
Abstract
Background
Clinical indications for ibrutinib reimbursement in Australia should consider the inclusion of patients with chronic lymphocytic leukemia (CLL) harboring prognostically unfavorable TP53/IGHV genomic aberrations. This study assessed the cost effectiveness of five first-line treatment strategies in CLL for young (aged ≤ 65 years), fit patients without significant comorbidities: (1) no testing (fludarabine, cyclophosphamide and rituximab [FCR] for all), (2) test for del(17p) only, (3) test for TP53 gene mutation status, (4) test for TP53 and IGHV gene mutation status and (5) no testing (ibrutinib for all).
Method
A decision analytic model (decision tree and partitioned survival model) was developed from the Australian healthcare system perspective with a lifetime horizon. Comparative treatment effects were estimated from indirect treatment comparisons and survival analysis using several studies. Costs, utility and adverse events were derived from public literature sources. Deterministic and probabilistic sensitivity analyses explored the impact of modeling uncertainties on outcomes.
Results
Strategy 1 was associated with 5.69 quality-adjusted life-years (QALYs) and cost 458,836 Australian dollars (AUD). All other strategies had greater effectiveness but were more expensive than Strategy 1. At the willingness-to-pay (WTP) threshold of 100,000 AUD per QALY gained, Strategy 1 was most cost effective with an estimated probability of 68.8%. Strategy 4 was cost effective between thresholds 155,000–432,300 AUD per QALY gained, and Strategy 5 >432,300 AUD per QALY gained.
Conclusion
Population targeting using mutation testing for TP53 and IGHV when performed with del(17p) testing specifically in the context of frontline ibrutinib choice does not make a cost-ineffective treatment into a cost-effective treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
A health economic model was developed to project clinical and economic outcomes of TP53 and IGHV gene mutation testing for the treatment selection of chronic lymphocytic leukemia (CLL) for young (aged ≤65 years), fit patients without significant comorbidities. |
Testing for TP53 and IGHV gene mutation status improved health outcomes for patients with CLL through improved treatment targeting. |
Testing for TP53 and IGHV gene mutation status could be cost effective at a WTP threshold of 155,000 AUD per QALY gained or if per-cycle treatment cost of ibrutinib based on list prices was reduced by 41.8%. |
1 Introduction
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia in Western countries [1]. Global burden from the disease has increased over the past 30 years and in 2019 there were an estimated 103,467 new cases of CLL with 44,613 CLL-related deaths reported [1]. Clinical course of CLL varies extensively between patients; some are asymptomatic and may not need therapy, while others experience rapidly progressive disease and require early intervention. Conventional systemic multi-agent chemoimmunotherapy (CIT) such as the combination of fludarabine, cyclophosphamide and rituximab (FCR) have historically been the standard first-line treatment for CLL in patients with disease requiring treatment [2].
Deletions in chromosome 17p (del(17p)) and/or mutations of the tumor suppressor p53 (TP53) gene (herein collectively referred to as TP53 aberrations) and unmutated immunoglobulin heavy chain (IGHV-U) are strong prognostic factors associated with inferior response rates, early progression and development of refractory disease following CIT [3,4,5,6,7,8]. The TP53 aberrations and/or IGHV-U confer a high-risk CLL disease, and the clinical predictive value for these biomarkers have been recognized by the International Workshop on Chronic Lymphocytic Leukemia (iwCLL) 2018 guidelines and the European Society for Medical Oncology (ESMO) 2021 guidelines [9, 10]. In these guidelines, testing of TP53 and IGHV mutational status is recommended before treatment to establish the presence of high-risk CLL, which, in turn, will inform clinical treatment of the patients using novel agents beyond CIT.
Ibrutinib was among the first novel agents for CLL to be developed and revolutionized treatment options for patients with high-risk CLL. On March 2016, the US Food and Drug Administration (FDA) approved ibrutinib, an oral, once-daily Bruton’s tyrosine kinase (BTK) inhibitor, for first-line CLL treatment [11]. The pivotal phase III RESONATE-2 study demonstrated a survival benefit for single-agent ibrutinib compared with chlorambucil in previously untreated patients with CLL, with prolonged progression-free survival (PFS) (hazard ratio (HR) 0.16; 95% confidence interval [CI] 0.09–0.28; p < 0.001) and overall survival (OS) (HR 0.16; 95% CI 0.05–0.56; p = 0.001) [12]. Importantly, subgroup analyses from the long-term follow-up data of the RESONATE-2 study support superior survival with ibrutinib for patients with the TP53 aberrations and IGHV-U [13, 14]. Subsequent clinical trials have also shown a significant survival advantage for ibrutinib-based regimens compared with CIT in previously untreated patients, particularly in high-risk CLL [15,16,17]. Furthermore, the ECOG-ACRIN E1912 trial, which compared ibrutinib with rituximab to FCR, showed a reduced rate of grade ≥ 3 treatment-related adverse events (AEs) in the ibrutinib with rituximab group (73.0%) compared with the FCR group (83.5%) (odds ratio (OR) 0.53; 95% CI 0.32–0.88; p = 0.0097) [16].
Since ibrutinib, other second-generation BTK agents (e.g., acalabrutinib with or without obinutuzumab) and oral B-cell lymphoma-2 (BCL2) inhibitors (e.g., venetoclax with obinutuzumab) have also been approved for previously untreated CLL. More treatment choice is afforded where patient preferences, concomitant medications and comorbidities may influence decisions between these targeted therapies until there is further evidence to support an optimal first-line treatment regimen beyond CIT. In light of these considerations, for the CLL patient population of young (aged ≤ 65 years), fit patients without significant comorbidities who have a high-risk molecular profile, ibrutinib is recommended as the preferred choice of first-line therapy (unless contraindicated) for many countries owing to extensive follow-up data for this patient group compared with other novel-based agents [18,19,20]. Conversely, in the same CLL patient population with a low-risk molecular profile (TP53 wildtype and mutated immunoglobulin heavy chain (IGHV-M)), FCR is still an available treatment option.
