
Abstract
CD4+CD25highFoxP3+ T regulatory cells (Tregs) are immunodominant suppressors in the immune system. Tregs use various mechanisms to control immune responses. Preclinical data from animal models have confirmed the huge therapeutic potential of Tregs in many immune-mediated diseases. Hence, these cells are now on the road to translation to cell therapy in the clinic as the first clinical trials are accomplished. To date, clinical research has involved mainly hematopoietic stem cell transplantations, solid organ transplantations, and autoimmunity. Despite difficulties with legislation and technical issues, treatment is constantly evolving and may soon represent a valid alternative for patients with diseases that are currently incurable. This review focuses on the basic and clinical experience with Tregs with adoptive transfer of these cells, primarily from clinical trials, as well as on perspectives on clinical use and technical problems with implementing the therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cells characterized by the CD4+CD25highFoxP3+ phenotype are responsible for maintaining immune tolerance and suppression of excessive immune responses. |
Preclinical animal studies have confirmed the therapeutic potential of T regulatory cells (Tregs) and pave the way for their use in therapy in humans. |
Clinical trials of this therapy continue in many research centers worldwide, mainly in hematopoietic stem cell transplantation, the induction and maintenance of tolerance to solid organ allotransplants, and the treatment of autoimmune diseases. |
1 Introduction
Regulatory T cells (Tregs) are a population of lymphocytes whose role is to regulate and suppress excessive responses from other immune cells. Tregs are able to control a variety of other subsets, such as activated effector cells [T conventional (Tconv) cells], and inhibit antigen-presenting cells (APCs), natural killer (NK) cells, B cells, and innate immunity [1,2,3]. In the 1970s, Gershon and Kondo [4] introduced an hypothesis of a cell population that regulated the immune system. However, it was not until 1995 that Sakaguchi et al. [5] presented the first firm evidence that the hypothesis was true. They used a mouse model to prove that a lack of cluster of differentiation (CD)4+CD25+ T cells resulted in autoimmune-mediated multiple organ dysfunction [5]. This syndrome was also later associated with mutation of the foxp3 gene, a master regulator of Tregs, defined as “scurfy” in mice and IPEX (immune dysfunction, polyendocrinopathy, and enteropathy, X-linked) syndrome in humans [6]. Tregs responsible for the syndrome are characterized by a CD4+CD25highFoxP3+ phenotype, originate from the thymus, and are often called natural Tregs (nTregs or tTregs). Other regulatory subsets also exist within CD4+ T cells: primarily so-called induced or peripheral Tregs (iTregs or pTregs, respectively) with Tr1 cells and T-helper (Th)-3 cells, which are generated by the conversion of conventional CD4+ T cells at the periphery [7]. However, nTregs are drawing attention as a potential cellular medicine because of their stability and pronounced suppressive effects when administered in vivo [8].
2 Biology of T Regulatory (Treg) Cells
nTregs have several modes of action at the periphery, but they primarily recognize self-antigens and self-like antigens released from damaged tissues, actively migrate to such sites, and switch off the activity of other immune cells to inhibit inflammation [9]. Thus, Tregs protect from potential or ongoing auto-aggression and damage to tissues; this activity is limited to within very close proximity of the inflammation site [10]. This suppressive mode of action has led Tregs to be called “intelligent steroids” as they have the immunosuppressive power of glucocorticoid-based medicines and lack the associated adverse effects these hormonal drugs have because of their more generalized influence on the whole body. Moreover, Tregs play an important role in the induction of tolerance to allotransplants of solid organs and can control allergy [11,12,13,14]. Even more interesting is that much research suggests the therapeutic effect of many routinely used immunosuppressive drugs depends on the stimulation of Tregs [15, 16].
The suppressive effect of Tregs on Tconvs is executed mainly via cell-to-cell contacts, for example via programmed cell death (PD)-1-PD-ligand (L) coupling but also via the transfer of cyclic adenosine monophosphate (cAMP) through the membrane gap junctions and adenosine produced in the paracrine fashion by the CD39 and CD73 receptors expressed on Tregs [17, 18]. Another mode of action is “control by starvation/theft” of interleukin (IL)-2. The CD25 molecule (a high-affinity receptor for IL-2) is highly expressed on nTregs and thus Tregs win the competition with Tconv cells for this cytokine. The deficit of IL-2 stops the proliferation of other cells and induces apoptosis by granzyme and perforin [19]. As well as direct suppression of activated Tconv cells, nTregs prevent the activation of these lymphocytes via the inhibition of APCs. In the cell-to-cell contact dependent on CTLA-4-CD80/CD86 interactions, Tregs induce expression of indoleamine 2,3-dioxygenase (IDO) in dendritic cells, which in turn results in the suppression of helper and cytotoxic Tconv populations [20]. The inhibition of autoreactive B cells by Tregs is partially governed by the mechanisms described for Tconv cells. It involves interaction between surface molecules—PD-1 expressed by B cells and PD-L1 ligands on Tregs. Tregs suppress the production of autoantibodies and inhibit B-cell proliferation and induce their apoptosis [21]. In the case of innate immunity, more distant regulation is engaged, involving suppressive cytokines secreted by Tregs. The inhibition of monocytes/macrophages partially depends on IL-10, IL-4, and IL-13 [22]. Tregs suppress the production of reactive oxygen intermediates (ROI) and the cytokines produced by neutrophils. The cytokine IL-10, transforming growth factor (TGF)-β, and direct cell-to-cell contacts all take part in this process. Moreover, granzymes and perforin secreted by Tregs are able to induce apoptosis of neutrophils and other cells in the inflammation site [14].
