
Abstract
The XprESS multi-sinus dilation system (XprESS) is a minimally invasive alternative to functional endoscopic sinus surgery (FESS) used in the treatment of people with chronic or recurrent acute sinusitis refractory to medical treatment. The manufacturer of XprESS, Entellus Medical, claims the technology is as effective as FESS in improving quality of life and is associated with quicker recovery times and reduced costs. The Medical Technologies Advisory Committee (MTAC) at the National Institute for Health and Care Excellence (NICE) selected XprESS for evaluation. Nine trials published in 13 papers were correctly identified by the company as relevant to the decision problem, including one randomised controlled trial (REMODEL study). From this evidence, the company concluded that XprESS is as beneficial as FESS for a range of clinical endpoints. The External Assessment Centre (EAC) agreed with the company’s conclusion in a subgroup of patients, but judged that the evidence did not generalise to patients within the NHS fully. The company constructed a de novo costing model. XprESS generated cost-savings of £1302 per patient compared with FESS. The EAC critiqued and updated the model’s inputs, with differences in results driven by changes in assumptions on procedure duration, length of hospital stay and the proportion of procedures undertaken in an outpatient setting under local anaesthetic. Although cost-incurring in the base case, XprESS generated cost savings under certain scenarios. The MTAC reviewed the evidence and supported the case for adoption, issuing positive draft recommendations. After public consultation NICE published this as Medical Technologies Guidance 30.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The XprESS multi-sinus dilation system is an example of a technology that aims to shift healthcare from an inpatient to an outpatient setting and thus save resources. |
The case for adopting XprESS for treating uncomplicated chronic sinusitis after medical treatment has failed is supported by the evidence. Treatment with XprESS leads to a rapid and sustained improvement in chronic symptoms, fewer acute episodes and improved quality of life, which is comparable to functional endoscopic sinus surgery (FESS). |
XprESS should be considered in patients with uncomplicated chronic sinusitis who do not have severe nasal polyposis. In these patients, XprESS works as well as FESS, is associated with faster recovery times, and can more often be done under local anaesthesia. |
Cost modelling indicates that XprESS is cost saving compared with FESS when treatment is administered using local anaesthetic in an outpatient setting. |
1 Introduction
The National Institute for Health and Care Excellence (NICE) produces evidence-based medical technologies guidance to evaluate and, where appropriate, encourage adoption of novel and innovative medical devices and diagnostics within the National Health Service (NHS) in England. Companies notify their potentially eligible products to NICE’s Medical Technologies Evaluation Programme (MTEP). Devices are selected for evaluation as part of NICE medical technologies guidance by MTEP if they have the potential to offer clinical benefits to patients and the NHS, and/or to reduce costs, compared with standard care.
Guidance is developed by the Medical Technologies Advisory Committee (MTAC) after the clinical and economic evidence submitted by the company is independently assessed by an External Assessment Centre (EAC) and following a public consultation period. Campbell and Campbell described the MTEP methodology in detail [1].
In December 2016, NICE issued final guidance on the XprESS multi-sinus dilation system (XprESS) for treating chronic sinusitis (also known as rhinosinusitis and sinus infection). XprESS is a sterile single-use re-shapeable balloon-tipped device. It is inserted through the nose into the bony sinus outflow tracts (ostia) in the maxillary, frontal or sphenoidal sinuses. Once in the ostia, the balloon is inflated with saline in order to reshape and open the sinuses, allowing them to drain more effectively [2]. The EAC critiquing the evidence was the Newcastle upon Tyne Hospitals NHS Foundation Trust and York Health Economics Consortium partnership. Clinical experts, identified using NICE’s published processes, provided advice to the EAC and MTAC.
This article is one of a series of Medical Technology Guidance summaries being published in Applied Health Economics and Health Policy. It provides an overview of the clinical and economic benefits of XprESS as demonstrated by the company’s submission, the EAC’s assessment report and the subsequent development of the NICE guidance. Full documentation of the process, supporting evidence and the final guidance is on the NICE website [2].
2 Background to the Indication and Device
Sinusitis is defined as inflammation of the nose and paranasal sinuses. Episodes of the disease lasting 12 weeks or less are categorised as acute sinusitis and longer episodes are classed as chronic sinusitis (CS). Acute sinusitis usually has an infective aetiology. The aetiology of CS is more complex and likely to be multifactorial, with inflammation, infection and obstruction of sinus ventilation (blockage of sinus entrances) contributing to the pathology. A third classification of sinusitis, recurrent acute rhinosinusitis (RARS), refers to frequent episodes of acute symptomatic rhinosinusitis requiring long-term management similar to CS [3, 4].
The prevalence of CS in Europe and North America has been estimated to be between 6% (physician-led diagnosis) and 11% (patient-led diagnosis) [5]. Up to two-thirds of patients presenting to secondary care with CS have nasal polyps [6]. Both CS and RARS are debilitating diseases that have a high negative impact on patient quality of life (QoL) and pose a large financial burden to the National Health Service (NHS) [5].
People with CS within the NHS are treated initially in primary care, typically through saline irrigation and intranasal corticosteroid (ICS). For patients with visible nasal polyps, a course of oral corticosteroids may be considered before ICS. Patients should be reassessed after 3 months and treatment may be continued if symptoms are mild and responsive. Referral to an ear, nose and throat (ENT) specialist should be considered where patients have frequent recurrent episodes of acute sinusitis, unremitting or progressive facial pain, nasal polyps causing significant nasal obstruction or an ineffective 3-month trial of ICS [7].
