is a high-affinity monoclonal antibody that selectively targets interleukin (IL)-17A and is approved for the treatment of moderate-to-severe psoriasis in adult patients [1]. The approved dose for patients with psoriasis is 160 mg [two subcutaneous (SC) 80-mg injections] at week 0, followed by 80 mg at weeks 2, 4, 6, 8, 10, and 12, then 80 mg every 4 weeks.
Psoriasis is a chronic immune-mediated disease of the skin that may require long-term treatment. As most people will receive or require vaccinations during their lifetime, it is important to understand if vaccines can be safely and efficaciously administered alongside long-term treatments for chronic diseases. In addition, treatment of psoriasis or other immune-mediated diseases (e.g., rheumatoid arthritis, irritable bowel disease) with agents that may suppress the immune system has been associated with increased risk of infections that may be prevented by vaccinations, including but not limited to tetanus, pertussis, and influenza [2]; therefore, vaccination rates in the psoriasis patient population would be expected to be at least as high as the general population. However, although non-live vaccinations for patients with immune-mediated inflammatory diseases (IMIDs), such as psoriasis, are recommended [2], the percentage of vaccinated patients is unexpectedly low compared to the general population [2, 3]. Only 9 and 45% of IMID patients have received pneumococcal or tetanus vaccinations [3], respectively, compared with 20.3 and 62.2% of the general population [4]. The low rates of vaccination in adults with IMIDs may be attributed to a lack of knowledge in vaccine recipients and concerns from healthcare providers that a vaccine could induce disease flares or potentially even trigger the onset of an autoimmune disease. Further, there is insufficient data on whether IMID patients treated with immunomodulatory therapies would have a response to vaccination that provides sufficient protection [2, 4].
The humoral response to non-live vaccinations has been previously studied with a variety of monoclonal antibodies that target T- and B-cell cytokines. In some cases, biologic therapies have been shown to alter the immunologic response to vaccines in patients with IMIDs [5,6,7]. Most notably, rituximab showed a measureable decrease in vaccine antibody production to tetanus and pneumococcal vaccinations in patients with rheumatoid arthritis [6]. In contrast to the rituximab data, multiple biologic therapies have been shown not to affect vaccine antibody production, for instance, abatacept, a selective T-cell co-stimulation modulator (using tetanus and pneumococcal vaccines) [5], tofacitinib, an oral Janus kinase (JAK) inhibitor (using influenza and pneumococcal vaccines) [7], the tumor necrosis factor alpha (TNF-α) inhibitor golimumab (using pneumococcal vaccine) [8], the IL-12/IL-23 antagonist ustekinumab (using tetanus and pneumococcal vaccines) [9], and the IL-17A antagonist, secukinumab (using influenza and meningococcal vaccines) [10]. Further, the vaccines chosen for these studies demonstrate T-cell-dependent (tetanus or influenza) and T-cell-independent (pneumococcal or meningococcal) humoral antibody responses following vaccine administration, since both are important components of the humoral immune response to non-live vaccines.
As with these other IMID chronic therapies, it is important to evaluate the potential for IXE to impact the ability to produce adequate antibody production to non-live vaccines. This study evaluated the immune response to two routinely recommended non-live vaccines, tetanus (Boostrix®; GlaxoSmithKline Biologicals, Belgium) and pneumococcus (Pneumovax 23®; Merck & Co, NJ, USA), for which clinical laboratory assays for antibody response are available in clinical practice. Immune response (antibody levels) to these vaccines was studied in the presence or absence of IXE in healthy subjects. Tolerability and pharmacokinetics (PK) of IXE were also evaluated. Live vaccines were not studied [1].
2 Methods
2.1 Participants
Healthy males or females, 18–65 years old, with a body mass index (BMI) of 18–32 kg/m2 were included in the study. Subjects were excluded if they had a known allergy or hypersensitivity to any biologic therapy or vaccine; had a compromised immune system or had other comorbidities (such as having an active infectious disease, a history of Guillain–Barre Syndrome or lymphoma, leukemia, or any malignancy within the past 5 years); had received IXE or IL-17 antagonists; had a live vaccination within 1 year prior to screening; had received a tetanus toxoid–containing vaccine within the last 5 years; or had been immunized with the pneumococcal vaccine. Since the study was conducted in the USA, the baseline assumption was the tetanus vaccine would likely be a “booster” vaccine rather than the subjects’ first tetanus vaccination. While a tetanus booster is recommended at least once every 10 years [11], the restriction of a subject having no booster within the last 5 years was selected based on dose and schedule information in the Boostrix label [12] and published studies [5, 6, 9].