In the context of NK cells, the main mechanism of action is through membrane-bound TGF-β and latency-associated peptide (LAP) on Tregs. Tregs inhibit interferon (IFN)-γ production and proliferation and the cytotoxicity of NK cells [10]. Finally, Tregs can inhibit FcεRI-dependent degranulation of mast cells and therefore inhibit allergy and anaphylaxis. This has been shown to be mediated by a surface molecule—OX40—on the surface of Tregs and its ligand (OX40L) on the surface of mast cells [9]. All these mechanisms guard the body from autoimmunity but may also tip the balance of the immune system to cancer. Nevertheless, recent studies have revealed that tumor-homed Tregs are distinct from Tregs localized in normal tissues [23]. Interestingly, novel activities of Tregs have been described recently. For example, these cells appear to have a major role in tissue repair and maintenance [24]. Tregs have also been reported to modulate the progression of muscular dystrophies [25]. While knowledge on the activity of Tregs is extensive, it should be highlighted that at least some of the mechanisms, mostly those recently described, are still speculative and require more study (Fig. 1).

Chosen mechanisms used by T regulatory cells (Tregs). I suppression of antigen presentation, induction of expression of IDO in DCs via the CTLA-4; II inhibition of activation of Th and cytotoxic T effector via cell-to-cell interactions, extracellularly produced adenosine via CD39, CD73 receptors; transferred cAMP and consumption of IL-2; III induction of apoptosis of mono/mac; IV inhibition of B-cell proliferation and induction of apoptosis via PD-1; V induction of apoptosis of neutrophils; VI inhibition of function and proliferation of NK cells; VII inhibition of degranulation of mast cells. cAMP cyclic adenosine monophosphate, CD cluster of differentiation, DCs dendritic cells, IDO indoleamine 2,3-dioxygenase, IL interleukin, mono/mac monocytes/macrophages; NK natural killer, PD-1 programmed cell death-1, Tc cytotoxic T effector, Th T helper
2.1 Preclinical Models of Treg Therapy
The use of Tregs to treat disease has been tempting clinicians from the first reports on the immunoregulatory activity of these cells. The idea was initially verified in animal models through adoptive transfer of cells between animals. Early reports proved that the transfer of Tregs associated with hematopoietic stem cell transplantation (HSCT) in mice protected from graft versus host disease (GvHD) and promoted the graft versus leukemia effect (GvL) [26]. Unfortunately, this simple transfer between donor and recipient cannot be translated to humans as the clinical effect requires the administration of a high number of Tregs [27]. The low number of Tregs in peripheral blood, which is a natural feature of this subset, is therefore a technical challenge. The search for an effective method of Treg expansion has begun. Initial trials in an allotransplant setting in mice showed that in vivo conversion of Tconv to induced Treg is possible but was not feasible for human clinics [28]. Therefore, manufacturing procedures to allow expansion of Tregs in vitro before administration was developed. In brief, a small number of Tregs isolated from a donor were cultured in vitro in specific conditions to impose proliferation before transfer to a recipient. This method of ex vivo expansion has the advantage that the product can be analyzed on an ongoing basis in terms of functional and phenotypic activity and the dose can be precisely controlled. Tests in animal models have shown that such ex vivo expansion is possible [29]. Both polyclonal and recipient-specific cells prepared ex vivo were able to induce a GvHD-free state after bone marrow transplantation [29]. Ex vivo manufactured Tregs were also confirmed as having good suppressive abilities in solid organ transplantation in animal models, including non-human primates [30,31,32].
Apart from the transplant setting, which implies specific alloantigen mismatches and a very clear beginning of the immune reaction starting from the transplantation procedure, the therapy has also been tested in animal models of autoimmune diseases. In this case, the initiating event and antigens responsible for triggering the response are not always clear, and the animal models therefore less closely mimic human diseases. However, some solid evidence has been collected. For example, it has been confirmed that transferring autoimmune anti-islet T cells from diabetic animals to previously healthy animals induced insulitis and type 1 diabetes mellitus (T1DM) [33]. It was subsequently suggested that the accumulation of Tregs in local lymph nodes around the pancreas may protect mice from diabetes [34]. Moreover, so-called non-obese diabetic (NOD) mice, which spontaneously acquire T1DM because of Treg impairment [35], can be treated with adoptive transfer of Tregs, which traffic to the pancreas and suppress islet-reactive Tconv cells [36].
Multiple sclerosis (MS) is another well-defined autoimmune syndrome with good evidence of Treg involvement [37]. The autoimmunity in MS is easy to follow because of the principal autoantigens linked to the disease—proteins building myelin [38]. Sensitization of mice with these proteins results in the development of experimental autoimmune encephalomyelitis (EAE), an equivalent of human MS. Interestingly, remission or prevention of EAE was associated with the induction of CD4+CD25+ Tregs [39]. This hypothesis was further confirmed with the adoptive transfer of Tregs: transfer before EAE induction prevented EAE, and transfer to animals that already had EAE relieved symptoms [40]. Similar observations on the curative role of the adoptive transfer of Tregs were reported in animal models of multi-organ inflammation [41].
Humanized animal models are the final proof of concept that the adoptive transfer of Tregs has a suppressive effect on the immune system. Humanized animals are immunocompromised animals homed with a human immune system. Such animals reject transplanted human allogeneic tissues when human Tregs are depleted from the body and accept the tissues when Tregs are adoptively transferred together with other human immune cells to the animal [42, 43]. Nevertheless, it must be mentioned that adoptive transfers failed in some animal trials. For example, the cells were minimally effective in a collagen-induced arthritis model [44] and completely failed to inhibit glomerulonephritis and sialadenitis in mice with lupus [45].