Despite the current clinical evidence, the definition of who is eligible for treatment reimbursement of first-line ibrutinib is inconsistent with its clinical use. For instance, the Pharmaceutical Benefits Advisory Committee (PBAC) in Australia recommended reimbursement of first-line ibrutinib in CLL for patients with del(17p) only (as opposed to both deletions and mutations of TP53) [21]. By contrast, the National Institute for Health and Care Excellence (NICE) in the UK recommended reimbursement for TP53 aberrations [22] whereas the Canadian Agency for Drugs and Technologies in Health (CADTH) recommended reimbursement for those in whom fludarabine-based treatment is inappropriate (which may include TP53 aberrations and/or IGHV-U depending on the country’s province) [23]. Not only are the funding policies different from the clinical guidelines, but as highlighted, the policies vary depending on the health system and can lead to a subgroup of high-risk patients without access to effective treatment.
Reimbursement policies concerning ibrutinib in Australia require revision to ensure that patients with CLL have appropriate access to the most effective available treatment. Therefore, in accordance with the clinical guidelines, testing for TP53 aberrations (including deletions and mutations) alongside IGHV mutations should be considered before making treatment decisions for patients with CLL. Health economic evidence is a considerable component of reimbursement decisions. In this respect, cost-effectiveness studies to assess the health benefits and costs of molecular testing for TP53 and IGHV to guide first-line treatment decisions with ibrutinib in CLL compared with current standard of care are useful for decision makers. However, these studies are limited. To address this evidence gap, the objective of this study was to evaluate the cost utility of several testing strategies for the molecular characterization of TP53 and IGHV mutations for the frontline treatment of either ibrutinib or CIT in young, fit patients with CLL from the Australian healthcare system perspective.
2 Methods
2.1 Model Overview
The patient population was a hypothetical cohort of young, fit patients with active CLL disease. The perspective was the Australian healthcare system over a lifetime horizon. Both health and economic outcomes were discounted at an annual rate of 5% as per Australian guidelines [24]. The model cycle length was 4 weeks to coincide with a typical CLL treatment cycle. Australia does not have an explicit willingness-to-pay (WTP) threshold, but the analysis assumed a threshold of 100,000 AUD per quality-adjusted life-year (QALY) gained for cancer drugs (two times greater than the commonly used 50,000 AUD per QALY gained) [25]. Outcomes included the incremental cost-effectiveness ratio (ICER) and net monetary benefit (NMB). The ICER was the difference in cost in Australia dollars (AUD) divided by the difference in QALY gained between alternative strategies using standard methods for economic evaluations. The NMB was the total QALY multiplied with the WTP threshold subtracted by the total cost for each strategy. The strategy with the highest NMB was considered the most cost effective. Face validity of the model structure was confirmed through ongoing consultations with two clinical hematologists. These experts were consulted to judge the suitability of the model structure in Australian practice. They were also specifically asked to assess the appropriateness of the identified clinical data sources for populating the model, identify any potential issues of using published clinical trials in the analysis and provide feedback on the sensibility of the model outcomes. The model was developed and analyzed in R Statistical Software version 4.0.2 (R Foundation for Statistical Computing, Vienna, Austria) following a pre-specified health economic analysis plan (available upon request from author).
2.2 Model Strategies and Structure
A decision analytic model was developed to compare five mutually exclusive strategies consisting of a decision tree for molecular testing and first-line treatment selection, as well as a partitioned survival model (PSM) to assess the health economic consequences of these treatment decisions (Fig. 1).

Graphical diagram of the decision analytic model consisting of a (A) decision tree and (B) PSM, and the five strategies. CLL chronic lymphocytic leukemia, FCR fludarabine, cyclophosphamide and rituximab, IGHV immunoglobulin heavy chain, M mutated, PSM partitioned survival model, TP53 tumor suppressor p53, U unmutated
The decision tree reflected the allocation of first-line treatment following each strategy. These strategies were designed to support policy decision makers to consider a range of potential test-treatment scenarios for patients with CLL, which included the following:
-
Strategy 1: no testing (FCR for all). Patients in Strategy 1 all received FCR.
-
Strategy 2: test for del(17p) only. Patients in Strategy 2 were tested for del(17p). Patients with del(17p) received ibrutinib, while patients without deletions received FCR. Moreover, health economic outcomes were modeled separately for a subset of patients without deletions but with underlying TP53 inactivation by mutation (approximately 30–40% of patients with TP53 aberrations) due to known inferior outcomes with FCR [6, 26].
-
Strategy 3: test for TP53 gene mutation status. Patients in Strategy 3 were tested for TP53 aberrations (both del(17p) and TP53 mutations). Patients with TP53 aberrations received ibrutinib, while patients with TP53 wildtype received FCR.
-
Strategy 4: test for TP53 and IGHV gene mutation status. Patients in Strategy 4 were first tested for TP53 aberrations (both del(17p) and TP53 mutations), receiving ibrutinib if TP53 aberrant. For patients with TP53 wildtype, there was a second testing for IGHV mutations, receiving ibrutinib if IGHV-U. Only patients who were TP53 wildtype and IGHV-M received FCR.
-
Strategy 5: no testing (ibrutinib for all). Patients in Strategy 5 all received ibrutinib.
Choice of FCR as the CIT regimen is consistent with the clinical management of young, fit patients in Australia outlined by the Australian PBAC [21]. The prevalence of TP53 aberrations and IGHV-U among treatment-naïve patients for the decision tree was estimated using published literature sources [16, 26].