2.2 Study Design and Treatment
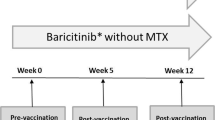
In this phase I, multicenter (three study centers in the USA), open-label, parallel-group study (NCT02543918) (Fig. 1), healthy subjects were randomized to IXE (N = 41) or control (N = 43). At randomization (week 0), IXE subjects received an SC 160-mg IXE starting dose, and at week 2, an 80-mg IXE dose. The 160-mg IXE dose at week 0 and the 80-mg IXE dose at week 2 have been administered extensively to subjects with psoriasis in phase III studies and are the start of the approved dosing regimen for moderate-to-severe plaque psoriasis. The dosing in this study is designed to achieve therapeutic levels by week 6, the time of the vaccine assessment.

Study design (NCT02543918). *Vaccine antibody samples taken prior to IXE administration. AB antibody, IXE ixekizumab
Vaccinations for tetanus (Boostrix) [12] and pneumococcus (Pneumovax 23) [13] were administered 2 weeks after the IXE 160-mg dose (week 0) and on the same day as the 80-mg dose of IXE (week 2). The primary vaccine response endpoint measurement was at week 6 (4 weeks post-vaccination) and was based on the performing laboratory’s validated assay protocols for both vaccines. Published data also indicated that this time period includes the time from vaccination in which the greatest interference with response to the vaccine (2 weeks) is likely to occur [5].
2.3 Endpoints and Assessments
The primary endpoints are the percentages of patients with a response to the tetanus and pneumococcal vaccines. Response to tetanus vaccination was defined as anti-tetanus antibodies ≥ 1.0 IU and a ≥ 1.5-fold increase if baseline was ≤ 1.0 IU or a ≥ 2.5-fold increase if baseline was > 1.0 IU. Response to pneumococcal vaccination was defined as a ≥ 2-fold increase from baseline in anti-pneumococcal antibodies against > 50% of the 23 serotypes. These responses were based on validated laboratory assay protocols and published data [6, 9, 14]. Immune response to vaccinations, specifically antibody production to the tetanus and pneumococcal vaccines, was measured using validated quantitative multiplex bead-based immunoassays performed for routine clinical testing in a Clinical Laboratory Improvement Amendments (CLIA) certified laboratory (ARUP Laboratories, Salt Lake City, UT, USA). The serotypes for analysis of the pneumococcal vaccine were as follows: 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F, and 33F.
Additional exploratory endpoints were assessed based on various published criteria and post hoc assessments in response to a regulatory request to further understand the impact of IXE on the ability to produce an adequate antibody response [14,15,16]. These endpoints were (1) the percentage of subjects with either an increase to protective anti-tetanus antibody (ATAb) or anti-pneumococcal antibody (APAb) levels from non-protective baseline levels; (2) the change from baseline in geometric mean antibody levels at 2 and 4 weeks post-vaccination; (3) the number and percentage of subjects with a > 4-fold increase from baseline in ATAb level; and (4) the percentage of subjects with a ≥ 2-fold increase in APAb to at least 70% of serotypes for pneumococcal vaccine, those with a ≥ 4-fold increase from baseline in APAb to at least 50% of serotypes for pneumococcal vaccine, and those with a ≥ 4-fold increase from baseline in APAb to at least 70% of serotypes for the pneumococcal vaccine.
The tolerability and PK of IXE in healthy subjects were secondary and exploratory endpoints, respectively. Safety parameters assessed included adverse events (AEs), laboratory parameters, vital signs, and electrocardiogram parameters, and the Quick Inventory of Depressive Symptomatology–Self-Report (QIDS-SR16). Serum samples for IXE PK analysis were obtained during the study at the following times: prior to administration of the 160-mg IXE dose on day 1 (week 0), on days 3, 5, 8, and 11 following the 160-mg IXE dose, and prior to administration of the 80-mg IXE dose on day 15 (week 2) and then at weeks 4, 6, and 12. Serum samples were analyzed for IXE using a validated enzyme-linked immunosorbent assay (ELISA) (Intertek Pharmaceutical Services, San Diego, CA, USA). The lower limit of quantification was 7.5 ng/mL, and the upper limit of quantification was 300.0 ng/mL. A 1:5 minimum required dilution was applied to all samples. Samples above the limit of quantification were diluted to yield results within the calibrated range. The inter-assay precision (percentage relative standard deviation) during validation ranged from 11.8 to 17.3%.