Patients referred to secondary care should have a full history taken and examination to assess causes and co-morbidities. Patients’ QoL should be assessed using the disease-specific Sino-nasal outcome test (SNOT)-22 scale [4]. A previous version of this tool, the SNOT-20 scale, is also used. Medical treatment in secondary care is maximal to prevent the need for surgery where possible. Treatment from primary care will typically continue with the addition of oral corticosteroids (in patients with nasal polyps) and oral antibiotics. Patients will typically be reviewed after 3 months of maximal medical treatment. If moderate or severe symptoms persist, surgery will be considered following a mandatory computed tomography (CT) scan [4]. Functional endoscopic sinus surgery (FESS) is typically conducted. The principal purpose of FESS is to re-establish ventilation and enhance mucociliary clearance of the sinuses. This is achieved through endoscopic removal of diseased tissue, including in the anterior ethmoid sinus, sphenoid sinus, maxillary sinus and middle meatus [8].
Balloon dilation, through devices such as XprESS, is a minimally invasive intervention with the principal aims of improving sinus ventilation and mucociliary clearance. Unlike FESS, balloon dilation does not excise soft tissue. Instead, the balloon is inflated under high pressure, which causes displacement and micro-fractures of the bony lamina surrounding the entrances to the sinuses such as the maxillary ostia. The subsequent remoulding of the bone is reported to improve sinus drainage with minimal disruption to the mucosal lining [9]. As the soft tissue is relatively unaffected, reduced patient discomfort and quicker recovery times compared with FESS are claimed. XprESS has the potential to be used under local, rather than general, anaesthetic once operators have experience using the device. Therefore, the company claimed in their evidence submission to NICE that, for some patient groups, this may reduce the risks associated with general anaesthesia.
XprESS was considered by MTAC as an alternative to FESS or other balloon systems available within the NHS, as defined by the scope produced by NICE [10]. The decision problem addressed by the company and EAC is described in detail in the following section.
3 Decision Problem (Scope)
3.1 Population
The population described in the scope was people with CS, including RARS, in whom all medical therapy has failed. A number of subgroups were also specified within the scope including people with uncomplicated CS, people with CS with and without nasal polyps, people with CS with orbital or intracranial involvement, and those with CS affecting the anterior ethmoid sinus. Both patients with anatomical variants and children were also specified.
3.2 Intervention (XprESS)
The intervention specified in the scope was ‘The XprESS Multi-Sinus Dilation System’. However, several studies included by the company used the predecessor FinESS system. The FinESS system employs a trans-antral approach for balloon insertion, which is more invasive than the trans-nasal approach used by XprESS, and limits its indication to the treatment of the maxillary ostia or infundibula of the anterior ethmoid only (in contrast to XprESS, which can be used on frontal ostia and recesses and the sphenoid sinus ostia). The company informed the EAC that there were no functional differences between devices in terms of their action on the ostia.
3.3 Comparator (Current NHS Practice)
Two comparators were specified in the scope: FESS and other balloon dilation systems available in the NHS. Expert advice received by the EAC during the assessment process indicated that the primary other balloon system was a device manufactured by Acclarent, which was withdrawn from the UK market in December 2015. As the company did not report evidence pertaining to other balloon systems, FESS was considered the sole comparator of interest.
3.4 Outcomes
Outcomes specified in the scope were divided into three categories: patient outcomes, healthcare system outcomes and adverse effects. The company provided evidence on, at least to some extent, all eight of the patient outcomes listed. These were: change in rhinosinusitis symptoms, number of post-procedure rhinosinusitis episodes requiring medication, number of post-operative debridements, change in ostial patency, duration of analgesic medication, patient-reported tolerance of the procedure and/or patient reported severity of pain and number/type of sinuses treated. Three of the seven healthcare system outcomes were reported in the clinical evidence and addressed by the company (success rates of maxillary sinus ostial cannulation, rate of revision surgery and number of sinus-related follow-up appointments). The remaining four healthcare system outcomes (length of hospital stay, procedure time and treatment room time, rate of readmission, and number and grade of staff required) were not reported within the clinical evidence, but were considered within the economic submission. Both adverse effect-related outcomes (rate and severity of nasal bleeding and device-related adverse events) were addressed by the company.
4 Review of Clinical and Economic Evidence
Clinical and economic evidence, including a de novo economic model, was submitted by the company in line with the process set out by NICE [11]. The EAC critically appraised the submission and economic model against the scope. Section 4.1 summarises the clinical evidence submitted, the EAC’s critique and the EAC’s additional work. Section 4.2 provides the same detail for the economic evidence.
4.1 Clinical Effectiveness Evidence
4.1.1 Company’s Review of Clinical Effectiveness Evidence
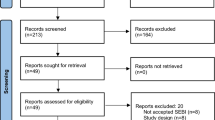
The company searched for clinical evidence relating to the decision problem, i.e. studies of patients with CS or RARS using XprESS or its predecessor, FinESS. Studies reporting clinical efficacy or safety outcomes with any or no comparator were included. The company’s search identified 395 studies, of which the company included 11 published and two unpublished papers, reporting on nine unique trials all set within the USA (Fig. 1). Three of the included papers reported on the REMODEL trial, a randomised controlled trial (RCT) comparing XprESS or FinESS balloon dilation with FESS in adult patients with maxillary sinus disease, with or without anterior ethmoid disease [12,13,14]. The remaining papers reported on single-armed observational studies that reported the use of XprESS or FinESS balloon dilation in various populations of patients suffering from CS or related sinus disease. Three papers reported on the first published observational study, the BREATHE study [15,16,17]. One paper reported on the RELIEF study [18]. A further paper by Eloy et al. [19] reported on the only retrospective study included by the company [19]. The XprESS Maxillary Pilot Study by Gould et al. [20] was published as a white paper (not peer reviewed) [20]. The XprESS registry by Brodner et al. [21] and XprESS multi-sinus study by Gould et al. [6] were both prospective observational studies [22].