3 Completed and Ongoing Clinical Trials
With confirmation of the therapeutic potential of Tregs in a number of animal studies, the first clinical trials commenced [46] (Table 1). The most tempting factor in the translation of Tregs to the clinic was that Tregs seem to retain all the advantages of standard immunosuppression without the adverse effects [47]. Treg therapy has been somewhat introduced with the drugs abatacept and belatacept, which are fusion proteins containing the moiety of receptor CTLA-4, the receptor responsible for the major suppressive abilities of Tregs. The effectiveness of these drugs as maintenance immunosuppression after solid organ transplantation and in the treatment of autoimmune diseases has already been confirmed [48,49,50]. As mentioned, clinical potentiation of many other drugs through increased Treg activity has been reported [15, 16].
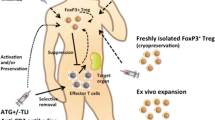
Intrinsic therapy with Tregs administered to patients has also already occurred. Tregs can be prepared from allogeneic donors or as an autologous preparation from the patient. They can either be directly administered as freshly isolated cells or expanded under Good Manufacturing Practice (GMP) conditions before administration. They can be used as polyclonal or antigen-specific cell preparations. These tolerogenic cells have been tested as prophylaxis and/or treatment in a variety of indications that can be categorized into four main streams: after HSCT, in solid organ transplants, in autoimmune diseases, and in allergic syndromes (Table 1).
3.1 Hematopoietic Stem Cell Transplantation
Tregs as cell therapy has mostly been tested in the treatment or prophylaxis of GvHD after HSCT. The first-in-man administration of ex vivo expanded nTregs was performed by our group (Trzonkowski et al. [52]) in patients with ongoing GvHD. Cells were harvested from respective donors, expanded ex vivo, and infused into HSCT recipients who had GvHD. The program is still continuing, mainly as compassionate use in patients with GvHD unresponsive to other forms of pharmacological immunosuppression. To date, we have applied the therapy in 13 patients and observed a good safety profile. However, it has been effective only in the chronic form of GvHD, in which we have observed alleviation of symptoms despite weakened immunosuppression after Treg administration. On the other hand, we have observed no effects in acute GvHD, mainly because the time between the decision to use the therapy and administration of Tregs is too long: it takes approximately 2 weeks to manufacture the cell product, which is too long for a patient with heavy (grade III–IV) acute GvHD whose disease progresses continuously [52].
Since then, a number of trials with either nTregs or Tr1 cells in the prophylaxis or treatment of GvHD have been performed in different centers around the world [53,54,55,56,57,58]. In 2011, Brunstein and colleagues [54, 59] reported a reduction in the incidence of acute GvHD rates and no toxicities after administration of cord blood (CB)-derived nTregs. However, this lack of toxicities has since been corrected as patients treated with Tregs experienced an increased incidence of viral infections [60]. Neoplasms were also reported in patients treated with Tregs [58], but patients in both trials were also treated with other forms of heavy immunosuppression, so the adverse effects could not be attributed to Tregs alone. A group from Perugia reported very intriguing results; they found a lower incidence of GvHD and relapse rates in leukemic patients after treatment with Tregs at the time of HSCT [53, 55]. In other words, the strategy, which consisted of Treg administration together with Tconv in different ratios, seemed not only to prevent GvHD but also to facilitate the GvL effect [53, 55]. Importantly, recent studies with Tregs expanded in vivo confirmed these observations [61]. Not much is known about the kinetics of in vivo expansion of Tregs after HSCT, but existing data suggest that infused Tregs quickly disappear from the peripheral blood and change the repertoire of T-cell receptors (TCRs) [63, 64]. It also appears that particular transferred clones have a different lifespan in the body [62]. In a positive scenario, this correlates with clinical improvement. It is possible that Tregs traffic to the tissues and appropriate clones guard Tconvs from GvHD [65]. At the same time, some Treg clones might control proliferation of residual tumor cells facilitating GvL executed by Tconvs [66]. If true, the dependency of efficacy on particular clones highlights the need for antigen-specific Treg preparations.
3.2 Solid Organ Transplantation
Tregs are also important in the induction and maintenance of tolerance to solid organ allotransplants. Studies using Tregs derived from patients in the context of the prevention of organ rejection and reduced immunosuppression after kidney or liver transplantations are ongoing. Both expanded polyclonal nTregs and antigen-specific nTregs are being tested [67, 68], but results are not yet available. In a separate study, a group from Japan has recently provided results from a pilot study on Treg therapy in liver transplantation in which the administered suppressive cells consisted of recipient T cells enriched in Tregs after ex vivo co-culture with irradiated donor cells. The results from this trial showed the therapy was safe and that it was possible to obtain effective drug minimization and operational tolerance to the allograft [69].
3.3 Autoimmunity
Studies on the link between autoimmune diseases and Tregs have demonstrated a significantly reduced number and/or function of Tregs in the initiation and progression of these diseases [5, 6]. T1DM is a classical autoimmune disease that is a natural target for Treg therapy. No treatment is available to stop disease progression, and the fact that it affects mainly children provides further motivation for scientists and doctors worldwide to look for novel effective agents to stop the disease. In 2012, we presented the first promising results of treatment with Tregs expanded ex vivo in children with recently diagnosed T1DM [63]. Importantly, the therapy appeared to be safe in this group and did not compromise general immunity, as verified by stable post-immunization antibody titers examined in the follow-up [62]. Safety was also confirmed in another trial testing nTregs in adults with T1DM [64]. Since then, we have treated over 50 children in two different trials and found improved long-term survival of pancreatic islets. Compared with non-treated controls, significantly higher levels of functional pancreatic islets secreting insulin were found in treated subjects 2–3 years after commencing therapy.
