
Abstract
Background
Janus kinase (JAK) inhibitors, including upadacitinib, have been recently approved for the treatment of moderate-severe atopic dermatitis (AD) and real-world data on upadacitinib effectiveness and safety are limited. This interim analysis aimed to assess effectiveness and safety of upadacitinib throughout 48 weeks of observation in a real-world adult AD population.
Methods
This prospective study collected data on adult patients affected by moderate-to-severe AD and treated with upadacitinib at the dosage of either 15 mg or 30 mg daily based on the physician decision. Upadacitinib was prescribed in the context of a national compassionate use programme. In this interim analysis, within patient comparisons of continuous scores of different scales (namely Eczema Area and Severity Index [EASI], body surface area [BSA], Dermatology Life Quality Index [DLQI], Patient Oriented Eczema Measure [POEM], Numeric Rating Scale [NRS] subtests) were performed. The percentage of patients achieving EASI 75, EASI 90 and EASI 100 at Week 16, 32 and 48 was also evaluated.
Results
One hundred and forty-six patients were included in the analysis. Upadacitinib 15 mg or 30 mg daily was prescribed as monotherapy in most cases (127/146, 87.0%). Upadacitinib was initially prescribed at the dosage of 30 mg daily in 118 of 146 (80.8%) patients and 15 mg daily in 28/146 (19.2%) patients. A significant improvement in the clinical signs and symptoms of AD was detected by Week 16 and throughout the study period. EASI 75, EASI 90 and EASI 100 responses were achieved by 87.6%, 69.1% and 44.3% at Week 48, associated with a sustained reduction in the mean values of all physician-reported (EASI and BSA) and patient-reported (Itch- Sleep- and Pain-NRS, DLQI, and POEM) disease severity outcomes, up to 48 weeks of treatment. Treatment response observed in 15 mg upadacitinib-treated patients was comparable with that detected in 30 mg upadacitinib-treated patients, revealing no statistical difference between the two patient sub-cohorts. Through the observation period, dose reduction or escalation was observed in 38/146 (26%) of treated cases. Overall, 26 of 146 (17.8%) patients experienced at least one adverse event (AE) during the treatment period. In total, 29 AEs were recorded and most of them were evaluated as mild to moderate, while in 4 cases the occurrence of AE led to drug discontinuation, for a total of 7/146 (4.8%) dropouts.
Conclusion
This study provides strong evidence of a sustained response obtained by upadacitinib in AD patients, who had failed to respond to conventional or biological systemic agents, through 48 weeks of observation. Upadacitinib was also demonstrated to be advantageous in terms of flexibility in dose reduction or escalation as upadacitinib dose was shaped on clinical needs that, in a real-world setting, might frequently change.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Real-world data on the effectiveness and safety of upadacitinib in the treatment of moderate-severe atopic dermatitis are limited. |
This interim analysis investigated effectiveness and safety of upadacitinib 15 mg or 30 mg daily in a large atopic dermatitis real-world population throughout 48 weeks of observation. |
A significant improvement in the clinical signs and symptoms of atopic dermatitis was detected in the overall population by Week 16 and throughout the study period. |
The percentage of patients treated with either 15 or 30 mg upadacitinib daily and achieving EASI 75, EASI 90 and EASI 100 responses was: 78.2%, 47.6% and 28.2% of patients at 16 weeks and 87.6%, 69.1% and 44.3% at Week 48. |
1 Introduction
Atopic dermatitis (AD) is a chronic inflammatory skin disease associated with intense itch and a negative impact on patients’ quality of life (QoL) [1]. Disease prevalence is increasing in adulthood reaching an estimation of 8.1% in Italy [2]. The therapeutic management of AD is mostly based on topical and/or systemic immunosuppressive/immunomodulant therapies and can be challenging in the long-term period, particularly for moderate-to-severe AD [3].
Beside conventional systemic agents, such as cyclosporine, targeted therapies approved for the treatment of AD are currently available [3]. Approved biological agents include dupilumab, a subcutaneous monoclonal antibody inhibiting the signalling of two pathogenic type 2 inflammatory cytokines, interleukin (IL)-4 and IL-13 [3,4,5,6], and tralokinumab, a subcutaneous monoclonal antibody selectively neutralising IL-13 [7]. In addition, a new class of oral small molecule drugs targeting the intracellular signal transducers belonging to the Janus kinase (JAK) family, obtained indication for the treatment of moderate-to-severe AD [8]. Janus kinase inhibitors show different selectivity in blocking JAK isoforms with abrocitinib and upadacitinib representing JAK 1 selective inhibitors while baricitinib is identified as a JAK1/2 inhibitor [8].