2.4 Statistical Analyses
Data analysis was performed using SAS® version 9.3. Antibody vaccine analyses included all randomized subjects receiving that vaccine who had a baseline and at least one evaluable post-baseline value. Safety analyses included all randomized subjects. PK analyses included all randomized subjects who received at least one dose of IXE and had sufficient evaluable PK data.
For the primary analysis, the difference between the two groups (IXE group minus control) in the proportion of responders to each vaccine at 4 weeks post-vaccination together with the 90% confidence interval (CI) of the difference was calculated for the tetanus and pneumococcal vaccines. Noninferiority of the IXE group to the control group for each vaccine was established if the lower limit of the 90% CI of the difference between the two groups (IXE minus control) in the proportion of responders to each vaccine at 4 weeks post-vaccination was greater than 40%. This noninferiority margin has been used previously [10]. No multiplicity adjustment was used. CIs for the differences in proportions between the two groups were calculated using the Newcombe method based on the Wilson score [17, 18]. This method was also used to compare the proportions of patients between the two groups for post hoc analyses. A mixed-effect model was used to analyze the differences from baseline in natural log-transformed serotype levels with treatment arm (IXE or control), visit, and visit-by-treatment interaction as fixed effects and subject as a random effect. Geometric mean ratios (GMRs) for each serotype level to the baseline at 2 and 4 weeks post-vaccination (week 4 and week 6, respectively) with their 95% CI were reported for both the IXE and control groups. In addition, GMRs of serotype levels at 2 and 4 weeks post-vaccination (study week 4 and week 6, respectively) in the IXE group to the control group were provided with their 95% CI.
The proportion of subjects showing an increase from a non-protective level at baseline (≤ 1 IU) to a protective level (> 1 IU) of ATAb at 4 weeks post-vaccination was calculated. In addition, the proportion of subjects showing an increase from a non-protective level at baseline (≤ 1.3 µg/mL) to a protective level (> 1.3 µg/mL) of APAb to at least 50% of serotypes at 4 weeks post-vaccination was identified. CIs for the proportion of subjects that showed an increase from a non-protective level to a protective level for each group were calculated using the Wilson method [19]. Additional exploratory, non-prespecified immune response statistical analyses were conducted to compare the proportion of responders between groups using the alternate responder definitions described previously in Sect. 2.3. The differences between the two groups (IXE group minus control) in the proportion of responders based on these criteria at 4 weeks post-vaccination together with the 95% CI of the difference were calculated.
The incidence of AEs for the IXE versus control group is presented. AEs reported during the study were not necessarily caused by the therapy. Therefore, the reported frequencies do not reflect causality as evaluated by the investigator.
PK parameters for IXE were calculated by standard non-compartmental methods of analysis using Phoenix WinNonlin version 6.2.1 and included the maximum concentration (C max), time of maximum concentration (t max), area under the concentration time curve from time zero to time t last (AUC0–tlast), where t last is the last time point with a measurable concentration after the 160-mg dose administered on day 1 (week 0), and the terminal elimination half-life (t ½) after administration of the 80-mg dose at week 2. The PK parameters were summarized using standard descriptive statistics.
3 Results
3.1 Demographics
Demographic characteristics were similar across both IXE and control groups. Of the 84 randomized healthy subjects, 46 were male and 38 were female, ages ranged between 21 and 65 years, and subjects had a mean BMI of 26.69 kg/m2 and body weight of 76.97 kg (Table 1). The majority of subjects were white (49/84; 58.3%) or black/African American (32/84; 38.1%).
A total of 84 subjects were randomized; 41 to IXE and 43 to control. Of these, 83 received treatment (either IXE and/or vaccines); one subject randomized to the control group withdrew prior to the administration of vaccines due to scheduling conflicts. The remaining 42 subjects in the control group received vaccinations and 41 completed the study (week 6). One subject from the control group received vaccinations, but was subsequently lost to follow-up before data collection. Of the 41 subjects randomized to the IXE group, all 41 received at least one dose of IXE and 38 also received vaccines and completed the study (week 6). Three subjects discontinued from the IXE group after the 160-mg dose (one each due to injection site erythema, voluntary withdrawal, and lost to follow-up).