The PSM estimated the long-term costs and health outcomes of different treatment sequences. The PSM consisted of three mutually exclusive health states common in oncology: progression free (PF), progressive disease (PD) and death (Fig. 1). Two treatment sequences were modeled: (1) first-line ibrutinib in PF followed by venetoclax in combination with rituximab (VR) in PD (PF ibrutinib → PD VR) and (2) first-line FCR in PF followed by ibrutinib in PD (PF FCR → PD ibrutinib). Outcomes for treatment sequences were modeled conditional on the molecular profile of the corresponding patient population. Ibrutinib was an ongoing therapy until PD, whereas FCR was a fixed duration of six cycles and VR was a fixed duration of 26 cycles including an initial titration cycle. Given the different costs and utility associated with treatment regimens as either ongoing or fixed therapy, two additional substates designated as ‘with treatment’ and ‘without treatment’ were modeled for PF and PD. Patients enter the PSM in the PF state beginning ‘with treatment’ and may remain until ‘without treatment’, or transition to either the PD or death state over time but cannot revert to a former state. The proportion of patients within each health state over time (i.e., state membership) was calculated by modeling the relative survival of each biomarker-defined treatment group included in the model using an area under the curve approach [27]. The costs and health outcomes accrued over time were calculated based on the proportion of patients in each model state. Due to the limited capacity to monitor the number of treatment cycles that a patient may receive in the PD state, patients entering the PD state from the PF state on ibrutinib (i.e., PF ibrutinib → PD VR) incurred a once-off total average expected discounted cost and QALY for VR estimated based on the proportion of patients completing full-course VR treatment reported in the MURANO trial [28]. More specifically, tracking treatment cycles for patients transitioning from PD ‘with treatment’ to PD ‘without treatment’ when receiving VR treatment was particularly challenging in cohort models. To address this challenge, we estimated a once-off total average expected discounted cost for VR by aggregating costs across the 26 model cycles (equivalent to 2 years of treatment) considering the proportion of patients with VR treatment across each cycle, assuming 67% of patients completed a full course of treatment. Additionally, we aggregated the discounted utility and disutility of VR treatment over the 26 model cycles, accounting for the proportion of patients receiving VR treatment in each cycle to estimate the once-off total average expected QALY for VR. Further details outlining the transition of patients are outlined in Electronic Supplemental Materials 2 (ESM 2).
2.3 Clinical Data
The PFS and OS curves for each biomarker-defined treatment group were derived from secondary analyses of published literature. Further details are provided in ESM 1 and ESM 2.
Firstly, in the absence of direct head-to-head comparisons of FCR and ibrutinib in patients with either low-risk or high-risk molecular biomarkers, a review of PFS and OS data from phase III or II trials was conducted to estimate the relative HRs with 95% confidence intervals (95% CI) through an indirect treatment comparison (ITC) using the frequentist approach of a fixed effect model. Trials were included if the trial population comprised previously untreated patients with CLL and reported PFS and OS outcomes. From this, several different treatment groups were considered but assumptions were made to enable a connected evidence network (Supplementary Table S1 in the ESM). The evidence network for the ITC of first-line treatment in patients with CLL is presented in Fig. 2. The impact of these assumptions on model outcomes was explored through sensitivity analyses. For the model, FCR in patients with TP53 wildtype irrespective of IGHV mutational status was selected as the reference comparator. This choice reflected the proposed clinical management of first-line CLL treatment by the Australian PBAC [21]. The analyses were conducted using the netmeta package in R [29]. The results of the ITC are presented in Table 1.

Evidence network for the indirect treatment comparison of first-line treatment in patients with chronic lymphocytic leukemia. Each link between modeled biomarker-defined treatment groups represents a direct comparison from a clinical trial study. FCR fludarabine, cyclophosphamide and rituximab, IGHV immunoglobulin heavy chain, M mutated, TP53 tumor suppressor p53, U unmutated
Secondly, PFS and OS outcomes for the reference comparator were extrapolated and validated using published clinical trial data from the ECOG-ACRIN E1912 study and CLL8 study, respectively. The ECOG-ACRIN E1912 study was a phase III trial which enrolled 529 previously untreated patients with CLL between March 2014 and June 2016 to receive either a combination of ibrutinib and rituximab or FCR, with a median follow-up of 33.6 months [16]. The FCR trial arm of the study provided the best available evidence to model the reference comparator since patients with del(17p) were excluded from trial participation. Consequently, it was assumed that this would be generalizable to patients with TP53 wildtype receiving FCR. Although subsequent publications from the ECOG-ACRIN E1912 study reported four patients with TP53 mutations out of 134 patients with available sequencing results in the FCR trial arm (3.0%), relevant survival outcomes for secondary survival analysis of FCR in a patient cohort of both the del(17p) and TP53 mutations in literature is limited [30]. Conversely, the ECOG-ACRIN E1912 study published all relevant inputs (i.e., Kaplan–Meier curves and risk table) to replicate survival data.
The published Kaplan–Meier PFS and OS curves of the FCR trial arm in the ECOG-ACRIN E1912 study were digitized using a validated graphical digitizer (WebPlotDigitizer version 4.5; Ankit Rohatgi, CA, USA). Next, individual patient time-to-event data were reconstructed using an algorithm developed by Guyot et al. [31]. The following parametric distributions were considered for extrapolating PFS and OS: exponential, Gompertz, log-logistic, log-normal and Weibull (in its proportional hazard parameterization) [32]. Candidate distributions were visually assessed for extrapolation plausibility, after which the final selection between plausible distributions was made on likelihood-based goodness-of-fit measures (i.e., Akaike Information Criterion and Bayesian Information Criterion) (Supplemental Table 3 and Supplemental Figs. 1, 2, see ESM). Additional internal validation from a comparison of yearly survival probabilities between the selected distribution and ECOG-ACRIN E1912 trial data, and external validation from the CLL8 study of a similar treatment group, were examined (Supplemental Table 4, see ESM). Although the Gompertz distribution was the best candidate for the PFS data, no distribution appropriately modeled the OS data. To address this issue, PFS and OS were modeled jointly using one Gompertz distribution, with a shared shape parameter but separate rate parameters. Fig. 3 presents the extrapolated survival curve of the reference comparator.