Short- and long-term efficacy and safety of upadacitinib in the treatment of moderate-to-severe AD have been described in clinical trials and found confirmation in a few real-world experiences [9,10,11,12,13,14,15,16,17,18,19]. One of these derived from the Italian data collection platform is named ERUDA, which prospectively collects demographic and clinical data related to the national compassionate use of upadacitinib in AD patients [16].
This interim analysis assessed effectiveness and safety of upadacitinib throughout 48 weeks of observation in a real-world adult AD population.
2 Methods
This prospective study collected data on adult patients affected by moderate-to-severe AD and treated with upadacitinib from October 2020 to September 2022. Patients referred to 15 Italian dermatological centres in the context of a national compassionate use programme authorised by the Italian Medical Agency (AIFA) that allows the use of either 15 mg or 30 mg upadacitinib based on physician decision.
Adult patients (aged ≥ 18 years) affected by moderate-to-severe AD (Eczema Area and Severity Index, EASI ≥ 16), who were unresponsive, intolerant or had contraindications to the approved therapies for moderate-to-severe AD at the time of protocol definition, in June 2020 (cyclosporine and dupilumab were the only agents with indication for AD in Italy) were eligible for upadacitinib therapy, either alone or combined with topical/systemic corticosteroids.
2.1 Assessment Tools for Disease Severity and Safety
Baseline characteristics included age, sex, smoking habits (smoker, former smoker, or non-smoker), AD history and severity, prior treatments, atopic and non-atopic comorbidities, and concomitant therapies. At baseline and at each follow-up visit (performed every 16 weeks with slight variation according to the appointment timetable scheduled at each centre), disease severity was assessed by EASI whilst skin involvement was assessed by body surface area (BSA), itch severity by a 0–10 numeric rating scale (itch-NRS), sleep disturbances/sleeplessness by a 0–10 NRS scale (sleep-NRS), pain intensity by a 0–10 NRS (pain-NRS), patient’s QoL by the Dermatology Life Quality Index (DLQI) and global patient-oriented disease severity by the Patient-Oriented Eczema Measure (POEM).
Safety was assessed by physical examination and laboratory tests (i.e., complete blood count, transaminases, creatinine, blood glucose, prothrombin time, activated partial thromboplastin time, international normalised ratio, creatine phosphokinase). Adverse events (AEs) were defined as any abnormal physical condition, symptom, or blood test alteration collected by the physicians throughout the study period every 16 weeks or more tightly based on clinical needs.
2.2 Statistical Analysis
Patients who started treatment with upadacitinib were included in this interim analysis, though not all of them potentially achieved a 48-week observation period. The data analysis was carried out to address both descriptive and inferential aims. Treatment groups were identified based on initial dosage (intention to treat) and all comparisons were performed with respect to the baseline values. As for the descriptive analysis, data were expressed as mean and standard deviation (SD), median and interquartile range (IQR), and absolute number and percentage, as appropriate. Inferential analysis was based on within patient comparisons of continuous scores of different scales (namely EASI, BSA, DLQI, POEM, NRS subtests), through Wilcoxon test for the overall population and by group. The percentage (and 95% confidence interval [CI]) of patients achieving EASI 75, EASI 90 and EASI 100 at Week 16, 32 and 48 was also evaluated. The effect of different doses of upadacitinib 15 mg or 30 mg daily on EASI 75, EASI 90 and EASI 100 responses at different times was investigated by logistic regression analysis. To finalise this analysis, only the p value of the Wald test was reported. Statistical analysis was performed by R version 3.6.3.
3 Results
Overall, 146 patients (89 males and 57 females; mean age: 37.83 ± 14.4 years) were included in the analysis, with 125 patients achieving 16 weeks of treatment, whilst 113 and 97 patients achieved 32 and 48 weeks of treatment, respectively. Baseline demographic and clinical data of the study population are summarised in Table 1. Twenty-four percent (35/146) of the study population registered as smokers. Hypertension was the most common non-atopic comorbid conditions to be recorded (11%, 16/146, of patients) whereas diabetes was reported in 3 cases. No history of cardiovascular disorders (neither cardiovascular risk factors nor risk factors for venous thromboembolism) were reported.