3.2 Immune Response
The primary objective was met. The IXE group was determined to be noninferior to the control group based on a prespecified noninferiority margin of 40% for the difference in proportions of responders at 4 weeks post-vaccination (week 6) between IXE and control groups (Fig. 2). For the tetanus vaccine, response was seen in 52.6 and 51.2% of subjects in the IXE and control groups, respectively, yielding a difference of 1.4 with 90% CI − 16.6 to 19.2. For the pneumococcal vaccine, response was seen in 89.5 and 90.2% of subjects in the IXE and control groups, respectively, yielding a difference of − 0.8 with 90% CI − 12.9 to 11.0.

Noninferiority plot [90% confidence interval (CI)] of the treatment difference of responders to the tetanus (Boostrix) and pneumococcal (Pneumovax 23) vaccines 4 weeks after vaccination (week 6). CIs for the difference in proportions between the two groups calculated using the Newcombe method based on the Wilson score. A responder to tetanus (Boostrix) vaccine is defined as having a post-vaccination anti-tetanus antibody (ATAb) level of ≥ 1 IU and a ≥ 1.5-fold increase (50% increase) from baseline if the baseline pre-vaccination level is ≤ 1.0 or a ≥ 2.5-fold increase (150% increase) from baseline if the pre-vaccination level is > 1.0 IU. A responder to the pneumococcal (Pneumovax 23) vaccine is defined as having a ≥ 2-fold increase (100% increase) from baseline in anti-pneumococcal antibody (APAb) levels against > 50% of the 23 serotypes
The proportion of responders between the two groups was similar across the responder definitions used for analysis (i.e., prespecified definition and alternative post hoc published and regulatory requested definitions) (Table 2).
The GMRs to baseline for the IXE and control groups were similar and had overlapping 95% CIs for antibody levels at 2 and 4 weeks post-vaccination (week 4 and week 6, respectively) for tetanus and all of the pneumococcal serotypes (see the Electronic Supplementary Material, Online Resource 1; Supplementary Table 1 and Supplementary Fig. 1). For the tetanus vaccine, the GMRs to baseline were 2.94 and 4.01 at week 6 for the control and IXE groups, respectively. The GMR of IXE to control was close to 1, with the 95% CI including 1. For the pneumococcal vaccine, the GMR to baseline varied from 1.73 (serotype 5) to 8.83 (serotype 8) for the control group and from 1.81 (serotype 5) to 10.9 (serotype 2) for the IXE group. The 95% CIs for the GMRs of the IXE group to control included 1 for all but two of the 23 serotype antibody levels at 2 and 4 weeks post-vaccination (week 4 and week 6 time points). For these two serotypes (6B and 11A), the IXE group showed a higher increase from baseline than the control group at both time points of the post-vaccine measurement.
The proportion of subjects showing an increase from a non-protective level to protective ATAb and APAb levels at week 6 was evaluated. Only five subjects (one in the control group and four in the IXE group) had non-protective tetanus levels at baseline. All of these subjects increased to a protective level at week 6. For the pneumococcal vaccine, 29 subjects in each group had non-protective levels at baseline. A protective immune response to the pneumococcal vaccine at week 6 was achieved for 89.7% (90% CI 76.8–95.8) and 93.1% (90% CI 81.2–97.7) of the subjects, respectively, in the control and IXE groups.
For the tetanus vaccine, 29.3% of subjects in the control group and 42.1% of subjects in the IXE group had a > 4-fold increase from baseline in ATAb levels. The percentage of subjects with a ≥ 2-fold increase in at least 70% of serotypes for the pneumococcal vaccine, as well as those with a ≥ 4-fold increase for at least 50 and 70% of serotypes for the pneumococcal vaccine, was similar (e.g., 0 was contained in each CI of the difference between groups and the lower bound was greater than the prespecified 40% noninferiority margin) across the IXE and control groups (Table 2).