Modeled PFS and OS curves of the ECOG-ACRIN E1912 study for chronic lymphocytic leukemia patients with TP53 aberrations irrespective of IGHV mutational status receiving first-line FCR therapy, with data from the CLL8 trial presented as external validation. FCR fludarabine, cyclophosphamide and rituximab, IGHV immunoglobulin heavy chain, OS overall survival, PFS progression-free survival, TP53 tumor suppressor p53
Thirdly, the PFS and OS survival functions for modeled treatments were estimated by multiplying the HRs from the ITC analysis to the hazard function of the reference comparator extrapolated from the ECOG-ACRIN E1912 study, assuming proportional hazards. To ensure that OS of patients in the model could not exceed that of the general population, it was assumed that the treatment effect duration in the PF state was 10 years. After given time t > 10 years, the hazard functions were elicited as the maximum value of either the hazard rate of the reference comparator or the hazard rate of the general population mortality derived from the Human Mortality Database [33]. Survival models for each treatment group are presented in Figs S5 and S6 (see ESM).
2.4 Costs
Details of the unit cost inputs are provided in Table 2, and the full itemized cost calculations are provided in ESM 3. All costs were indexed to 2021 AUD. The cost of testing for del(17p) using fluorescence in situ hybridization (FISH) was sourced from the Australian Medical Benefits Schedule (MBS) [34] whereas the cost of testing for TP53 mutations using targeted amplicon sequencing and cost of testing for IGHV mutations using Lymphotrack IGHV Leader Somatic Hypermutation Assay (Invivoscribe) were sourced from the Molecular Haematology Laboratory at the Peter MacCallum Cancer Centre, where testing for TP53 and IGHV mutations are currently performed. Costs of drug therapy were estimated using prices from the Australian Pharmaceutical Benefits Scheme (PBS) [35] and dosing information from national treatment guidelines developed by eviQ and clinical inputs, assuming an average body surface area of 1.8 m2 for drug dose calculations [36]. Relevant data sources from the MBS [34] and the Australian Refined Diagnosis Related Group (AR-DRG) [37] were used to estimate the costs of patient administration of chemotherapy. The costs of cancer management and associated laboratory investigations were also considered with the frequency of routine review derived from clinical input and prices from the MBS [34] and PBS [35]. The cost of AE hospitalization was derived from the AR-DRG data [37]. All patients who transitioned to the death state incurred a once-off cost for terminal care [38].
2.5 Utilities
Utility scores for health states were obtained from Kosmas et al. [39], who used a time trade-off methodology to elicit utility values for the different health states associated with the disease and treatment of CLL amongst members of the UK general population. Utility scores were allocated to each health state in the model, with considerations to whether patients are actively receiving treatment and route of treatment administration (Table 2). Disutility associated with AEs were sourced from literature [22, 40].
2.6 Adverse Events
Major grade 3/4 AEs that are likely to result in hospitalization as reported in the ECOG-ACRIN E1912 trial [16] for ibrutinib and FCR, and in the MURANO trial [41] for VR, were considered in the model. A one-off cost and disutility of AE was weighted according to AE incidence rates reported in the trial and the average length of hospitalization for AEs was estimated based on the consensus of two clinical hematologists.
2.7 Model Analyses
Deterministic sensitivity analyses explored the impact of each model parameter on the NMB model output, which is less ambiguous than interpretation of unilateral changes to ICER. Parameters were varied independently, with HRs and health state utilities tested at the upper and lower of their respective 95% CIs and other parameters using a range of ± 20%. Further, a minimum value of 3% and maximum value of 7% were imposed for the discount rate. A probabilistic analysis quantified the joint impact of parameter uncertainty on the uncertainty in the model outcomes. This was achieved by randomly sampling model parameter values from specified probability distributions for each parameter to generate 5000 cost and effectiveness estimates for each molecular diagnostic testing strategy. The decision to use 5000 iterations in the probabilistic analysis was supported by observing the stability of the mean model outcomes (e.g., ICER and NMB) and CI as a function of the number of runs in the analysis. To incorporate uncertainty in the parameters, standard parametric distributions were used, except for costs (Table 2). The cost estimates were fixed in the analysis because there was limited uncertainty associated with these parameters. Additional sensitivity analyses assessed the uncertainty of ITC assumptions (ESM 1 and ESM 4). Subgroup analyses were also performed to explore the health economic impact of ibrutinib versus FCR in the setting of two high-risk treatment groups based on Strategy 4 in Fig. 1 (patients with TP53 aberrations and IGHV irrespective and patients with TP53 wildtype and IGHV-U). The purpose of this was to assess the cost effectiveness if these patients were instead to receive FCR.
3 Results
The cost-effectiveness results comparing the lifetime average discounted costs and QALYs of each strategy for the hypothetical cohort of patients with CLL based on the probabilistic analysis are described in Table 3 and the outcomes illustrated on a cost-effectiveness plane in Fig. 4. The efficiency frontier in Fig. 3 demonstrates the ICER between alternative strategies. Strategy 1 (no testing (FCR for all)) had the lowest cost per patient (458,836 AUD [95% CI 236,792–786,314]) and was the least effective (5.69 QALY [95% CI 2.85–10.22]). All other strategies were more effective but more expensive than Strategy 1. The ICERs of Strategy 2 (test for del(17p) only), Strategy 3 (test for TP53 gene mutation status), Strategy 4 (test for TP53 and IGHV gene mutation status) and Strategy 5 (no testing (ibrutinib for all)) were 138,698 AUD, 140,013 AUD, 164,462 AUD and 1,124,983 AUD per QALY gained, respectively, when compared with the next most effective strategy. Despite the health benefits, none of the ICERs were below the 100,000 AUD per QALY gained threshold. Additionally, the 95% confidence ellipsis for the strategies revealed considerable uncertainty of the mean costs and QALYs for each strategy. Strategy 1 had the highest NMB at 110,454 AUD.