The company’s PRISMA diagram
The primary evidence reported by the company was derived from three papers reporting on the REMODEL study [12,13,14]. Cutler et al. [14] provided baseline (n = 92) and 6-month follow-up data (n = 91) [14]. Bikhazi et al. [12] provided data up to 12 months for 89 patients from the original cohort [12]. Finally, Chandra et al. [13] reported on data up to 24 months for 25 patients from the original cohort, and also reported on more recently recruited patients, resulting in a total baseline population of n = 133. Within the same publication, a meta-analysis combining six published studies of XprESS was reported [13]. The primary outcome of the review was change in SNOT-20 score from baseline, whereby the SNOT-20 is a disease-specific patient-reported outcome measure. Secondary outcomes included debridement frequency, technical success, nasal bleeding, recovery and adverse events. The company critically appraised the REMODEL trial, finding it to be at low risk of bias.
All observational studies followed patients up to 6, 12 or 24 months after surgery, with change in either SNOT-20 or SNOT-22 scores over time being the primary outcome [15,16,17,18,19,20,21,22,23,24]. Other reported outcomes included technical success of the device, medication use, recovery time, productivity and revision rates.
The company accurately reported results from the REMODEL study and the observational studies. The results from the REMODEL study demonstrated that the improvement in mean SNOT-20 scores compared with baseline was statistically significant in both groups at all time points (p < 0.0001). Furthermore, in all cases this difference exceeded 0.8, a magnitude of effect that has been previously reported as clinically meaningful [25]. There was no statistical difference between treatments at any time point, with the exception of 1 week where there was a significantly greater, but not clinically important, improvement in the XprESS arm. Postoperative debridement was not required in 92.0% (46/50) of patients in the balloon dilation arm, compared with 26.2% (11/42) of FESS patients. There was a mean of 0.1 ± 0.6 (SD) postoperative debridements in the balloon arm, compared with 1.2 ± 1.0 in the FESS arm (p < 0.0001). FESS was associated with a statistically significant increased recovery time (return to normal activities, 1.6 ± 11 vs. 4.8 ± 6.2 days, p = 0.002) and duration of prescription pain medication (0.9 ± 1.4 vs. 2.8 ± 2.7 days, p < 0.001) compared with balloon dilation. No significant differences were reported in post-discharge nausea, duration of over-the-counter pain medication, technical success, ostial patency, complications or revision surgery [12,13,14].
The observational studies provided supplementary longitudinal data that were largely consistent with the results reported in the REMODEL trial. A published meta-analysis reported that there was no statistical difference in SNOT-20 outcomes between studies (REMODEL FESS, REMODEL balloon dilation or pooled observational studies), measured at 6, 12 and 24 months. There were significant reductions (p < 0.0001) from baseline compared with 12 months in the standalone balloon studies in: work/school missed due to nasal problems (5.0 ± 9.5 days); homebound due to nasal problems (6.3 ± 11.3 days); number of physician/nurse visits due to nasal problems (4.5 ± 11.5 visits); number of infections of nose/sinuses (3.9 ± 4.5 episodes); and number of antibiotic courses (2.9 ± 3.1). Revision rates at 12 months were 1.7% for the FESS arm of the REMODEL trial, 1.4% for the balloon dilation arm of the REMODEL trial and 3.2% for the pooled analysis (p = 0.628). However, this analysis was based on a small number of events (for example, a single patient in each of the REMODEL arms) [13].
The company reported that one serious procedure-related adverse event was reported in the selected studies; this was in a patient enrolled to the BREATHE study who received treatment with the FinESS system [15]. The patient suffered from subcutaneous emphysema (facial swelling) after resuming continuous positive airway pressure following the procedure, and recovered within a week. No other adverse events were reported in any of the studies.
The company concluded that the evidence shows that balloon dilation improves health outcomes and is as beneficial as FESS for a range of clinical endpoints. Further, they concluded that the evidence is directly relevant to the scope of the MTEP evaluation and that the results of the studies presented should apply to clinical practice where included patients meet the criteria for FESS after failing maximal medical management for CS.
4.1.2 Critique of Clinical Effectiveness Evidence
The EAC critiqued the company’s literature search, evidence selection and quality assessment of included studies. The company’s searches were limited in scope, but replicated as far as possible, yielding a similar number of studies. A de novo literature search was undertaken by the EAC, which aimed to be more sensitive and included a wider range of databases than that conducted by the company. This search aimed to identify evidence on the XprESS MSDS or the FinESS system for treatment of patients with CS or recurrent acute rhinosinusitis, and was carried out on a number of databases including Medline, EMBASE, the Cochrane library and the grey literature (strategies are described in the EAC assessment report) [2]. It comprised two concepts (rhinosinusitis and balloon catheter dilation) and identified 545 unique records. The records were assessed by two reviewers and no relevant studies were identified as missed by the company within their evidence submission to NICE. One study included by the company, Eloy et al. [19], was excluded by the EAC given that it investigated the use of standalone XprESS balloon dilation in patients (n = 5) who had previously undergone a failed frontal sinusotomy. Revision surgery was not listed in the population or subgroup domains of the scope [19]. This study was not included in the meta-analysis and thus its exclusion does not alter the results reported by the company [13]. The studies deemed eligible for inclusion by the EAC are summarised in Table 1.