Nevertheless, the major limitation of this therapy is that only around 20% of pancreatic islets remain at the time of T1DM diagnosis and therefore only this margin of pancreatic function can be spared. Probably for that reason, all patients eventually became insulin dependent. Still, the gain of the therapy is the preserved marginal secretion of insulin, which in the long term can control glycemia and reduce T1DM-related complications. We found that not only the dose but also the number of injections with nTregs is important. We also found that the disease may significantly influence the population of nTregs and it is therefore important to initiate this therapy as early as possible to obtain the least affected Tregs for expansion and the best possible preparation for infusion. Finally, as IL-2 is crucial for the survival of nTregs, we have verified the need for concomitant administration of this cytokine with the cells. IL-2 levels in serum in vivo as well as interaction between nTregs and other lymphocytes were good enough to keep nTregs viable in treated patients without exogenous IL-2 [72]. nTreg therapy is also being tested in other autoimmune and allergic diseases, such as MS, lupus erythematosus, asthma, and autoimmune uveitis [73]. Tr1 cells are being studied in a separate track. In 2012, TxCell company published results of therapy with Tr1 cells in refractory Crohn’s disease. The treatment has proven safe and efficacious. There were as many as 75% responders, and remission was noted in 38% of the patients 5 weeks after the treatment. The therapy is currently under phase IIb clinical development [74].
4 Perspectives for Future Clinical Use
While Tregs have crossed the threshold of hospital wards, challenges remain in the development of therapy with these cells.
4.1 Biology
Despite the recent exponential growth in the number of research papers on Tregs, the biology of these cells is still not fully described. The history of major milestones in the characterization of these cells provides a good lesson in respect for nature. The need to modify the phenotype from initial CD4+CD25+ T cells to CD4+ T cells with the highest expression of CD25 receptors [75], the discovery of the plasticity of T cells with possible links between Tregs and Th17 cells [76], and findings around the expression of the foxP3 gene and its epigenetic changes in Tregs have all provided knowledge that must be taken into account when manufacturing medicinal Tregs [77, 78].
The best classification for Tregs is still under debate, particularly for clinical applications. For example, should we consider them analogous to Tconv cells (naïve, memory, and effector Treg cells) or does splitting them into tissue-resident CD44+CD62L Tregs and central CD44–CD62L+ Tregs better indicate the nature of the suppressive activity of these cells? [79]. Furthermore, should the idea of tissue-specific Tregs, based on the repertoire of expressed receptors that allow Tregs to home specifically to the inflamed tissues, be emphasized in clinical research? [80]. Theoretically, therapy with these tissue-specific cells can limit the activity of medicinal Tregs to the site of infection and reduce further possible adverse effects elsewhere [81]. Indeed, what is the suppressive nature of the cells? Are they precise antigen-specific regulators or maybe polyclonal infiltrates are better in the inflammation site because of infectious tolerance, bystander activation, and suppression of innate immunity?
4.2 Legislation
The biological features of Tregs must be translated by the clinical laboratory, tissue establishment, or the facility producing them to either cellular preparations or medicinal products. The transfer of Treg therapy from a scientific model to the clinic requires cooperation between scientists, clinicians, and regulatory authorities as the therapy must be both efficacious and safe. In the EU, Treg treatments are classified either as cell transplantation or the administration of a new category: an advanced therapy medicinal product (ATMP). The former are governed by the transplantation Acts adherent to EU tissue and cell directive 2004/23/EC [82], and the latter are regulated by the EU directive on cell-based medicinal products 1394/2007/EC on ATMPs and the amendment of the directive 2001/83/EC, which defines standards for pre-clinical and clinical cell preparations and equalizes ATMPs to other categories of drugs [83, 84]. In brief, Tregs (and other kinds of cells) are classified based on the purpose of use and the extent of modifications while manufactured in vitro. Tregs can be classified as cells for transplantation when they are both (1) intended to be used for the same essential function in the recipient as in the donor (so-called homologous use) and (2) in vitro modification of the cells is not substantial, that is, the biological characteristics, physiological functions, or structural properties do not change (the directives contain a list of such non-essential modifications). If either of these two conditions are not fulfilled, the cells are defined as ATMPs. Finally, the manufacturing of the cells for both transplantation and ATMPs for human use is tightly regulated by GMP [85]. All this needs to be taken into account while translating the science to clinical investigation.