3.3 Safety
Twenty subjects [14 (34.1%) IXE, 6 (14.3%) control] reported 43 treatment emergent adverse events (TEAEs); all were mild in severity. No deaths or serious AEs occurred during the study. Of these 43 TEAEs, 24 were reported by the investigator to be related to treatment with IXE or vaccines. Twelve subjects [10 (24.4%) IXE and 2 (4.8%) control] reported 24 treatment-related AEs, with 21 and three AEs in the IXE and control groups, respectively; all were mild in severity. A treatment-related AE (injection site erythema) led to discontinuation of one subject following 160 mg IXE. Headache, injection-site erythema, and fatigue were the most common TEAEs in all subjects (reported in up to 5% of subjects). The most common treatment-related AE was injection site erythema, reported in four subjects in the IXE group (Table 3). All reported TEAEs and treatment-related AEs are listed in Table 3.
There were no clinically relevant effects on clinical laboratory chemistry values, hematology, urinalysis, vital signs, or the QIDS-SR16 scores. No clinically significant alterations in vital sign values of systolic blood pressure, diastolic blood pressure, pulse rate, and oral cavity temperature were observed during the study. Low-grade transient reductions in neutrophil counts were observed in subjects in both groups (IXE = 5; control = 4) and were not associated with infection. Of these subjects, one subject in the IXE group and one subject in the control group each had low neutrophil counts consistent with grade 2 neutropenia (defined as an absolute neutrophil count of 1000 to < 1500 per cubic millimeter) at week 6. By the week 12 visit, the neutrophil count in the IXE subject improved from grade 2 to grade 1 neutropenia (a count of 1500 to < 2000 per cubic millimeter). No laboratory data were collected for control subjects after week 6.
3.4 Pharmacokinetics
Data regarding the PK of IXE in healthy subjects following a single SC dose of 160 mg IXE on day 1 (week 0) suggest that the mean C max and AUC0–tlast were 17.2 µg/mL and 180 µg·day/mL, respectively. The geometric mean (range) elimination t ½ was 14.7 (10.6–24.1) days, estimated after the 80-mg dose administered at week 2.
4 Discussion
Treatment with IXE did not affect the humoral response to non-live vaccines, as assessed using tetanus and pneumococcal vaccines. IXE was noninferior to the control group based on the difference in the proportion of responders to tetanus and pneumococcal vaccines 4 weeks post-vaccination. A similar proportion of healthy subjects treated with IXE and control subjects were able to mount an adequate immune response in antibody levels to both vaccines. Our results were consistent with findings with other systemic biologic agents for psoriasis with varying mechanisms of action, such as TNF-α inhibitors etanercept [20], infliximab [21], or adalimumab [22], the IL-17A antagonist secukinumab [10], and the IL-12/IL-23 inhibitor ustekinumab [9], which showed no clinically important effect on humoral responses to non-live vaccines.
A protective immune response to the pneumococcal vaccine was also evident 4 weeks post-vaccination. A protective immune response to the pneumococcal vaccine was achieved for 93 and 90% of the subjects (29 subjects in each group) with non-protective levels at baseline in the IXE and control groups, respectively. For the tetanus vaccine, even though all patients with non-protective levels achieved protective levels, there were only five subjects for IXE (N = 4) and control (N = 1) groups that had non-protective levels at baseline.
The conclusion of noninferiority in the immune response to non-live vaccines in healthy subjects administered IXE compared to control subjects was further supported by exploratory and post-hoc analyses. With tetanus, for example, an increase in antibody level of at least 4-fold in response to a booster vaccination has been used as a primary endpoint [6] or from observations by the World Health Organization (WHO) [23], although other endpoints have included a 2-fold increase [5]. This analysis was conducted and showed that a comparable percentage of subjects in the control group (approximately 29%) and in the IXE group (approximately 42%) had at least a 4-fold increase in antibody levels in response to the booster vaccine.