The entire study population had been treated with at least one systemic agent prior to upadacitinib; in particular, a previous treatment with dupilumab was reported in 136/146 (93.2%) of patients and with cyclosporine in 110/146 (75.3%). Upadacitinib was initially prescribed at the dosage of 30 mg daily in 118 of 146 (80.8%) patients and 15 mg daily in 28/146 (19.2%) patients. The two patient sub-cohorts did not differ in terms of baseline disease severity (Table 2). Dose variation was observed in 38 of 146 patients (26%) for a total of 48 treatment courses. Dose reduction from 30 to 15 mg was frequently prescribed (n = 33 treatment courses) whereas dose escalation from 15 to 30 mg daily dosage was detected in 15 treatment courses. A temporary suspension of upadacitinib treatment occurred in 12 cases (12/146, 8.2%), most commonly due to anti-SARS-CoV-2 vaccination. Upadacitinib was prescribed as monotherapy in most cases (127/146, 87.0%), while in 16/146 (11.0%) of patients upadacitinib was combined with topical corticosteroids (more frequently mid-potency or super potent corticosteroids) and/or oral corticosteroids in 5/146 (3.4%) of cases.
3.1 Therapeutic Response
The mean baseline EASI score was 25 ± 11.2, which significantly dropped to 5.4 ± 7.9 at Week 16 (mean percentage EASI reduction: 78.4%), with a sustained response observed at 32 and 48 weeks (p < 0.001 for comparisons between baseline EASI score and EASI values at Week 32 or Week 48; Table 2).
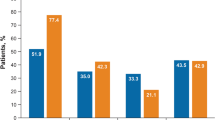
Eczema area severity index (EASI) 75, EASI 90 and EASI 100 responses were achieved by 78.2%, 47.6% and 28.2% of patients at 16 weeks and by 87.6%, 69.1% and 44.3% at Week 48, respectively (Fig. 1). The percentage of patients obtaining these therapeutic goals increased until Week 32 with a subsequent plateau (Fig. 1). A significant reduction of mean sleeplessness, skin pain, and itch NRS was observed from baseline to Week 16, 32 and 48 (p < 0.001 for all, Table 2).

Percentages of upadacitinib-treated patients achieving Eczema Area and Severity Index (EASI) 50, EASI 75, EASI 90, and EASI 100 responses through 48 weeks of treatment
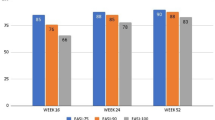
The improvement of AD manifestations was accompanied by a significant amelioration of patient’s QoL and patient’s perception of disease severity (significant reduction of DLQI and POEM, respectively, through 48 weeks) as early as Week 16 with a mean percentage DLQI reduction from baseline of 74.2%. The re-treatment after a mean period of 18.8 (± 12.4) days of suspension was not associated with loss of effectiveness in most patients, preserving the therapeutic response in 8/12 (66.7%) of cases. Overall, upadacitinib decreased disease severity with both dosages, showing significant reductions of mean EASI, DLQI, BSA, POEM, itch-NRS, pain-NRS, and sleep-NRS (Table 2). Treatment response observed in 15 mg upadacitinib-treated patients was comparable with that detected in 30 mg upadacitinib-treated patients, revealing no statistical difference between the two patient sub-cohorts. Along these lines, the percentage of patients achieving EASI 75, EASI 90, and EASI 100 response was similar between the two patient sub-cohorts (Fig. 2) with no significant differences in terms of likelihood in achieving treatment response (p value ranging from 0.19 to 0.90).

Treatment response was similar between 15 mg versus 30 mg upadacitinib dosage. EASI 75, EASI 90, and EASI 100 responses in patients treated with an initial dose of 15 mg (A) or 30 mg (B) upadacitinib. EASI Eczema Area and Severity Index
3.2 Safety
Overall, 26 of 146 (17.8%) patients experienced at least one AE during the treatment period. In total, 30 AEs were recorded, with a 2-fold higher frequency of AEs (20/30, 66.7%) than that detected within the first 16 weeks of treatment compared to the late period of observation (from Week 16 to Week 48), albeit the reports of serious AEs were equally distributed throughout the study period.