4.3 Technical Issues
The first problem is the low number of nTregs: they account for no more than 5–10% of peripheral blood CD4+ T cells. Hence, alternative sources have been developed, such as bone marrow blood or umbilical blood [73]. Recently published work reported the isolation of nTregs from discarded pediatric thymuses [86]. Regardless of the source, the material should be purified to obtain a pure population of Tregs. Purification is performed using an immunomagnetic method or fluorescence-activated cell sorting (FACS). The former is more feasible as it occurs in closed vessels with no contact between cells and the external environment. However, a major disadvantage of immunomagnetic sorting is the low purity of the Treg preparation. It is still acceptable in applications with fresh cells. Nevertheless, when the procedure includes in vitro expansion, impurities with other cells almost always overgrow the expansion cultures as Tregs are characterized by a low proliferation index. As a result, the final purity of the product is worse than the initial product. The solution might be to include rapamycin or other agents in the culture to preferentially promote proliferation of Tregs and inhibition of Tconv cells [87, 88]. Another possibility is the use of FACS, which produces extremely pure post-sort Tregs. However, this method is challenging as the sort occurs in the air and therefore clinical sorting requires a special clean room environment. New generations of sorters are equipped with single-use sterile sample lines and high efficiency particulate air (HEPA) enclosures, which provide the necessary standard of cleanliness. New methods also attempt to merge the advantage of the closed environment with the precision of FACS [73, 89]. Given the low number of Tregs available after purification, they must be multiplied under GMP conditions before administration. Disadvantages of in vitro expansion include a risk of contamination and a decline in the immunosuppressive abilities of these cells. For this reason, the manufacturing of Tregs is performed in clean room facilities in which the entire environment is sterile and continuously surveilled; Tregs under expansion are sampled and checked for sterility and activity throughout the process [90]. There is still much debate over which phenotypic and functional in vitro tests best predict the in vivo activity of the cells. This becomes increasingly important as attempts are made to direct cultured Tregs against particular antigens [91, 92]. Moreover, attempts are also being made to genetically modify medicinal Tregs to make them antigen-specific. Currently, the main idea is to obtain antigen specificity through gene transduction of a chimeric antigen receptor (CAR) [93,94,95,96]. For obvious reasons, this technique in Tregs is most advanced in transplantation applications where antigens triggering pathology are the best characterized. Other bioengineering attempts with Tregs include the generation of artificial organoids by fusion of Tregs with other cells, as we did with pancreatic islets coated with Tregs as a functional capsule protecting the islets from rejection [97, 98].
Along with the quality of the manufactured preparations, a proper assessment of the efficacy of the therapy constitutes a problem. In general, it seems that prophylaxis with Tregs is much better than treatment of ongoing diseases. GvHD is a good example. Treatments already exist for many of the tested syndromes, and Treg therapy should be compared with these treatments so the best option can be chosen for patients. The scarce evidence from existing human trials indicates that Tregs are not a ‘magic bullet’ for all immunopathologies, and good efficacy is seen only in some diseases. In others, a combination of several different agents, including Tregs, might be superb. For example, in our ongoing trial (EudraCT 2014-004319-35), we are testing Tregs combined with anti-CD20 antibody to target two different arms of the immune response involved in the progression of T1DM.
4.4 Academia Versus Commercial Development
The development of this therapy is also hampered by inconsistencies between the scientific nature of the early phase of the research and GMP and good clinical practice (GCP) requirements that force specific modes of clinical trials to obtain the marketing authorization necessary to offer the medications commercially. In brief, support for scientific studies is provided by periodic grants that are too short and provide too small a budget to support the whole registration process. The solution to this is the selling of research results to commercial companies; however, doing that in an early phase of the research poses a risk of misinterpretation of data and erroneous processing of further studies by sponsors. By definition, a commercial development expects a good income relatively quickly; therefore, many scientific ideas are often abandoned because of the long distance to profits. This is either because the therapy is at too early a phase of development or an inappropriate business model has been applied that very often generates enormous costs that cannot be compensated by available forms of reimbursement or directly by patients. Such a commercial approach towards cellular therapies affects the number of therapies offered routinely to patients in Europe. Treating cellular medications with the business solutions applied routinely for other categories of drugs has resulted in a very low number of registrations of ATMPs in Europe: around ten in almost 10 years from the introduction of the 1394/2007EU directive. Therefore, regulators need to rethink the approach of over-representation of commercial sponsors in obtaining marketing authorization, particularly for cellular medications. An extreme option would be to cancel the idea of ATMPs and revert to the unified model of cells as a transplantation material that cannot be commercialized. Academia should be much more involved in the later stages of registration and market authorization. Conversely, academia should also change the attitude towards more flexible thinking around studies with cellular medicines. For example, the simple idea of a ‘dose’ when referring to cells totally differs from that with other drug forms. The number of patients recruited for studies or the definition of placebo must also differ because of the specificity of these medicines. This specificity should also be accepted by editors of scientific journals; it is very common for a submitted report to be rejected when the study differs from a classical scheme and even more common when the researchers are not from major academic sites. It simply delays the development of effective treatments. We all should be aware there is no monopoly on or boundaries for good ideas.
5 Conclusions
For all these reasons, one should be glad that relatively complex treatment with Tregs has advanced so much in recent years. It provides hope for the many patients with a variety of often incurable diseases. There are many other subsets of regulatory cells on the horizon, but therapies using them will definitely use solutions worked out during studies with Tregs. It is therefore in the common interest to support future clinical research with Tregs and help researchers avoid possible hurdles on the path towards offering these cells to patients.
References
Chen X, Du Y, Lin X, Qian Y, Zhou T, Huang Z. CD4+ CD25+ regulatory T cells in tumor immunity. Int Immunopharmacol. 2016;34:244–9.
Trzonkowski P, Dukat-Mazurek A, Bieniaszewska M, Marek-Trzonkowska N, Dobyszuk A, Juścińska J, et al. Treatment of graft-versus-host disease with naturally occurring T regulatory cells. BioDrugs. 2013;27(6):605–14.
Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8(7):523–32. doi:10.1038/nri2343.
Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. 1970;18:723–37.
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–64.
Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27(1):18–20.
Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30(5):636–45. doi:10.1016/j.immuni.2009.04.010.
Edinger M. Regulatory T cells for the prevention of graft-versus-host disease: professionals defeat amateurs. Eur J Immunol. 2009;39(11):2966–8.
Mekori YA, Hershko AY. T cell-mediated modulation of mast cell function: heterotypic adhesion-induced stimulatory or inhibitory effects. Front Immunol. 2012;3:6. doi:10.3389/fimmu.2012.00006.