Incremental cost-effectiveness plane from the probabilistic sensitivity analysis. AUD Australian dollars, FCR fludarabine, cyclophosphamide and rituximab, IGHV immunoglobulin heavy chain, QALY quality-adjusted life-years, TP53 tumor suppressor p53
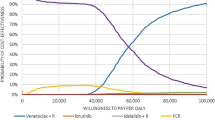
The cost-effectiveness acceptability curve summarizing the uncertainty in the model outcomes and probability of cost effectiveness is depicted in Fig. 5. The estimated probability of cost effectiveness for Strategy 1 was 68.8% at the 100,000 AUD per QALY gained threshold. From the cost-effectiveness acceptability frontier in Fig. 4, Strategy 4 was the most cost-effective strategy between a WTP threshold of 155,000 AUD and 432,300 AUD per QALY gained whereas Strategy 5 was the most cost-effective strategy at a WTP threshold > 432,300 AUD per QALY gained.

Cost-effectiveness acceptability curve showing the probability for each strategy of being cost effective as a function of the WTP per QALY gained. Thick border line corresponds to the cost-efficient frontier. AUD Australian dollars, FCR fludarabine, cyclophosphamide and rituximab, IGHV immunoglobulin heavy chain, QALY quality-adjusted life-years, TP53 tumor suppressor p53, WTP willingness-to-pay
The tornado diagram illustrating the five single model parameters that had the greatest impact on model outcomes for each strategy is shown in Fig. 6. The parameters with substantial influence for each strategy included the per-cycle treatment cost of ibrutinib and health state utility scores. In these instances, a lower treatment cost for ibrutinib per cycle or higher utility score was associated with an increase in NMB compared with the base-case outputs. Other model parameters with varying influence on the strategies included per-cycle treatment cost of FCR and VR, relative treatment survival and discount rates.

Tornado diagram of the deterministic sensitivity analysis for the five parameters with the greatest impact on NMB across each strategy. AUD Australian dollars, FCR fludarabine, cyclophosphamide and rituximab, HR hazard ratio, IGHV immunoglobulin heavy chain, NMB net monetary benefit, PD progressive disease, PF progression free, PFS progression-free survival, QALY quality-adjusted life-years, TP53 tumor suppressor p53, U unmutated, VR venetoclax and rituximab, WTP willingness-to-pay
The additional sensitivity, threshold and subgroup analyses are presented in ESM 4. The additional sensitivity analyses assessed the impact of ITC analysis assumptions, which revealed that there was minimal effect on overall model conclusions (i.e., Strategy 1 as the preferred intervention). The threshold analysis demonstrated that if the cost of ibrutinib was < 4800 AUD per cycle (41.8% decrease from base-case value), the ICERs for Strategy 2, Strategy 3 and Strategy 4 would be below the 100,000 AUD per QALY gained threshold. Ibrutinib is unlikely to be cost effective compared with FCR for patients with TP53 aberrations and IGHV irrespective and patients with TP53 wildtype and IGHV-U.
4 Discussion
This study assessed the health economic impact of targeting first-line treatment with FCR or ibrutinib in young, fit patients with CLL. The results demonstrate that biomarker testing for TP53 and IGHV improved patient’s health outcomes but incurred a relatively higher cost than providing FCR treatment for all patients regardless of risk group. At the WTP threshold of 100,000 AUD per QALY gained, this strategy was unlikely to be cost effective, but could be at a higher threshold value (e.g., 155,000 AUD per QALY gained). Although ibrutinib treatment for all patients was the most effective strategy, a much higher WTP threshold (e.g., > 432,300 AUD per QALY gained) will need to be adopted for it to substantiate value for money. Several factors were identified that could improve the cost effectiveness of TP53 and IGHV, including health state utility scores and relative treatment survival, but notably the per-cycle treatment cost of ibrutinib. The cost of ibrutinib was based on list prices from Australia’s national health insurance scheme, but if the cost of ibrutinib was to decrease from 8244 AUD to 4800 AUD per cycle (41.8%), the ICER of molecular testing with TP53 and IGHV would be < 100,000 AUD per QALY gained.
To ensure better access to effective treatments for high-risk patients, it is essential for these health technology assessment (HTA) organizations (e.g., PBAC, NICE and CADTH) to reassess and harmonize their reimbursement policies based on the clinical evidence supporting the inclusion of TP53 and IGHV testing. It is evident that incorporating biomarker testing for TP53 and IGHV improves patient health outcomes but at a higher cost compared with a uniform approach of providing FCR treatment for all patients, regardless of risk group. At the same time, testing for TP53 and IGHV is more cost effective than administering ibrutinib for all patients. Perhaps the most tangible implication for HTA and industry is that reducing the cost of ibrutinib could potentially enhance the cost effectiveness of TP53 and IGHV. It is important to note that our analysis was conducted using the list price of CLL treatment, and it is possible that the true cost of treatment including ibrutinib may be lower.
The cost effectiveness of upfront ibrutinib (without molecular testing) has been previously investigated amongst several treatment regimens and is unlikely cost effective in CLL despite health benefits gained [42,43,44,45,46]. There are important differences between these studies that limit their direct comparability, such as the choice of comparator, the patient population (i.e., older patients or patients with comorbidities), treatment sequences and the health system under evaluation. Nevertheless, relatively fewer cost-effectiveness studies have assessed molecular diagnostic tools to match high-risk patients (specifically TP53 aberrations) with targeted treatment. A Markov model published by Buchanan et al. [47] showed that genomic testing to stratify patients with CLL in receiving first-line ibrutinib improved health outcomes, but that this was not cost effective for the UK healthcare system. Of note, similar to our study, the cost of ibrutinib was also identified as a factor that may lead to favorable cost effectiveness of targeted CLL therapies if the cost was lowered. Our study, to our knowledge, is the first to consider IGHV gene mutations in view of recent CLL guidelines and although testing for TP53 and IGHV did not necessarily translate to ibrutinib being cost effective, it did at least make such therapies more economical. Specifically, the incremental benefit from providing ibrutinib for all patients compared with the TP53 and IGHV testing strategy as the next best alternative was marginal (0.11 QALY) but the additional cost to deliver treatment was substantial (119,356 AUD). By reason, a greater investment or a higher WTP threshold will need to be adopted to make the uniform ibrutinib treatment worthwhile. This finding could be of particular importance for future studies assessing the health economic impact of second-generation BTK agents, BCL2 inhibitors and other newer therapies as the evidence continues to emerge for young, fit patients with CLL [42, 46].