For pneumococcal vaccine response, a review of published studies [6, 8, 9] revealed variability in the serotypes that were evaluated. Nonetheless, the endpoint used in those studies was the same as the primary endpoint in this study, that is, a ≥ 2-fold increase in antibody levels in 50% of the serotypes assessed. Published data also indicated that increased antibody levels for 50–70% of the serotypes are thought to represent a normal humoral response to the pneumococcal vaccine; therefore, an exploratory analysis was done relative to a 70% response [24, 25]. Although the conclusion of Daly and Hill [25] indicates the endpoint of a > 2-fold increase in antibody levels is generally the appropriate measure for evaluating the immune response to the pneumococcal vaccine, a post hoc analysis with a ≥ 4-fold criterion was also evaluated by request. The results of all the additional analyses for pneumococcus were consistent with the primary endpoint that the response to vaccination in the IXE group compared to the control group was similar across both groups. The percentage of subjects with a ≥ 2-fold increase in at least 70% of serotypes for the pneumococcal vaccine, as well as those with a ≥ 4-fold increase for at least 50 and 70% of serotypes for the pneumococcal vaccine, was similar. Thus, the results of this study support the conclusion of Daly and Hill [25] that the 2-fold increase criterion used to test the primary objective for the pneumococcal vaccine represents a definition of adequate antibody production to vaccination. Finally, while other studies, to evaluate whether a medicinal product could affect the response to the pneumococcal vaccine, typically prespecified only six to 12 of the 23 vaccine serotypes for evaluation of the response, all 23 serotypes were assessed in this study. By evaluating a subset of serotypes, it could be argued that the data can be analyzed using the most prevalent serotypes, accounting for disease in the population of interest. If this study had evaluated only these subgroups of serotypes, the conclusion of noninferiority of IXE to control would also have been achieved.
All AEs were mild in severity. The most commonly reported AEs in subjects in the study were headache, injection site erythema, and fatigue. With the exception of fatigue, this is consistent with those reported in phase III trials with IXE in patients with psoriasis [26, 27]. In clinical trials of patients with plaque psoriasis, adverse reactions that occurred in the first 12 weeks of treatment in ≥ 1% of patients and more frequently than those in the placebo group also included injection-site reactions, as well as upper respiratory infections (includes nasopharyngitis and rhinovirus), tinea infections, and nausea [1]. Neutropenia observed in healthy subjects treated with IXE was not associated with infection, and the low-grade transient neutrophil changes were consistent with observations from previous clinical studies with IXE in patients with psoriasis [28] as well as other anti-IL-17 biologics [29, 30].
The PK parameters in healthy subjects were within the range observed in patients with psoriasis. Thus, clinically relevant concentrations of IXE were achieved in this study [1, 31].
While patients with chronic diseases, such as psoriasis, are likely to need vaccinations while on therapy, it is preferable, but not always practical, to vaccinate prior to initiating therapy. Routine vaccinations for patients with psoriasis are recommended prior to treatment with biologics; however, concurrent vaccination with live vaccines is not recommended [2, 32]. Healthcare professionals are encouraged to assess the immunization status of their patients as part of their initial consultation in order to optimize their treatment plan [2].
4.1 Limitations
Although this study included both T-cell-dependent and T-cell-independent vaccines commonly used by healthcare providers, these are only two of the routine non-live vaccines that are recommended by the Advisory Committee on Immunization Practices, the American College of Obstetrics and Gynecologists, and the American Academy of Family Physicians [11]. Due to the prevalence of use of the tetanus vaccine in the US population, the majority of subjects at baseline in both the IXE and control groups had protective levels of anti-tetanus antibodies, despite the exclusion criterion that precluded subjects who had received a tetanus toxoid-containing vaccine within the previous 5 years. However, this potential limitation was mitigated by using the change from baseline for the primary analysis. While the multiplex immunoassay format has been associated with more interlaboratory variability and higher antibody levels when compared to ELISA [33], these subject samples were all assayed at the same laboratory. Additionally, these differences do not adversely impact interpretation of the protective result of vaccination [14, 34]. Lastly, this evaluation was performed in healthy subjects, and vaccine response was not studied in patients who had immune-modulated disease.
5 Conclusions
IXE does not suppress the humoral immune response to non-live, T-cell-dependent or T-cell-independent vaccines as assessed using tetanus and pneumococcal vaccines in healthy subjects. There were no unexpected safety findings in healthy subjects, with a PK response similar to patients with psoriasis.
References
Eli Lilly and Company. Taltz (ixekizumab) [prescribing information]. 2016. http://pi.lilly.com/us/taltz-uspi.pdf. Accessed 15 May 2017.
Wine-Lee L, Keller SC, Wilck MB, Gluckman SJ, Van Voorhees AS. From the Medical Board of the National Psoriasis Foundation: vaccination in adult patients on systemic therapy for psoriasis. J Am Acad Dermatol. 2013;69:1003–13.
Rahier JF, Moutschen M, Van Gompel A, Van Ranst M, Louis E, Segaert S, et al. Vaccinations in patients with immune-mediated inflammatory diseases. Rheumatology (Oxford). 2010;49:1815–27.