The EAC conducted a detailed critical appraisal of the internal and external validity of the pivotal REMODEL study, which was a non-inferiority RCT comparing balloon dilation with the XprESS or FinESS system with FESS in patients with CS in the USA. This study exhibited relatively high methodological quality, with appropriate randomisation and concealment of allocation and reporting of a relevant and adequately powered pre-specified primary outcome (SNOT-20 score) with suitable statistical analysis. This reduced the risk of both selection and reporting bias. As blinding was not feasible and a subjective primary outcome was selected, there was a moderate to high risk of performance bias. However, the important factor affecting the internal validity of the trial was the high dropout rate exhibited in the FESS arm following randomisation and immediately prior to surgery, and the subsequent need for post hoc modified intention-to-treat (ITT) analysis, which has been associated with bias [26]. An additional limitation was the reduced number of patients analysed at later follow-up (for instance n = 25 at 24 months), which increased uncertainty in the outcomes at these time points.
The EAC identified some limitations in the external validity of the REMODEL trial in its application to the NHS:
-
A reduction in debridement was reported with XprESS compared with FESS. Experts advised that debridement is not common practice in the NHS, hence this benefit would likely not be realised in practice.
-
It was unclear whether the definition of ‘maximal medical treatment’ in the trial was equivalent to that used within NHS practice, where it is possible that medical treatment may be pursued for longer.
-
A lack of data available on patients with nasal polyps compared with those without reduces the generalisability of the REMODEL study. It has been reported that two-thirds of patients undergoing FESS within the NHS have nasal polyps [27].
-
Possible differences in efficacy and safety of the XprESS MSDS and the FinESS system may exist (with about half of the interventions used in the REMODEL trial being FinESS). Following advice from the company, these interventions were assumed to be equivalent. However, there was only weak indirect evidence from the published literature to support this assumption [13].
The REMODEL trial was considered, on balance, to likely comprise a subgroup of the full spectrum of patients treated within the NHS. The EAC considered that, in general, longitudinal data from the observational studies supported the results of the REMODEL trial, with the same caveats as described in the bullet points above.
The EAC agreed with the company’s reporting of results, on the whole. The changes in SNOT-20 score reported by Chandra et al. [13] are reported in Table 2. Overall, the data showed that in a selected patient population, the use of balloon dilation is associated with non-inferior QoL improvements compared with FESS, and that this effect is immediate and continues for at least 2 years. The evidence also showed that balloon dilation is associated with improved patient recovery times and reduced requirement for analgesia.
Regarding safety, a search of the US Food and Drug Administration (FDA) Manufacturer and User Facility Device Experience (MAUDE) database was conducted by the EAC to identify device-related adverse events. Twelve reports were identified, two of which had been specifically referenced by the company in their evidence submission to NICE. The majority of the MAUDE reports described cerebral spinal fluid leaks with no long-term adverse health effects. There was also one case of death unrelated to the use of the device and one device malfunction of the XprESS LoProfile Multi-Sinus Dilation System.
4.1.3 Conclusions from Clinical Effectiveness Evidence
The EAC considered that the company provided evidence that the XprESS MSDS or FinESS system provides non-inferior patient benefits compared with FESS in selected patients with refractory CS of RARS of the maxillary sinus, with or without anterior ethmoid disease. This comparative evidence was supported by more extensive observational data that showed comparable longitudinal results in a broader population of patients [13]. However, as this patient population represents a subgroup of those treated within the NHS, there is currently uncertainty as to whether the procedure would be as effective in all patients indicated for surgery within the NHS, and in particular there is uncertainty as to the efficacy of the procedure in patients with nasal polyps. Additionally, evidence on the effectiveness of treatment other than the maxillary and anterior ethmoid sinuses was limited to a single uncontrolled observational study [22]. To reduce this uncertainty, further prospective research is required. Nevertheless, the EAC considered that, these reservations aside, the company had substantiated their claims of clinical effectiveness as specified in the scope within the selected patient population.
4.2 Economic Evidence
4.2.1 Company’s Economic Submission
The company searched for economic evidence from studies of patients with CS. Six studies were included, four of which compared balloon therapy (either as hybrid surgery with FESS or standalone) with FESS [28,29,30,31]. The remaining two studies were systematic reviews of cost studies [32, 33]. As none of the included studies considered XprESS, the company concluded that no relevant high-quality evaluation or cost-effectiveness studies comparing XprESS to either FESS or other balloon dilations systems were available.