Still, the question of how to standardize patient access to molecular tests and ibrutinib treatment to ensure clinical evidence for a CLL treatment algorithm is reflected in policy remains. While the role of FCR diminishes to a selected subset of low-risk patients, there are not many alternative therapies in the Australian frontline setting that are also reimbursed [48]. There needs to be a reimbursement pathway driven by the clinical unmet need for effective treatment in high-risk patients, particularly IGHV-U. Further, this study emphasizes a cost reduction for ibrutinib and motivates a price negotiation between funders and manufacturers to assist with achieving cost effectiveness. Alternatively, an outcomes-based payment scheme could be considered, which is a cost-sharing arrangement that establishes the price of drug reimbursement based on the delivery of pre-agreed outcomes in real-world clinical practice [49].
This study has certain limitations. Firstly, there was a lack of direct clinical evidence to inform the comparison of biomarker-defined treatment groups in the model for the ITC analysis, and assumptions were made to form a connected evidence network. This study utilized the best available public literature sources to inform a range of plausible clinical parameters in the model, but further effectiveness evidence may be required to improve the long-term clinical outcome estimates. Yet, such data will remain scarce unless clinical trials are designed to directly investigate the clinical benefit of treatment for predictive groups categorized by TP53 wildtype/aberrations and IGHV-U/IGHV-M. These clinical trials are costly to conduct, and it is unlikely future patients with TP53 aberrations and/or IGHV-U enrolled in clinical trials will receive FCR with the shift away from this treatment for these patients [50]. Nonetheless, the trials included in the ITC analysis were comparable based on the study design, patient population and outcome measures (Table S2 in the ESM). Assumptions for the ITC analysis were discussed and validated by clinical experts, with justifications provided in Table S1 (see ESM), and their impact was explored in sensitivity analyses. Secondly, uncertainty in extrapolating the long-term OS in the PSM owing to immature long-term follow-up data for the reference comparator in the model could influence outcomes of the cost-effectiveness analysis. However, several efforts ensured the choice of projecting OS was plausible from the perspective of internal and external validation. Thirdly, utility and disutility scores were from a health state utility study and other public sources in a UK population, which may be different to an Australian population and could limit the generalizability of study findings. Fourthly, only the major grade 3/4 AEs leading to hospitalization were included where all other AEs were excluded from the evaluation. However, findings of the deterministic analysis suggest that the cost of AEs may not majorly contribute to the cost effectiveness of evaluated strategies. Lastly, upon completion of study conceptualization and analysis, venetoclax in combination with obinutuzumab is now reimbursed for CLL patients in Australia but was not considered in the model [51]. Ibrutinib may still be the preferred treatment option over venetoclax with obinutuzumab depending on the comorbidities profile [52]. Nevertheless, findings of our study are still relevant from a health policy perspective to highlight the potential to enhance eligibility of the current use of ibrutinib through the adoption of a risk-based approach, which includes reducing the cost of ibrutinib treatment.
References
Yao Y, Lin X, Li F, Jin J, Wang H. The global burden and attributable risk factors of chronic lymphocytic leukemia in 204 countries and territories from 1990 to 2019: analysis based on the Global Burden of Disease study 2019. Biomed Eng Online. 2022;21:4. https://doi.org/10.1186/s12938-021-00973-6.
Smolej L, Vodárek P, Écsiová D, Šimkovič M. Chemoimmunotherapy in the first-line treatment of chronic lymphocytic leukaemia: dead yet, or alive and kicking? Cancers (Basel). 2021;13:3134. https://doi.org/10.3390/cancers13133134.
Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376:1164–74. https://doi.org/10.1016/S0140-6736(10)61381-5.
Fischer K, Bahlo J, Fink AM, Goede V, Herling CD, Cramer P, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127:208–15. https://doi.org/10.1182/blood-2015-06-651125.
Crombie J, Davids MS. IGHV mutational status testing in chronic lymphocytic leukemia. Am J Hematol. 2017;92:1393–7. https://doi.org/10.1002/ajh.24808.
Campo E, Cymbalista F, Ghia P, Jäger U, Pospisilova S, Rosenquist R, et al. TP53 aberrations in chronic lymphocytic leukemia: an overview of the clinical implications of improved diagnostics. Haematologica. 2018;103:1956–68. https://doi.org/10.3324/haematol.2018.187583.
Giudice ID, Foà R. Another step forward in the 20-year history of IGHV mutations in chronic lymphocytic leukemia. Haematologica. 2019;104:219–21. https://doi.org/10.3324/haematol.2018.207399.
Moia R, Boggione P, Mahmoud AM, Kodipad AA, Adhinaveni R, Sagiraju S, et al. Targeting p53 in chronic lymphocytic leukemia. Expert Opin Ther Targets. 2020;24:1239–50. https://doi.org/10.1080/14728222.2020.1832465.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745–60. https://doi.org/10.1182/blood-2017-09-806398.
Eichhorst B, Robak T, Montserrat E, Ghia P, Niemann CU, Kater AP, et al. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32:23–33. https://doi.org/10.1016/j.annonc.2020.09.019.
Abbvie. IMBRUVICA® (ibrutinib) approved by U.S. FDA for the first-line treatment of chronic lymphocytic leukemia. 2016. https://news.abbvie.com/news/imbruvica-ibrutinib-approved-by-us-fda-for-first-line-treatment-chronic-lymphocytic-leukemia.htm#:~:text=NORTH%20CHICAGO%2C%20Ill.%2C%20March,chronic%20lymphocytic%20leukemia%20(CLL). Accessed 9 Jun 2022.