Trzonkowski P, Szmit E, Myśliwska J, Dobyszuk A, Myśliwski A. CD4+CD25+ T regulatory cells inhibit cytotoxic activity of T CD8+ and NK lymphocytes in the direct cell-to-cell interaction. ClinImmunol. 2004;112(3):258–67.
Sagoo P, Perucha E, Sawitzki B, Tomiuk S, Stephens DA, Miqueu P, et al. Development of a cross-platform biomarker signature to detect renal transplant tolerance in humans. J Clin Invest. 2010;120(6):1848–61.
Newell KA, Asare A, Kirk AD, Gisler TD, Bourcier K, Suthanthiran M, et al. Immune Tolerance Network ST507 Study Group. Identification of a B cell signature associated with renal transplant tolerance in humans. J Clin Invest. 2010;120(6):1836–47.
Palomares O, Martin-Fontecha M, Launer M, Traidl-Hoffmann C, Cavkaytar O, Akdis M, et al. Regulatory T cells and immune regulation of allergic diseases: roles of IL-10 and TGF-β. Gene Immun. 2014;15(8):511–20.
Alberu J, Vargas-Rojas MI, Morales-Buenrostro LE, Crispin JC, Rodríguez-Romo R, Uribe-Uribe NO et al. De novo donor-specific HLA antibody development and peripheral CD4(+)CD25(high) cells in kidney transplant recipients: a place for interaction? J Transplant 2012;1–8.
Trzonkowski P, Szaryńska M, Myśliwska J, Myśliwski A. Ex vivo expansion of CD4(+)CD25(+) T regulatory cells for immunosuppressive therapy. Cytometry A. 2009;75(3):175–88. doi:10.1002/cyto.a.20659.
Wang XJ, Leveson-Gower D, Golab K, Wang LJ, Marek-Trzonkowska N, Krzystyniak A, et al. Influence of pharmacological immunomodulatory agents on CD4(+)CD25(high)FoxP3(+) T regulatory cells in humans. Int Immunopharmacol. 2013;16(3):364–70.
Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med. 2007;204(6):1303–10.
Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol. 2014;5:304.
Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188(2):287–96.
Mellor AL, Chandler P, Baban B, Hansen AM, Marshall B, Pihkala J, et al. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol. 2004;16(10):1391–401.
Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. 2011;17:983–8.
Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci USA. 2007;104:19446–51.
Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity. 2016;45(5):1122–34.
Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell. 2015;162(5):1078–89.
Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG, Margeta M, Spencer MJ, Bluestone JA. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci Transl Med. 2014;6(258):258ra142.
Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003;9(9):1144–50.
Tang Q, Lee K. Regulatory T-cell therapy for transplantation: how many cells do we need? Curr Opin Organ Transpl. 2012;17:349–54.
Karim M, Kingsley CI, Bushell AR, Sawitzki BS, Wood KJ. Alloantigen-induced CD25+CD4+ regulatory T cells can develop in vivo from CD25—CD4+ precursors in a thymus-independent process. J Immunol. 2004;172(2):923–8.
Trenado A, Sudres M, Tang Q, Maury S, Charlotte F, Grégoire S, et al. Ex vivo-expanded CD4+CD25+ immunoregulatory T cells prevent graft-versus-host-disease by inhibiting activation/differentiation of pathogenic T cells. J Immunol. 2006;176(2):1266–73.
Duran-Struuck R, Sondermeijer HP, Bühler L, Alonso-Guallart P, Zitsman J, Kato Y, et al. Effect of ex vivo-expanded recipient regulatory T cells on hematopoietic chimerism and kidney allograft tolerance across MHC barriers in cynomolgus macaques. Transplantation. 2017;101(2):274–83.
Kingsley CI, Karim M, Bushell AR, Wood KJ. CD25+CD4+ regulatory T cells prevent graft rejection: CTLA-4- and IL-10-dependent immunoregulation of alloresponses. J Immunol. 2002;168(3):1080–6.
Karim M, Feng G, Wood KJ, Bushell AR. CD25+CD4+ regulatory T cells generated by exposure to a model protein antigen prevent allograft rejection: antigen-specific reactivation in vivo is critical for bystander regulation. Blood. 2005;105(12):4871–7.
Bendelac A, Carnaud C, Boitard C, Bach JF. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates. Requirement for both L3T4+ and Lyt-2+ T cells. J Exp Med. 1987;166(4):823–32.
Green EA, Gorelik L, McGregor CM, Tran EH, Flavell RA. CD4+CD25+ T regulatory cells control anti-islet CD8 + T cells through TGF-beta-TGF-beta receptor interactions in type 1 diabetes. Proc Natl Acad Sci USA. 2003;100(19):10878–83.
Tritt M, Sgouroudis E, D’Hennezel E, Albanese A, Piccirillo CA. Functional waning of naturally occurring CD4+ regulatory T-cells contributes to the onset of autoimmune diabetes. Diabetes. 2008;57:113–23.
Tonkin DR, Haskins K. Regulatory T cells enter the pancreas during suppression of type 1 diabetes and inhibit effector T cells and macrophages in a TGF-beta-dependent manner. Eur J Immunol. 2009;39(5):1313–22.
Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199(7):971–9.
Elong Ngono A, Pettré S, Salou M, Bahbouhi B, Soulillou JP, Brouard S, et al. Frequency of circulating autoreactive T cells committed to myelin determinants in relapsing-remitting multiple sclerosis patients. Clin Immunol. 2012;144(2):117–26.
Jee Y, Piao WH, Liu R, Bai XF, Rhodes S, Rodebaugh R, et al. CD4(+)CD25(+) regulatory T cells contribute to the therapeutic effects of glatiramer acetate in experimental autoimmune encephalomyelitis. Clin Immunol. 2007;125(1):34–42.
Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169(9):4712–6.
Ono M, Shimizu J, Miyachi Y, Sakaguchi S. Control of autoimmune myocarditis and multiorgan inflammation by glucocorticoid-induced TNF receptor family-related protein(high), Foxp3-expressing CD25+ and CD25− regulatory T cells. J Immunol. 2006;176(8):4748–56.
Wu DC, Hester J, Nadig SN, Zhang W, Trzonkowski P, Gray D, et al. Ex vivo expanded human regulatory T cells can prolong survival of a human islet allograft in a humanized mouse model. Transplantation. 2013;96(8):707–16.
Nadig SN, Wieckiewicz J, Wu DC, Warnecke G, Zhang W, Luo S, et al. In vivo prevention of transplant arteriosclerosis by ex vivo-expanded human regulatory T cells. Nat Med. 2010;16(7):809–13.
Zhou X, Kong N, Wang J, Fan H, Zou H, Horwitz D, et al. Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J Immunol. 2010;185:2675–9.
Bagavant H, Tung KSK. Failure of CD25+ T cells from lupus-prone mice to suppress lupus glomerulonephritis and sialadenitis. J Immunol. 2005;175:944–50.
Juvet SC, Whatcott AG, Bushell AR, Wood KJ. Harnessing regulatory T cells for clinical use in transplantation: the end of the beginning. Am J Transplant. 2014;14(4):750–63.
St Clair EW, Turka LA, Saxon A, Matthews JB, Sayegh MH, Eisenbarth GS, et al. New reagents on the horizon for immune tolerance. Annu Rev Med. 2007;58:329–46.
Ford ML, Adams AB, Pearson TC. Targeting co-stimulatory pathways: transplantation and autoimmunity. Nat Rev Nephrol. 2014. doi:10.1038/nrneph.2013.183.
Durrbach A, Pestana JM, Florman S, Del Carmen RM, Rostaing L, Kuypers D, et al. Long-term outcomes in Belatacept- versus cyclosporine-treated recipients of extended criteria donor kidneys: final results from BENEFIT-EXT, a phase III randomized study. Am J Transplant. 2016;16(11):3192–201.
Orban T, Bundy B, Becker DJ, Di Meglio LA, Gitelman SE, Goland R, Type 1 Diabetes Trial Net Abatacept Study Group, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. 2011;378(9789):412–9.
EU Clinical Trials Register. https://www.clinicaltrialsregister.eu/. Accessed 01 May 2017.
Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+ CD127− T regulatory cells. Clin Immunol. 2009;133(1):22–6.
Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, Terenzi A, et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood. 2014;124(4):638–44.
Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117(3):1061–70. doi:10.1182/blood-2010-07-293795.
Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117(14):3921–8. doi:10.1182/blood-2010-10-311894.
Bacchetta R, Lucarelli B, Sartirana C, Gregori S, Lupo Stanghellini MT, Miqueu P, et al. Immunological outcome in haploidentical-HSC transplanted patients treated with IL-10-Anergized donor T cells. Front Immunol. 2014;5:16. doi:10.3389/fimmu.2014.00016.
Edinger M, Hoffmann P. Regulatory T cells in stem cell transplantation: strategies and first clinical experiences. Curr Opin Immunol. 2011;23(5):679–84. doi:10.1016/j.coi.2011.06.006.
Theil A, Tuve S, Oelschlägel U, Maiwald A, Döhler D, Oßmann D, et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy. 2015;17(4):473–86. doi:10.1016/j.jcyt.2014.11.005.
Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood. 2016;127(8):1044–51.
Brunstein CG, Blazar BR, Miller JS, Cao Q, Hippen KL, McKenna DH, et al. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol Blood Marrow Transplant. 2013;19(8):1271–3.
Wolf D, Barreras H, Bader CS, Copsel S, Lightbourn CO, Pfeiffer BJ, et al. Marked in vivo donor regulatory T cell expansion via interleukin-2 and TL1A-Ig stimulation ameliorates graft-versus-host disease but preserves graft-versus-leukemia in recipients after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2017;23(5):757–66.
Theil A, Wilhelm C, Kuhn M, Petzold A, Tuve S, Oelschlägel U, et al. T cell receptor repertoires after adoptive transfer of expanded allogeneic regulatory T cells. Clin Exp Immunol. 2017;187(2):316–24.
Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of CD4+CD25highCD127− regulatory T cellspreserves β-cellfunction in type 1 diabetes in children. Diabetes Care. 2012;35(9):1817–20.
Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. 2015;7(315):315ra189.
Kong Y, Wang YT, Cao XN, Song Y, Chen YH, Sun YQ, et al. Aberrant T cell responses in the bone marrow microenvironment of patients with poor graft function after allogeneic hematopoietic stem cell transplantation. J Transl Med. 2017;15(1):57.
Grygorowicz MA, Biernacka M, Bujko M, Nowak E, Rymkiewicz G, Paszkiewicz-Kozik E, et al. Human regulatory T cells suppress proliferation of B lymphoma cells. Leuk Lymphoma. 2016;57(8):1903–20.
Geissler EK, The ONE. Study compares cell therapy products in organ transplantation: introduction to a review series on suppressive monocyte-derived cells. Transplant Res. 2012;1(1):11. doi:10.1186/2047-1440-1-11.
The ONE study: a unified approach to evaluating cellular immunotherapy in solid organ transplantation. http://www.onestudy.org/. Accessed 01 May 2017.
Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. 2016;64:632–43.
Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127− regulatory T cells prolongs survival of pancreatic islets—results of one year follow-up. Clin Immunol. 2014;153(1):23–30.