The company submitted its own de novo cost model, which compared XprESS primarily to FESS and secondly to an alternative balloon system (Acclarent) over a 5-year time horizon. A fully executable model was submitted in Microsoft Excel®. The company described the model as following a cohort of average-risk patients attending for CS surgery in multiple sinuses. Costs incurred in the first year were modelled via a decision tree, whilst longer term costs were modelled via a two-state Markov model (Fig. 2). Within the decision tree element of the model, patients undergo sinus surgery, following which they have either a general practitioner (GP) follow-up (within the first 3 months) or sustained recovery (no GP visit). All patients, regardless of their GP visit status, are at risk of readmission to hospital. Furthermore, all patients, regardless of their readmission status, are at risk of revision surgery. The risk of revision surgery is not dependent on previous readmission to hospital or GP visits. Likewise, the risk of readmission to hospital is not dependent on prior GP visits. Within the Markov model element of the model there are two health states: surgery revision and surgery success. An annual risk of revision surgery is applied to patients and those patients having revision surgery have a cost applied. Patients are not able to have multiple revision surgeries. Within the longer term element of the model, patients also have a background risk of GP visits for the medical management of acute exacerbations. This risk is independent of their revision status.

Economic model schematic
To populate its economic analysis, the company utilised data from two key sources: the REMODEL study [13] and a national audit [27], in addition to advice from UK clinical experts. The inputs for surgical health states under general and local anaesthetic, GP follow-up, readmission and revision, and surgery success were referenced to relevant clinical studies and expert opinion. Values for unit costs were obtained from national datasets. The national audit comprises data on surgery for nasal polyposis and CS in 87 hospitals in England and Wales during a 6-month period in 2000 [27]. Patients (n = 3128) undergoing surgery were prospectively enrolled within the audit and the inclusion criteria were broad, meaning that a wide spectrum of patients was included. Outcomes were reported for patients with and without polyps separately, with the company making use of the without-polyp data within its model. All model input parameters are shown in Table 3.
Base-case results from the company’s model estimate that XprESS generates cost savings of £1302 per patient compared with FESS. This was based on costs of £2679 per patient treated with XprESS and £3981 per patient undergoing FESS. Univariate sensitivity analysis identified procedure time with XprESS and the cost of XprESS to be the key drivers of the analysis. Scenario analyses demonstrated cost savings over a range of time horizons and model assumptions. Breakeven analysis showed that XprESS was cost saving provided that the procedure time with XprESS was less than 80 min or the procedure time with FESS greater than 41 min.
The results of its de novo economic modelling led the company to conclude that XprESS produces cost savings arising from a reduction in time in theatre and faster recovery, as well as reduced healthcare utilisation and fewer surgical revisions.
4.2.2 Critique of Economic Evidence
The EAC re-ran the company’s searches as far as was possible based on the reporting provided. All six studies included by the company were excluded by the EAC given that XprESS was not an intervention in any of the included studies [28,29,30,31,32,33]. Rather, these studies considered alternative balloon therapies often in conjunction with FESS. The EAC’s search for clinical evidence did not specify study design; therefore, the results were sifted for any economic studies relating to XprESS. No studies were identified.
The EAC judged that it was appropriate for the company to develop its own de novo cost model given the lack of published evidence. The Acclarent device, the secondary comparator in the company’s model, was deemed not applicable given that the device is no longer available within the NHS. No data pertaining to any other balloon systems used within the NHS were available, and hence the comparison with FESS was deemed to be the only comparison of interest to the decision problem.
The EAC replicated the company’s calculations in order to confirm their accuracy and corrected a negligible error in the discounting applied in the scenario where patients under local anaesthetic were considered. The EAC sought advice from clinical experts around the model structure and assumptions made, and judged these to be reasonable on the whole. However, some specific points were highlighted. First, conflicting expert advice was received based upon the plausibility of post-surgery nasal bleeding as a predictor of GP visits post-surgery. Based on this conflicting opinion, the EAC judged it reasonable to build these assumptions into the model structure and explore their cost implications, ideally using several scenarios. Second, the company modelled a difference in risk of revision surgery over the time horizon of the model between XprESS and FESS. This difference was deemed by the EAC to be insufficiently supported by the evidence for inclusion in the model’s base case. Third, the EAC included costs associated with training surgeons on use of the device in the model.
All model inputs were validated by the EAC using expert advice and the literature. The EAC made changes to a number of model inputs, as shown in Table 4. Notably, the EAC assumed that the risk of revision surgery was the same in years 2–5 in both arms of the model. Further, the EAC reworked the bottom-up costing for the procedure cost both with XprESS and FESS based upon expert advice and published data. The cost estimated by the EAC for the FESS procedure was lower than that estimated by the company (£657 rather than £2594). This difference occurred primarily due to a large reduction in the duration of FESS used by the EAC (42.5 vs. 90 min). The EAC’s time estimate was based on expert advice concerning patients undergoing FESS who would be eligible for treatment with XprESS and supported by published evidence from a health technology assessment report and the national audit [27, 34]. The procedure cost associated with XprESS used by the EAC was also lower than that used by the company (Table 4).
Following these updates, the results estimated by the EAC changed direction, with XprESS costing £330 per patient more than FESS. This was largely driven by the change in FESS procedure time. The company’s univariate sensitivity analysis was re-run, involving each input parameter to be varied by ±20%. The direction of the results of the model did not change during this analysis. Threshold analysis showed that if FESS takes longer than 66 min (or around 24 min longer than XprESS), XprESS is cost saving. Threshold analysis of the unit cost of theatre time demonstrated that where this costs over £2040 per hour, XprESS becomes cost saving (compared with around £800 per hour used in the base case). The cost of the XprESS device was also a key driver of the analysis.