Most AEs were evaluated as mild to moderate, while in 4 cases the occurrence of AE led to drug discontinuation. Adverse events leading to drug discontinuation were represented by acute myocardial infarction (1 case), thrombophlebitis of lower limbs (1 case), heaviness and pain in the limbs associated with diffuse swelling and weight gain (1 case), haematuria associated with metaplasia of the bladder (1 case). Infections were the most frequently reported AEs (9/30, 30.0% of patients) with 3 cases of SARS-CoV-2 infection, 1 case of bronchopneumonia, 1 case of bronchitis, 1 case of recurrent upper respiratory tract viral infection, 1 case of urinary tract infection, 1 case of herpes zoster and 1 case of herpes simplex infection. Seven AEs (8/30, 26.7%) consisted of blood test abnormalities such as high levels of total cholesterol or creatine phosphokinase, anaemia, neutropenia, reduced platelet count. Folliculitis/acnes occurred in 5/146 patients (3.4%), 3 cases of thrombophlebitis or deep vein thrombosis (2.0%), and 1 case of acute myocardial infarction. Patients reporting thrombophlebitis were aged ≥65 years and males in 2 out of 3 cases. All were non-smokers and in one case AD was associated with hypothyroidism, Sjogren syndrome, and hypertension. Acute myocardial infarction occurred in a 69-year-old male, smoker, burdened by asthma and alcohol consumption habits. The patient sub-cohort aged ≥65 years was small (n = 5) and all presented at least one comorbid condition including diabetes, hypertension, hypothyroidism, Sjögren syndrome, asthma, allergic rhinitis, and allergic conjunctivitis. All patients were initially treated with upadacitinib 30 mg daily, with 2 out of 5 who reduced dosage to upadacitinib 15 mg daily during the observation period and 2 out of 5 who withdrew treatment because of AEs (acute myocardial infarction and deep vein thrombosis). Adverse events constituted the most frequent cause of treatment withdrawal (4/7). In one case, treatment withdrawal was due to both worsening of disease and patient’s decision. In addition, pregnancy led to treatment discontinuation in 2 cases (total of dropouts: 7/146 [4.8%] of treated patients).
4 Discussion
This interim analysis assessed effectiveness and safety of upadacitinib throughout 48 weeks of observation in a real-world adult AD population that, to our knowledge, consists of the largest reported upadacitinib-treated, real-world population observed through a 48-week period. The patient population included in our study consisted of patients with high disease burden (mean EASI and DLQI at baseline of 25 and 16.3, respectively) and high therapeutic need, as patients were unresponsive contraindicated or intolerant to the approved systemic therapies. Notably, this subset of patients, who were previously exposed to dupilumab, was not included in the upadacitinib clinical trials.
We detected a significant improvement in the clinical signs and symptoms of AD from Week 16 throughout the study period, with a sustained reduction in the mean values of all physician-reported (EASI and BSA) and patient-reported (Itch- Sleep- and Pain-NRS, DLQI, and POEM) disease severity outcomes, up to 48 weeks of treatment. The effectiveness detected in the overall population is in line with outcomes obtained in randomised clinical trials [9,10,11]. In particular, the percentage of EASI 75 (87.6% of patients at Week 48) and EASI 90 (69.1% of patients at Week 48) responses in our study were consistent with those reported in two large Phase III trials (MEASURE Up 1 and 2) testing upadacitinib in monotherapy, similar to our real-world study prescribing upadacitinib monotherapy in most cases (127/146, 87.0%) [10, 20]. Indeed, at Week 52, the proportion of patients achieving EASI 75 was 82.0% and 84.9% of patients treated with 15 mg and 30 mg, respectively, while EASI 90 response was achieved by 62.7% (15 mg upadacitinib) and 73.3% (30 mg upadacitinib) of patients in MEASURE Up 1 trial. Similarly in MEASURE Up 2, EASI 75 response was detected in 79.1% (15 mg upadacitinib) and 84.3% (30 mg upadacitinib) of patients, while EASI 90 was reported in 61.3% (15 mg upadacitinib) and 70.3% (30 mg upadacitinib) of patients [20].