Williams WW, Lu PJ, O’Halloran A, Kim DK, Grohskopf LA, Pilishvili T, et al. Surveillance of vaccination coverage among adult populations-United States, 2014. MMWR Surveill Summ. 2016;65:1–36.
Tay L, Leon F, Vratsanos G, Raymond R, Corbo M. Vaccination response to tetanus toxoid and 23-velent pneumococcal vaccines following administration of a single dose of abatacept: a randomized, open-label, parallel group study in healthy subjects. Arthritis Res Ther. 2007;9:R38.
Bingham CO 3rd, Looney RJ, Deodhar A, Halsey N, Greenwald M, Codding C, et al. Immunization responses in rheumatoid arthritis patients treated with rituximab: results from a controlled trial. Arthritis Rheumatol. 2010;62:64–74.
Winthrop KL, Silverfield J, Racewicz A, Neal J, Lee EB, Hrycaj P, et al. The effect of tofacitinib on pneumococcal and influenza vaccine responses in rheumatoid arthritis. Ann Rheum Dis. 2016;75:687–95.
Zhou H, Jang H, Fleischmann R, Bouman-Thio E, Xu Z, Marini JC, et al. Pharmacokinetic and safety of golimumab, a fully human anti-TNF-alpha monoclonal antibody, in subjects with rheumatoid arthritis. J Clin Pharmacol. 2007;47:383–96.
Brodmerkel C, Wadman E, Langley RG, Papp KA, Bourcier M, Poulin Y, et al. Immune response to pneumococcus and tetanus toxoid in patients with moderate-to-severe psoriasis following long-term ustekinumab use. J Drugs Dermatol. 2013;12:1122–9.
Chioato A, Noseda E, Stevens M, Gaitatzis N, Kleinschmidt A, Picaud H. Treatment with the interleukin-17A-blocking antibody secukinumab does not interfere with the efficacy of influenza and meningococcal vaccinations in healthy subjects: results of an open-label, parallel-group, randomized single-center study. Clin Vaccine Immunol. 2012;19:1597–602.
Center for Disease Control and Prevention. Adult immunization schedule. 2017. https://www.cdc.gov/vaccines/schedules/downloads/adult/adult-combined-schedule.pdf. Accessed 15 May 2017.
GlaxoSmithKline Biologicals. Boostrix (tetanus toxoid, reduced diphtheria toxoid and acellular pertussus vaccine) [package insert]. 2016. https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Boostrix/pdf/BOOSTRIX.PDF. Accessed 15 May 2017.
Merck & Co., Inc. Pneumovax 23 (pneumococcal vaccine polyvalent) [package insert]. 2015. http://www.merck.com/product/usa/pi_circulars/p/pneumovax_23/pneumovax_pi.pdf. Accessed 15 May 2017.
Daly TM, Pickering JW, Zhang X, Prince HE, Hill HR. Multilaboratory assessment of threshold versus fold-change algorithms for minimizing analytical variability in multiplexed pneumococcal IgG measurements. Clin Vaccine Immunol. 2014;21:982–8.
Tipples GA, Hamkar R, Mohktari-Azad T, Gray M, Parkyn G, Head C, et al. Assessment of immunoglobulin M enzyme immunoassays for diagnosis of measles. J Clin Microbiol. 2003;41:4790–2.
Moe CL, Sair A, Lindesmith L, Estes MK, Jaykus LA. Diagnosis of Norwalk virus infection by indirect enzyme immunoassay detection of salivary antibodies to recombinant Norwalk virus antigen. Clin Diagn Lab Immunol. 2004;11:1028–34.
Newcombe RG. Interval estimation for the difference between independent proportions: comparison of eleven methods. Stat Med. 1998;17:873–90.
Fagerland MW, Lydersen S, Laake P. Recommended confidence intervals for two independent binomial proportions. Stat Methods Med Res. 2015;24:224–54.
Agresti A, Coull BA. Approximate is better than ‘exact’ for interval estimation of binomial proportions. Am Stat. 1998;52:119–26.
Mease PJ, Ritchlin CT, Martin RW, Gottlieb AB, Baumgartner SW, Burge DJ, et al. Pneumococcal vaccine response in psoriatic arthritis patients during treatment with etanercept. J Rheumatol. 2004;31:1356–61.