Burger J, Tedeschi A, Barr P, Robak T, Owen C, Ghia P, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373:2425–38. https://doi.org/10.1056/NEJMoa1509388.
Barr PM, Robak T, Owen C, Tedeschi A, Bairey O, Bartlett NL, et al. Sustained efficacy and detailed clinical follow-up of first-line ibrutinib treatment in older patients with chronic lymphocytic leukemia: extended phase 3 results from RESONATE-2. Haematologica. 2018;103:1502–10. https://doi.org/10.3324/haematol.2018.192328.
Burger JA, Barr PM, Robak T, Owen C, Ghia P, Tedeschi A, et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia. 2020;34:787–98. https://doi.org/10.1038/s41375-019-0602-x.
Moreno C, Greil R, Demirkan F, Tedeschi A, Anz B, Larratt L, et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20:43–56. https://doi.org/10.1016/S1470-2045(18)30788-5.
Shanafelt T, Wang X, Kay N, Hanson C, O’Brien S, Barrientos J, et al. Ibrutinib-rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381:432–43. https://doi.org/10.1056/NEJMoa1817073.
Woyach JA, Ruppert AS, Heerema NA, Zhao W, Booth AM, Ding W, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379:2517–28. https://doi.org/10.1056/NEJMoa1812836.
Raa DGT, van der Straten L, van Gelder M, Kersting S, Levin MD, Mous R, et al. Diagnosis, treatment and supportive management of chronic lymphocytic leukemia: recommendations of the Dutch HOVON CLL working group. Leuk Lymphoma. 2022;63:2276–89. https://doi.org/10.1080/10428194.2022.2084731.
Kojima K, Burger JA. Treatment algorithm for Japanese patients with chronic lymphocytic leukemia in the era of novel targeted therapies. J Clin Exp Hematop. 2020;60:130–7. https://doi.org/10.3960/jslrt.20002.
Costello J, Kang M, Banerji V. Frontline treatment of the young, fit patient with CLL: a Canadian perspective. Curr Oncol. 2021;28:3825–35. https://doi.org/10.3390/curroncol28050326.
Pharmaceutical Benefit Schedule. Ibrutinib: capsule 140 mg; Imbruvica®. 2020. https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2019-11/ibrutinib-capsule-140-mg-imbruvica. Accessed 9 Jun 2022.
National Institute for Health and Care Excellence. Ibrutinib for previously treated chronic lymphocytic leukaemia and untreated chronic lymphocytic leukaemia with 17p deletion or TP53 mutation. 2017. https://www.nice.org.uk/guidance/ta429. Accessed 9 Jun 2022.
Canadian Agency for Drugs and Technologies in Health. Imbruvica for chronic lymphocytic leukemia (previously untreated)—details. 2016. https://www.cadth.ca/imbruvica-chronic-lymphocytic-leukemia-previously-untreated-details. Accessed 9 Jun 2022.
Australian Government Department of Health. Guidelines for preparing assessments for the Medical Services Advisory Committee. Canberra: Australian Government Department of Health; 2021.
Bae Y, Mullins CD. Do value thresholds for oncology drugs differ from nononcology drugs? J Manag Care Spec Pharm. 2014;20:1086–92. https://doi.org/10.18553/jmcp.2014.20.11.1086.
Gonzalez D, Martinez P, Wade R, Hockley S, Oscier D, Matutes E, et al. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol. 2011;29:2223–9. https://doi.org/10.1200/jco.2010.32.0838.
Woods BS, Sideris E, Palmer S, Latimer N, Soares M. Partitioned survival and state transition models for healthcare decision making in oncology: where are we now? Value Health. 2020;23:1613–21. https://doi.org/10.1016/j.jval.2020.08.2094.
Kater AP, Seymour JF, Hillmen P, Eichhorst B, Langerak AW, Owen C, et al. Fixed duration of venetoclax-rituximab in relapsed/refractory chronic lymphocytic leukemia eradicates minimal residual disease and prolongs survival: post-treatment follow-up of the MURANO phase III study. J Clin Oncol. 2019;37:269–77. https://doi.org/10.1200/jco.18.01580.
Rücker G, Krahn U, König J, Efthimiou O, Davies A, Papakonstantinou T, et al. netmeta: network meta-analysis using frequentist methods. 2022. https://cran.r-project.org/web/packages/netmeta/netmeta.pdf. Accessed 9 Jun 2022.
Shanafelt TD, Wang XV, Hanson CA, Paietta EM, O’Brien S, Barrientos J, et al. Long-term outcomes for ibrutinib–rituximab and chemoimmunotherapy in CLL: updated results of the E1912 trial. Blood. 2022;140:112–20. https://doi.org/10.1182/blood.2021014960.
Guyot P, Ades AE, Ouwens MJNM, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol. 2012;12:9. https://doi.org/10.1186/1471-2288-12-9.
Jackson C, Metcalfe P, Amdahl J, Warkentin M, Kunzmann K. Flexible parametric survival and multi-state models. 2021. https://cran.r-project.org/web/packages/flexsurv/flexsurv.pdf. Accessed 9 Jun 2022.
University of California, Berkeley and Max Planck Institute for Demographic Research. Human Mortality Database. 2022. www.mortality.org. Accessed 9 Jun 2022.
Medicare Benefit Schedule. MBS online downloads. 2022. http://www.mbsonline.gov.au/internet/mbsonline/publishing.nsf/Content/downloads. Accessed 9 June 2022.
Pharmaceutical Benefit Schedule. PBS publications archive. 2022. https://www.pbs.gov.au/info/publication/schedule/archive. Accessed 9 Jun 2022.
Cancer Institute NSW. Cancer treatments online. 2022. https://www.eviq.org.au/. Accessed 9 Jun 2022.
Independent Hospital Pricing Authority. National Hospital Cost Data Collection, AR-DRG cost weight tables V8.0x, round 21 (financial year 2016–17). 2019. https://www.ihpa.gov.au/publications/national-hospital-cost-data-collection-ar-drg-cost-weight-tables-v80x-round-21. Accessed 9 Jun 2022.