The achievement of EASI 100 response in our study was slightly higher than in Phase III trials and likely obtained with the use of oral corticosteroids and/or super-potent topical corticosteroids combined with upadacitinib that was not allowed in a clinical trial setting. The achievement of complete or almost complete clearance in a consistent proportion of treated patients distinguishes upadacitinib from dupilumab, as also suggested by a head-to-head trial revealing a superior efficacy of upadacitinib in achieving EASI 90 and EASI 100 responses at Week 16 and also at Week 24 compared to dupilumab [12]. Albeit dupilumab is highly effective and safe in treating AD, residual skin lesions and partial itch relief are described [4,5,6, 21,22,23]. Indeed, in a real-world experience including 149 AD patients under treatment with dupilumab, less than 14% experienced a complete or almost complete resolution of AD signs and symptoms (i.e., EASI ≤ 1, NRS ≤ 1, and DLQI ≤ 1) after 16 months of observation [22]. In another large sub-cohort of adult AD subjects, over 24% of subjects continued to experience transient relapses of disease during treatment [23]. The achievement of high levels of skin clearance (EASI 90 and 100 or absolute EASI score ≤ 1) is clinically meaningful as it is associated with a significantly greater reduction on the impact of AD on sleep, emotional state, daily activities, and overall QoL [16, 24]. In the current study, the intention-to-treat analysis revealed that these clinical goals were achieved by patients treated either with 15 mg or 30 mg upadacitinib, with no significant difference in terms of treatment response rates and likelihood of achieving them between the two treatment groups (EASI 75, EASI 90, and EASI 100 at the various time points), although the number of patients treated with upadacitinib 15 mg daily was low. The maintained effectiveness observed through the modulation of upadacitinib dosage is clinically relevant considering that the frequency of some AEs that might occur during upadacitinib therapy is dose-dependent (i.e., creatine phosphokinase elevation, folliculitis/acne, zoster infection) and physicians may have the opportunity to modulate dosage during the treatment course optimising the therapeutic risk/benefit ratio and potentially lowering direct drug costs.
Overall, 17.8% (26/146) of our patients experienced at least one AE during the treatment period, and most of these events were evaluated as mild and did not cause treatment discontinuation. Most AEs occurred within the first 16 weeks of treatment, while the reports of serious AEs were equally distributed throughout the study period. This finding might be affected by an under-reporting of any AE that has been described, whereas serious AEs were found to be reported regularly [25, 26].
Notably, folliculitis/acne was observed in a significantly lower proportion of patients in comparison with clinical trials (3.4% vs 7%–15%) that, contrary to our study, also enrolled adolescent patients, who may suffer from acne before starting treatment or have a higher risk of experiencing acneiform eruptions under upadacitinib treatment. Only 4.8% (7 out of 146) of treated subjects discontinued upadacitinib, whereas a temporary treatment suspension, commonly due to anti-SARS-CoV-2 vaccination, was reported in 10.3% of patients.
Re-treatment after treatment suspension was not associated with loss of efficacy and this finding may have important therapeutic implications considering that, in a real-world setting, treatment suspension because of vaccination, surgery or other conditions may commonly occur. In light of EMA recommendations on the use of JAK inhibitors issued on January 23rd 2023, some considerations about the safety profile of JAK inhibitors should be addressed. Based on safety data from ORAL Surveillance, a head-to-head study performed in a sub-cohort of rheumatoid arthritis patients enriched for CV risk, revealed that tofacitinib did not meet the non-inferiority criteria for major adverse cardiovascular (MACE) events and malignancies compared with TNF inhibitors [27]. European Medicines Agency (EMA) extended a warning for the use of anti-JAK molecules in patients aged ≥ 65 years, long-term smokers or ex-smokers, or those with an increased cardiovascular, venous thromboembolism, or malignancy risk [28].