ClinicalTrials.gov. https://clinicaltrials.gov/. Accessed 01 May 2017.
Marek-Trzonkowska N, Myśliwiec M, Iwaszkiewicz-Grześ D, Gliwiński M, Derkowska I, Żalińska M et al. Factors affecting long-term efficacy of T regulatory cell-based therapy in type 1 diabetes. J Transl Med. 2016;1;14(1):332.
Trzonkowski P, Bacchetta R, Battaglia M, Berglund D, Bohnenkamp HR, ten Brinke A, et al. Hurdles in therapy with regulatory T cells. Sci Transl Med. 2015;7(304):304ps18.
Desreumaux P, Foussat A, Allez M, Beaugerie L, Hébuterne X, Bouhnik Y, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology. 2012;143(5):1207–17. doi:10.1053/j.gastro.2012.07.116.
Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+ CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167(3):1245–53.
Kleinewietfeld M, Hafler DA. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin Immunol. 2013;25(4):305–12.
Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38(6):1654–63.
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–61.
Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med. 2014;211(1):121–36.
Engelhardt BG, Jagasia M, Savani BN, Bratcher NL, Greer JP, Jiang A, et al. Regulatory T cell expression of CLA or α(4)β(7) and skin or gut acute GVHD outcomes. Bone Marrow Transplant. 2011;46(3):436–42.
Lu J, Meng H, Zhang A, Yang J, Zhang X. Phenotype and function of tissue-resident unconventional Foxp3-expressing CD4(+) regulatory T cells. Cell Immunol. 2015;297(1):53–9. doi:10.1016/j.cellimm.2015.06.005.
Directive 2004/23/EC of The European Parliament and The Council of The European Union of 31 March 2004 on setting standards of quality and safety for donation, procurement, testing, processing, preservation, storage and distribution of human tissues and cells. Official Journal of the European Union 7.4.2004; L 102/48.
The European Parliament and the Council. Regulation (EC) No 1394/2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004. Off J Eur Union. 2007;324:121–37.
European Commission. Report from the Commission to the European Parliament and the Council in accordance with Article 25 of Regulation (EC) No 1394/2007 of the European Parliament and of the Council on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004. European Commission, Brussels, 28 March 2014 COM(2014) 188 final (2014).
Annex 1 ‘Manufacture of Sterile Medicinal Products’ to the DIRECTIVE 2003/94/EClaying down the principles and guidelines of good manufacturing practice in respect of medicinal products for human use and investigational medicinal products for human use. Official Journal of the European Union 14.10.2003; L 262/22.
Dijke IE, Hoeppli RE, Ellis T, Pearcey J, Huang Q, McMurchy AN, et al. Discarded human thymus is a novel source of stable and long-lived therapeutic regulatory T cells. Am J Transplant. 2016;16:58–71.
Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177(12):8338–47.
Golovina TN, Mikheeva T, Brusko TM, Blazar BR, Bluestone JA, Riley JL. Retinoic acid and rapamycin differentially affect and synergistically promote the ex vivo expansion of natural human T regulatory cells. PLoS One. 2011;6:e15868.
Hulspas R, Villa-Komaroff L, Koksal E, Etienne K, Rogers P, Tuttle M, et al. Purification of regulatory T cells with the use of a fully enclosed high-speed microfluidic system. Cytotherapy. 2014;14:00634-3.
Marek N, Bieniaszewska M, Krzystyniak A, Juścińska J, Myśliwska J, Witkowski P, et al. The time is crucial for ex vivo expansion of T regulatory cells for therapy. Cell Transplant. 2011;20(11–12):1747–58.
Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, Mintz MA, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transplant. 2013;13(11):3010–20.
Landwehr-Kenzel S, Issa F, Luu SH, Schmück M, Lei H, Zobel A, et al. Novel GMP-compatible protocol employing an allogeneic B cell bank for clonal expansion of allospecific natural regulatory T cells. Am J Transplant. 2014;14(3):594–606.
MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. 2016;126(4):1413–1424.1.
Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation. 2012;9:112.
Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. 2017;17(4):931–43. doi:10.1111/ajt.14185.
Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with a MHC-specific chimeric antigen receptor. Am J Transplant. 2016. doi:10.1111/ajt.14175.
Marek N, Krzystyniak A, Ergenc I, Cochet O, Misawa R, Wang LJ, et al. Coating human pancreatic islets with CD4(+)CD25(high)CD127(−) regulatory T cells as a novel approach for the local immunoprotection. Ann Surg. 2011;254(3):512–8.
Gołąb K, Kizilel S, Bal T, Hara M, Zielinski M, Grose R, et al. Improved coating of pancreatic islets with regulatory T cells to create local immunosuppression by using the biotin-polyethylene glycol-succinimidylvaleric acid ester molecule. Transplant Proc. 2014;46(6):1967–71.
Acknowledgements
The authors are supported by the National Centre for Research and Development, Poland: Grants STRATEGMED1/233368/1/NCBR/2014 and LIDER/160/L-6/14/NCBR/2015. MG, DIG, and PT are members of COST Action BM1305 A FACTT (www.afactt.eu) supported by COST (European Cooperation in Science and Technology). COST is part of the EU Framework Programme Horizon 2020.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
MG and DIG have no conflicts of interests. PT is a co-inventor of a patent related to the presented content and a stakeholder of the POLTREG venture. The Medical University of Gdańsk received payment for the license to the presented content.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gliwiński, M., Iwaszkiewicz-Grześ, D. & Trzonkowski, P. Cell-Based Therapies with T Regulatory Cells. BioDrugs 31, 335–347 (2017). https://doi.org/10.1007/s40259-017-0228-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-017-0228-3