Lafeuille M-H, Vekeman F, Wang S-T, Kerrigan M, Menditto L, Duh MS. Lifetime costs to Medicare of providing care to patients with chronic lymphocytic leukemia. Leuk Lymphoma. 2012;53:1146–54. https://doi.org/10.3109/10428194.2011.643405.
Kosmas CE, Shingler SL, Samanta K, Wiesner C, Moss PAH, Becker U, et al. Health state utilities for chronic lymphocytic leukemia: importance of prolonging progression-free survival. Leuk Lymphoma. 2015;56:1320–6. https://doi.org/10.3109/10428194.2014.961012.
Tolley K, Goad C, Yi Y, Maroudas P, Haiderali A, Thompson G. Utility elicitation study in the UK general public for late-stage chronic lymphocytic leukaemia. Eur J Health Econ. 2013;14:749–59. https://doi.org/10.1007/s10198-012-0419-2.
Seymour JF, Kipps TJ, Eichhorst B, Hillmen P, D’Rozario J, Assouline S, et al. Venetoclax–rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378:1107–20. https://doi.org/10.1056/NEJMoa1713976.
Chatterjee A, Shapouri S, Manzoor BS, Ravelo A, Sail K, Qendri V, et al. Cost-effectiveness of a 12-month fixed-duration venetoclax treatment in combination with obinutuzumab in first-line, unfit chronic lymphocytic leukemia in the United States. J Manag Care Spec Pharm. 2021;27:1532–44. https://doi.org/10.18553/jmcp.2021.27.11.1532.
Patel KK, Isufi I, Kothari S, Davidoff AJ, Gross CP, Huntington SF. Cost-effectiveness of first-line vs third-line ibrutinib in patients with untreated chronic lymphocytic leukemia. Blood. 2020;136:1946–55. https://doi.org/10.1182/blood.2020004922.
Sinha R, Redekop WK. Cost-effectiveness of ibrutinib compared with obinutuzumab with chlorambucil in untreated chronic lymphocytic leukemia patients with comorbidities in the United Kingdom. Clin Lymphoma Myeloma Leuk. 2018;18:e131–42. https://doi.org/10.1016/j.clml.2017.12.005.
Barnes JI, Divi V, Begaye A, Wong R, Coutre S, Owens DK, et al. Cost-effectiveness of ibrutinib as first-line therapy for chronic lymphocytic leukemia in older adults without deletion 17p. Blood Adv. 2018;2:1946–56. https://doi.org/10.1182/bloodadvances.2017015461.
Alrawashdh N, McBride A, Erstad B, Sweasy J, Persky DO, Abraham I. Cost-effectiveness and economic burden analyses on all first-line treatments of chronic lymphocytic leukemia. Value Health. 2022;25:1685–95. https://doi.org/10.1016/j.jval.2022.04.001.
Buchanan J, Wordsworth S, Clifford R, Robbe P, Taylor JC, Schuh A, et al. Using genomic information to guide ibrutinib treatment decisions in chronic lymphocytic leukaemia: a cost-effectiveness analysis. Pharmacoeconomics. 2017;35:845–58. https://doi.org/10.1007/s40273-017-0519-z.
Roeker LE, Thompson M, Mato AR. Current treatment of chronic lymphocytic leukemia: the diminishing role of chemoimmunotherapy. Drugs. 2022;82:133–43. https://doi.org/10.1007/s40265-021-01657-0.
Lorgelly P, Pollard J, Cubi-Molla P, Cole A, Sim D, Sussex J. Outcome-based payment schemes: what outcomes do patients with cancer value? Patient. 2020;13:599–610. https://doi.org/10.1007/s40271-020-00430-x.
van der Straten L, Hengeveld PJ, Kater AP, Langerak AW, Levin M-D. Treatment approaches to chronic lymphocytic leukemia with high-risk molecular features. Front Oncol. 2021. https://doi.org/10.3389/fonc.2021.780085.
Pharmaceutical Benefit Schedule. Venetoclax: pack containing 14 tablets venetoclax 10 mg and 7 tablets venetoclax 50 mg and 7 tablets venetoclax 100 mg and 14 tablets venetoclax 100 mg, tablet 10 mg, tablet 50 mg, tablet 100 mg; Venclexta®. 2020. https://www.pbs.gov.au/info/industry/listing/elements/pbac-meetings/psd/2020-07/venetoclax-pack-containing-14-tablets-venetoclax-10-mg. Accessed 28 Feb 2023.
Brem EA, O’Brien S. Frontline management of CLL in 2021. J Oncol Pract. 2021;18:109–13. https://doi.org/10.1200/OP.21.00258.
Acknowledgements
The authors wish to gratefully acknowledge funding support including the Snowdome foundation and the Wilson Centre for Blood Cancer Genomics. The model used in this study was provided to the journal’s peer reviewers for their reference when reviewing the manuscript.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
MV received a PhD (independent) scholarship from Illumina under the University of Melbourne/Illumina partnership.
Conflict of interest
The authors declare no competing financial interests.
Availability of data and material
The model inputs used to inform the decision analytic model and execute the cost-effectiveness analysis are included within the article with appropriate citations.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication from patients/participants
Not applicable.
Code availability
The custom codes for the model are available from the corresponding author upon reasonable request.
Author contributions
All authors contributed to the study conception and design. MV developed the economic model under supervision of KD. The initial draft of the manuscript was written by MV, and critically reviewed for scientific content by all other authors. All authors read and approved the final manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Vu, M., Degeling, K., Thompson, E.R. et al. Cost Effectiveness of Molecular Diagnostic Testing Algorithms for the Treatment Selection of Frontline Ibrutinib for Patients with Chronic Lymphocytic Leukemia in Australia. Appl Health Econ Health Policy 22, 107–122 (2024). https://doi.org/10.1007/s40258-023-00826-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40258-023-00826-4