In these patient subpopulations, JAK inhibitors should be used with caution and considered as valid therapeutic opportunity only if no suitable alternative treatment options are available. However, recent data from an integrated analysis of 12 Phase IIb/III upadacitinib trials, involving more than 6000 patients and 15,000 patient-years of exposure across five indications including AD, revealed an overall favourable safety profile with no significant differences in the rate of malignancies, MACE and thromboembolism events between upadacitinib and active comparators adalimumab and methotrexate [29]. No study structured as the ORAL surveillance has yet been conducted with upadacitinib, thereby, further studies to corroborate upadacitinib safety profile are needed. In our study an assessment regarding the causal relationship between serious AEs and the use of upadacitinib was not performed. Apparently, at baseline no risk factors or very low risk factors could be identified. Smoking habits were slightly more frequent than the overall Italian general population (19% prevalence) [30]. We started 30 mg upadacitinib daily treatment in patients aged ≥65 years prior to the issued EMA recommendations. The likely use of upadacitinib should be considered with caution in these patients, as well as in those with cardiovascular, venous thromboembolism, and malignancy risk factors, but consideran eventual opportunity for the lowest dosage (15 mg daily) of upadacitinib. Being an interim analysis, not all patients potentially achieved the 48 weeks of observation (97/146, 66.4% of patients achieved 48-week therapy), and this could limit data interpretation. In addition, a variability in terms of visit schedule among centres or missing follow-up visits may occur in a real-world setting, affecting data collection. Furthermore, the comparison between the two patient sub-cohorts (15 mg vs 30 mg) should be considered with caution as patients treated with 15 mg daily comprised a small percentage of patients (28/146, 19.2%) who, in a conspicuous number of treatment courses (n = 15), increased dosage during the observation period.
5 Conclusion
This study provides strong evidence of a sustained response obtained by upadacitinib in AD patients, through 48 weeks of observation. This satisfactory response was also obtained in patients who had failed to respond to conventional or biological systemic agents, preserving an acceptable safety profile. Upadacitinib was shown to be advantageous in terms of flexibility in dose reduction or escalation as in 26% of treated cases upadacitinib dose was shaped on clinical needs that, in a real-world setting, might frequently change.
Change history
19 August 2023
A Correction to this paper has been published: https://doi.org/10.1007/s40257-023-00813-4
References
Weidinger S, Beck LA, Bieber T, et al. Atopic dermatitis. Nat Rev Dis Primers. 2018;4:1.
Barbarot S, Auziere S, Gadkari A, et al. Epidemiology of atopic dermatitis in adults: results from an international survey. Allergy. 2018;73:1284–93.
Siegels D, Heratizadeh A, Abraham S, et al. Systemic treatments in the management of atopic dermatitis: a systematic review and meta-analysis. Allergy. 2021;76(4):1053–76.
Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335–48.
de Bruin-Weller M, Thaçi D, Smith CH, et al. Dupilumab with concomitant topical corticosteroid treatment in adults with atopic dermatitis with an inadequate response or intolerance to ciclosporin A or when this treatment is medically inadvisable: a placebo-controlled, randomized phase III clinical trial (LIBERTY AD CAFÉ). Br J Dermatol. 2018;178:1083–101.
Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389:2287–303.
Wollenberg A, Blauvelt A, Guttman-Yassky E, et al. Tralokinumab for moderate-to-severe atopic dermatitis: results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol. 2021;184:437–49.
Calabrese L, Chiricozzi A, De Simone C, et al. Pharmacodynamics of Janus kinase inhibitors for the treatment of atopic dermatitis. Expert Opin Drug Metab Toxicol. 2022;18:347–55.
Guttman-Yassky E, Thaçi D, Pangan AL, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol. 2020;145:877–84.
Guttman-Yassky E, Teixeira HD, Simpson EL, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet. 2021;397:2151–68.
Reich K, Teixeira HD, de Bruin-Weller M, et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): results from a randomised, double-blind, placebo controlled, phase 3 trial. Lancet. 2021;397:2169–81.
Blauvelt A, Teixeira HD, Simpson EL, et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2021;157:1047–55.
Kamphuis E, Loman L, Han HL, et al. Experiences from daily practice of upadacitinib treatment on atopic dermatitis with a focus on hand eczema: results from the BioDay registry. Contact Dermatitis. 2023. https://doi.org/10.1111/cod.14276.
Napolitano M, Potestio L, Hansel K, et al. Efficacy and safety of upadacitinib in adult patients affected by moderate to severe atopic dermatitis: a 16-week real-life dual-centre experience. Clin Exp Dermatol. 2023;48:247–9.
Gargiulo L, Ibba L, Cortese A, et al. Real-life effectiveness and safety of upadacitinib in adults and adolescents with moderate-to-severe atopic dermatitis: a single-center 16-week study. Dermatol Ther (Heidelb). 2023;13:651–60.