4.2.3 Additional Work Undertaken by EAC Relating to Economic Evidence
The EAC undertook a series of scenario analyses utilising different data sources. First, the EAC considered a scenario whereby XprESS is not used in a theatre setting, but rather in an office setting under local anaesthetic. Experts advised that the device is used in this manner in some NHS patients. However, based upon the EAC’s inputs XprESS remained cost incurring even where all XprESS procedures were carried out in this setting.
Second, one expert advised that patients undergoing FESS required an additional hospital appointment compared with those undergoing surgery with XprESS for debridement. This practice was supported by the clinical evidence; however, these US studies may not generalise to the NHS [13]. Under a scenario whereby all FESS patients required an additional follow-up appointment for debridement and over 85% of XprESS patients had the procedure in an office setting under local anaesthesia, XprESS generated cost savings.
Finally, based on information provided by experts and the company at the first MTAC meeting, the EAC considered a scenario in which the duration of FESS was 60 min, the company offer XprESS at a reduced cost of £820 and 80% of treatments with XprESS are conducted under local anaesthetic in an outpatient setting. Under this scenario, XprESS generated cost savings of £152 per patient.
5 NICE Guidance
5.1 Provisional Recommendations
The evidence submitted by the company and the EAC’s critique of this evidence was presented to MTAC, who provided draft recommendations relating to XprESS following their meeting in May 2016. These were as follows:
-
Evidence supports the use of the XprESS multi-sinus dilation system for treating chronic sinusitis after medical treatment has failed. Treatment with XprESS leads to a rapid and sustained improvement in chronic symptoms, fewer acute episodes and improved QoL which is comparable to functional endoscopic sinus surgery (FESS).
-
XprESS should only be used in patients with non-complex chronic sinusitis who do not have nasal polyps. In these patients, XprESS works as well as functional endoscopic sinus surgery, is associated with faster recovery times, and can more often be done under local anaesthetic.
-
Cost modelling indicates that savings are achievable as compared with FESS when XprESS treatment is performed under local anaesthetic in an office setting. If 80% of treatments are performed in this way, the estimated saving per patient is £152 with XprESS as compared with a 60-min FESS procedure and assuming a cost for XprESS of £820.
5.2 Consultation Response
During the consultation NICE received 25 consultation comments from eight consultees. The NICE guidance was updated to address a number of these comments. The recommendations were updated to provide clarification on the patient population in which the device should be used to align it with the company’s instruction for use and the evidence. This involved changes to its recommended use in patients with nasal polyps and the use of more appropriate terminology when describing patients with uncomplicated sinusitis in whom the device is recommended. These changes were augmented with additional explanatory text in the current management section to clarify the terminology used and the recommended population for use. NICE was facilitated in this by clinical experts with experience of using the technology.
Additionally changes were made to the evidence section concerning the reporting of the study population, and method of analysis used in the REMODEL trial.
6 Key Challenges and Learning Points
The key challenges faced by both the EAC and the company were the generalisability of the clinical data and the lack of comparative data pertaining to resource use in patients undergoing surgery with XprESS or FESS.
All of the included clinical studies were set in the USA rather than the NHS in England. Specifically, the comparative data from the REMODEL trial included a relatively highly selected group of patients who may not reflect those undergoing sinus surgeries within the NHS [12,13,14]. Three key issues relating to the population were identified. First, patients with frontal or sphenoid sinus disease, gross sinonasal polyposis or who had previously undergone sinus surgery were excluded, hence the comparative efficacy of balloon dilation and FESS in these patients in unknown. Second, within the NHS, patients undergoing surgery have exhausted medical management options. Whilst patients included within the REMODEL study were described as refractory to medical treatment, issues such as earlier access to surgical treatment within the USA may mean that trial patients were less maximally treated than those in the NHS. Third, the presence or absence of nasal polyps is an important aspect of the classification of CS and determinant of treatment according to European Position Paper on Rhinosinusitis and Nasal Polyps (EPOS) guidelines [3], with patients with nasal polyposis typically receiving more aggressive surgical management and benefitting more from this [27]. The REMODEL trial excluded patients with gross polyposis but otherwise did not report subgroup analysis on whether there were differences in response to treatment in patients with mild to moderate polyposis.
The EAC attempted to compare the trial population to those in the national audit [27] and also sought expert advice on its applicability to the NHS. On balance, it was concluded that the population enrolled in the REMODEL trial was likely to comprise a subgroup of the full spectrum of patients treated in the NHS, and therefore not be fully representative.
An additional challenge faced was the lack of comparative data reporting resource use with FESS and XprESS, specifically relating to duration of surgery and length of stay following surgery, which were key parameters within the economic analysis. As such, these parameters were populated either using expert opinion or data from non-comparative sources that did not fully represent the population included within the scope of the decision problem. Expert opinion indicated great variation in parameters, particularly the key driver of the analysis—the duration of FESS. The final guidance captured this uncertainty and its implication on the potential cost savings arising from the use of XprESS.
7 Conclusion
The MTEP evaluation process was followed for the development of medical technologies guidance on XprESS. This included a submission of clinical and economic evidence by the company, critical appraisal of this evidence by the EAC, additional work to address remaining uncertainties, drafting of recommendations by MTAC, and a subsequent public consultation. Following this process, MTAC judged that the evidence demonstrated sufficient potential benefits of XprESS to patients and the NHS to allow positive recommendations to be made for the device.
References
Campbell B, Campbell M. NICE medical technologies guidance: a novel and rigorous methodology to address a new health technology assessment challenge. Appl Health Econ Health Policy. 2012;10(5):295–7.