Visvanathan S, Keenan GF, Baker DG, Levinson AI, Wagner CI. Response to pneumococcal vaccine in patients with early rheumatoid arthritis receiving infliximab plus methotrexate plus methotrexate or methotrexate alone. J Rheumatol. 2007;34:952–7.
Kaine JL, Kivitz AJ, Birbara C, Luo AY. Immune responses following administration of influenza and pneumococcal vaccines to patients with rheumatoid arthritis receiving adalimumab. J Rheumatol. 2007;34:272–9.
World Health Organization. Annex 5. Recommendations to assure the quality, safety, and efficacy of tetanus vaccines (adsorbed). 2017. http://www.who.int/biologicals/vaccines/Tetanus_Recommendations_TRS_980_Annex_5.pdf?ua. Accessed 15 May 2017.
Paris K, Sorensen RU. Assessment and clinical interpretation of polysaccharide antibody responses. Ann Allergy Asthma Immunol. 2007;99:462–4.
Daly TM, Hill HR. Use and clinical interpretation of pneumococcal antibody measurements in the evaluation of humoral immune function. Clin Vaccine Immunol. 2015;22:148–52.
Gordon KB, Blauvelt A, Papp KA, Langley RG, Luger T, Ohtsuki M, et al. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345–56.
Griffiths CE, Reich K, Lebwohl M, van de Kerkhof P, Paul C, Menter A, et al. Comparison of ixekizumab with etanercept or control in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomized trials. Lancet. 2015;386:541–51.
Brown G, Malakouti M, Wang E, Koo JY, Levin E. Anti-IL-17 phase II data for psoriasis: a review. J Dermatol Treat. 2015;26:32–6.
Lebwohl M, Strober B, Menter A, Gordon K, Weglowska J, Puig L, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318–28.
van de Kerkhof PC, Griffiths CE, Reich K, Leonardi CL, Blauvelt A, Tsai TF, et al. Secukinumab long-term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75(83–98):e4.
Callis Duffin K, Bagel J, Bukhalo M, Mercado Clement IJ, Choi SL, Zhao F, et al. Phase 3, open-label, randomized study of the pharmacokinetics, efficacy and safety of ixekizumab following subcutaneous administration using a prefilled syringe or an autoinjector in patients with moderate-to-severe plaque psoriasis (UNCOVER-A). J Eur Acad Dermatol Venereol. 2017;31:107–13.
Menter A, Gottlieb A, Feldman SR, Van Voorhees AS, Leonardi CL, Gordon KB, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2017;58:826–50.
Whaley MJ, Rose C, Martinez J, Laher G, Sammons DL, Smith JP, et al. Interlaboratory comparison of the three multiplexed bead-based immunoassays for measuring serum antibodies to pneumococcal polysaccharides. Clin Vaccine Immunol. 2010;17:862–9.
Zhang X, Simmerman K, Yen-Lieberman B, Daly TM. Impact of analytical variability on clinical interpretation of multiplex pneumococcal serology assays. Clin Vaccine Immunol. 2013;20:957–61.
Acknowledgements
The authors wish to acknowledge Michael Turik and Wendy Komocsar (Eli Lilly and Company) for scientific discussion on study design and vaccine selection, Susan Ball for critical review of the clinical study report and the manuscript, Chin Hyok Lee (Eli Lilly and Company) for critical review of the manuscript, and Olawale Osuntokun (Eli Lilly and Company) for critical review of the clinical study report. The authors thank Gina Moore, MS (inVentiv Health Clinical) for editorial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was conducted in accordance with consensus ethics principles derived from international ethics guidelines, including the Declaration of Helsinki and Council for International Organizations of Medical Sciences (CIOMS) International Ethical Guidelines, the International Conference on Harmonization (ICH) GCP Guideline, and applicable laws and regulations. The study protocol and informed consent forms were approved by an investigational review board, and all patients signed informed consent before undergoing study-related procedures. This article does not contain any studies with animals performed by any of the authors.
Funding
The study was funded by Eli Lilly and Company.
Conflict of interest
The authors are employees and stockholders of Eli Lilly and Company.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gomez, E.V., Bishop, J.L., Jackson, K. et al. Response to Tetanus and Pneumococcal Vaccination Following Administration of Ixekizumab in Healthy Participants. BioDrugs 31, 545–554 (2017). https://doi.org/10.1007/s40259-017-0249-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-017-0249-y