National Institute for Health and Care Excellence. XprESS multi sinus dilation system for treating chronic sinusitis. 2016. https://www.nice.org.uk/guidance/mtg30. Accessed 22 Dec 2016.
Fokkens WJ, Lund VJ, Mullol J, Bachert C, Alobid I, Baroody F, et al. European position paper on rhinosinusitis and nasal polyps 2012. Rhinol Suppl. 2012;23:1–298.
Hopkins C, McCombe A, Philpott C, Hern J, Blythe J, Thomas T et al. Commisioning guide: rhinosinusitis. 2013. https://www.rcseng.ac.uk/healthcare-bodies/docs/published-guides/rhinosinusitus. Accessed 4 April 2016.
Philpott C, Hopkins C, Erskine S, et al. The burden of revision sinonasal surgery in the UK—data from the Chronic Rhinosinusitis Epidemiology Study (CRES): a cross-sectional study. BMJ Open. 2016;5:e006680.
Ah-See KL, MacKenzie J, Ah-See KW. Management of chronic rhinosinusitis. BMJ. 2012;345:e7054.
National Institute for Health and Care Excellence. Clinical knowledge summaries: sinusitis. 2013. http://cks.nice.org.uk/sinusitis. Accessed 6 April 2016.
Kennedy DW. Functional endoscopic sinus surgery: technique. Arch Otolaryngol. 1985;111(10):643–9. doi:10.1001/archotol.1985.00800120037003.
Levine H, Rabago D. Balloon sinuplasty: a minimally invasive option for patients with chronic rhinosinusitis. Postgrad Med. 2011;123(2):112–8. doi:10.3810/pgm.2011.03.2269.
National Institute for Health and Care Excellence (NICE). The XprESS Multi-Sinus Dilation System for the treatment of chronic rhinosinusitis: scoping document. National Institute for Health and Care Excellence, Online. 2016. https://www.nice.org.uk/guidance/GID-MTG10003/documents/final-scope. Accessed 21 March 2016.
National Institute for Health and Care Excellence. Medical technologies evaluation programme: process guide. 2011. http://www.nice.org.uk/Media/Default/About/what-we-do/NICE-guidance/NICE-medical-technologies/Medical-technologies-evaluation-programme-process-guide.pdf. Accessed 6 Feb 2015.
Bikhazi N, Light J, Truitt T, Schwartz M, Cutler J, Remodel Study Investigators. Standalone balloon dilation versus sinus surgery for chronic rhinosinusitis: a prospective, multicenter, randomized, controlled trial with 1-year follow-up. Am J Rhinol Allergy. 2014;28(4):323–9. doi:10.2500/ajra.2014.28.4064.
Chandra RK, Kern RC, Cutler JL, Welch KC, Russell PT. REMODEL larger cohort with long-term outcomes and meta-analysis of standalone balloon dilation studies. Laryngoscope. 2016;126(1):44–50. doi:10.1002/lary.25507.
Cutler J, Bikhazi N, Light J, Truitt T, Schwartz M, Investigators RS. Standalone balloon dilation versus sinus surgery for chronic rhinosinusitis: a prospective, multicenter, randomized, controlled trial. Am J Rhinol Allergy. 2013;27(5):416–22. doi:10.2500/ajra.2013.27.3970.
Cutler J, Truitt T, Atkins J, Winegar B, Lanier B, Schaeffer BT, et al. First clinic experience: patient selection and outcomes for ostial dilation for chronic rhinosinusitis. Int Forum Allergy Rhinol. 2011;1(6):460–5. doi:10.1002/alr.20069.
Stankiewicz J, Tami T, Truitt T, Atkins J, Winegar B, Cink P, et al. Impact of chronic rhinosinusitis on work productivity through one-year follow-up after balloon dilation of the ethmoid infundibulum. Int Forum Allergy Rhinol. 2011;1(1):38–45. doi:10.1002/alr.20008.
Stankiewicz J, Truitt T, Atkins J, Winegar B, Cink P, Raviv J, et al. Two-year results: transantral balloon dilation of the ethmoid infundibulum. Int Forum Allergy Rhinol. 2012;2(3):199–206. doi:10.1002/alr.21024.
Levine SB, Truitt T, Schwartz M, Atkins J. In-office stand-alone balloon dilation of maxillary sinus ostia and ethmoid infundibula in adults with chronic or recurrent acute rhinosinusitis: a prospective, multi-institutional study with-1-year follow-up. Ann Otol Rhinol Laryngol. 2013;122(11):665–71.
Eloy JA, Friedel ME, Eloy JD, Govindaraj S, Folbe AJ. In-office balloon dilation of the failed frontal sinusotomy. Otolaryngol Head Neck Surg. 2012;146(2):320–2. doi:10.1177/0194599811425885.
Gould J. In-office balloon dilation: procedure techniques and outcomes using a malleable multi-sinus dilation tool. Synergy ENT Spec. 2012. http://www.stlsinuscenter.com/Portals/1/Forms/In-Office%20Balloon%20Dilation.Procedue%20Technique%20and%20Outcome%20Dr.%20James%20D.%20Gould%20(2)%20copy.pdf.
Brodner D, Nachlas N, Mock P, Truitt T, Armstrong M, Pasha R, et al. Safety and outcomes following hybrid balloon and balloon-only procedures using a multifunction, multisinus balloon dilation tool. Int Forum Allergy Rhinol. 2013;3(8):652–8. doi:10.1002/alr.21156.
Gould J, Alexander I, Tomkin E, Brodner D. In-office, multisinus balloon dilation: 1-year outcomes from a prospective, multicenter, open label trial. Am J Rhinol Allergy. 2014;28(2):156.
Entellus Medical, Cartographer FinESS Registry Study. Bethesda: US National Library of Medicine; 2009.
Entellus Medical, Cartographer Sinus Balloon Dilation in Pediatric Patients. Bethesda: US National Library of Medicine; 2014.
Pynnonen MA, Kim HM, Terrell JE. Validation of the Sino-Nasal Outcome Test 20 (SNOT-20) domains in nonsurgical patients. Am J Rhinol Allergy. 2009;23(1):40–5. doi:10.2500/ajra.2009.23.3259.
Gupta SK. Intention-to-treat concept: a review. Perspect Clin Res. 2011;2(3):109–12. doi:10.4103/2229-3485.83221.
Browne J, Hopkins C, Slack R, Van der Meulen J, Lund VJ, Topham J et al. The national comparative audit of surgery for nasal polposis and chronic rhinosinusitis. London: Clinical Effectiveness Unit, Royal College of Surgeons of England; 2003. https://www.rcseng.ac.uk/surgeons/research/surgical-research/docs/the-national-comparative-audit-of-surgery-for-nasal-polyposis-and-chronic-rhinosinusitis. Accessed 4 April 2016.
Ference EH, Graber M, Conley D, Chandra RK, Tan BK, Evans C, et al. Operative utilization of balloon versus traditional endoscopic sinus surgery. Laryngoscope. 2015;125(1):49–56. doi:10.1002/lary.24901.
Ference EH, Schroeder JW Jr, Qureshi H, Conley D, Chandra RK, Tan BK, et al. Current utilization of balloon dilation versus endoscopic techniques in pediatric sinus surgery [Erratum appears in Otolaryngol Head Neck Surg. 2015 Aug;153(2):310–1; PMID: 26198863]. Otolaryngol Head Neck Surg. 2014;151(5):852–60. doi:10.1177/0194599814545442.
Holy C, Ellison J, Schneider C. Budget impact analysis of balloon dilation and functional endoscopic surgery: a us payer perspective. Value Health. 2013;16(3):A232. doi:10.1016/j.jval.2013.03.1180.
McElroy HJ, Belarbi S, Foteff C, Metz L, Chung L, Levine HL, et al. A systematic review of chronic rhinosinusitis in Asia-Pacific and the role of balloon sinuplasty. Value Health. 2011;14(7):A500. doi:10.1016/j.jval.2011.08.1455.
Smith KA, Orlandi RR, Rudmik L. Cost of adult chronic rhinosinusitis: a systematic review. Laryngoscope. 2015;125(7):1547–56. doi:10.1002/lary.25180.
Sorgeloose T, Demaire C, Stueve M. Budget impact analysis of balloon sinuplasty versus classic functional endoscopic sinus surgery. Using a budget impact model to identify country specific market access strategy. Value Health. 2012;15(7):A348. doi:10.1016/j.jval.2012.08.868.
Dalziel K, Stein K, Round A, Garside R, Royle P. Systematic review of endoscopic sinus surgery for nasal polyps. Health Technol Assess. 2003;7(17):iii, 1–159.
Joint Formulary Committee. British National Formulary (online). London: BMJ Group and Pharmaceutical Press; 2016. http://www.evidence.nhs.uk/formulary/bnf/current. Accessed March 2016.
Curtis L, Burns A. Unit Costs of Health and Social Care 2015. 2015. http://www.pssru.ac.uk. Accessed 21 March 2016.
Department of Health. NHS reference costs 2011–12. London: Department of Health; 2012.
Zilvetti M, Puttick M, Parameswaran R, McWhinnie D. Day surgery and national tariff: is there room for economic sense? J One Day Surg. 2009;19(3):64–7.
Department of Health. NHS reference costs 2014–15. London: Department of Health; 2015.
Author information
Authors and Affiliations
Contributions
The manuscript was prepared by MJ, with contributions from IW, EET, NH, MA, HC, JC and AS. Literature searching was undertaken by MA and evidence review by IW and HC with advice and quality assurance from JC and AS. The model critique was undertaken by MJ and EET with advice and quality assurance from JC and AS. NH reported the decision problem and the NICE recommendations. The guarantor for overall content is AS.
Corresponding author
Ethics declarations
Funding
The authors are grateful to the clinical experts identified by NICE, who provided expertise and clinical knowledge. Newcastle upon Tyne Hospitals and York Health Economics Consortium are funded by NICE to act as an EAC for the Medical Technologies Evaluation Programme. This summary of the Medical Technology Guidance was produced following publication of the final guidance report. This summary has not been externally peer reviewed by Applied Health Economics and Health Policy.
Conflict of interest
Three of the authors (IW, HC and AS) are NHS employees; the NHS has a financial interest in the guidance issued by NICE as a result of this work. Four of the authors (MJ, EET, MA and JC) work for the EAC but otherwise have no conflicts of interest. NH is an employee of NICE.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jenks, M., Willits, I., Turner, E.E. et al. The XprESS Multi-Sinus Dilation System for the Treatment of Chronic Sinusitis: A NICE Medical Technology Guidance. Appl Health Econ Health Policy 15, 567–582 (2017). https://doi.org/10.1007/s40258-017-0337-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40258-017-0337-7