Chiricozzi A, Gori N, Narcisi A, et al. Effectiveness and safety of upadacitinib in the treatment of moderate-severe atopic dermatitis: a multicentric, prospective, real-world, cohort study. Drugs R D. 2022;22:245–52.
Pereyra-Rodriguez JJ, Herranz P, Figuras-Nart I, et al. Upadacitinib for the treatment of atopic dermatitis in a Spanish cohort-real life: fifty-two-week follow-up results. Dermatitis. 2022;33:S124–7.
De Greef A, Ghislain PD, de Montjoye L, et al. Real-life effectiveness and tolerance of upadacitinib for severe atopic dermatitis in adolescents and adults. Adv Ther. 2023. https://doi.org/10.1007/s12325-023-02490-5.
Gori N, Ippoliti E, Antonelli F, et al. Successful response of upadacitinib in the treatment of atopic dermatitis lesions involving sensitive and visible areas resistant to dupilumab. Clin Exp Dermatol. 2023;2023:llad040. https://doi.org/10.1093/ced/llad040.
Simpson EL, Papp KA, Blauvelt A, et al. Efficacy and safety of upadacitinib in patients with moderate to severe atopic dermatitis: analysis of follow-up data from the measure up 1 and measure up 2 randomized clinical trials. JAMA Dermatol. 2022;158:404–13.
Gori N, Chiricozzi A, Malvaso D, et al. Successful combination of systemic agents for the treatment of atopic dermatitis resistant to dupilumab therapy. Dermatology. 2021;237:535–41.
Dal Bello G, Maurelli M, Schena D, et al. Drug survival of dupilumab compared to cyclosporin in moderate-to-severe atopic dermatitis patients. Dermatol Ther. 2020;33: e13979.
Patruno C, Fabbrocini G, Longo G, et al. Effectiveness and safety of long-term dupilumab treatment in elderly patients with atopic dermatitis: a multicenter real-life observational study. Am J Clin Dermatol. 2021;22:581–6.
Chiricozzi A, Gori N, Di Nardo L, et al. Therapeutic impact and management of persistent head and neck atopic dermatitis in dupilumab-treated patients. Dermatology. 2022;238:717–24.
Hazell L, Shakir SA. Under-reporting of adverse drug reactions: a systematic review. Drug Saf. 2006;29:385–96.
Lopez-Gonzalez E, Herdeiro MT, Figueiras A. Determinants of under-reporting of adverse drug reactions: a systematic review. Drug Saf. 2009;32:19–31.
Ytterberg SR, Bhatt DL, Mikuls TR, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386(4):316–26.
EMA/PRAC/68283/2022. https://www.ema.europa.eu/en/documents/referral/janus-kinase-inhibitors-jaki-article-20-procedure-prac-list-questions_en.pdf (2023).
Burmester GR, Cohen SB, Winthrop KL, et al. Safety profile of upadacitinib over 15 000 patient-years across rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis and atopic dermatitis. RMD Open. 2023;9(1):e002735. https://doi.org/10.1136/rmdopen-2022-002735.
https://www.salute.gov.it/portale/fumo/dettaglioContenutiFumo.jsp?lingua=italiano&id=5579&area=fumo&menu=vuoto#:~:text=Nel%202021%2C%20secondo%20dati%20ISTAT,donne%20il%2015%2C3%25. Accessed 30 May 2022.
Acknowledgements
The study and eCRF platform were promoted and supported by the Italian Society of Dermatology, SIDeMaST (Società Italiana di Dermatologia e Malattie Sessualmente Trasmesse). The authors received no external funding to perform this study. Study drug (upadacitinib) was provided by the manufacturer (AbbVie) through an expanded pre-approval access program. Medical writing was funded by SIDeMaST.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open access funding provided by Università Cattolica del Sacro Cuore within the CRUI-CARE Agreement.
Competing Interests
Alberto Maria Bertoldi has received honoraria for lectures for AbbVie and Sanofi. Andrea Chiricozzi has served as advisory board member and consultant and has received fees and speaker's honoraria or has participated in clinical trials for AbbVie, Almirall, Bristol Myers Squibb, Leo Pharma, Lilly, Janssen, Novartis, Pfizer and Sanofi Genzyme. Maria Concetta Fargnoli has served on advisory boards, received honoraria for lectures and/or research grants from AMGEN, Almirall, Abbvie, Boehringer-Ingelheim, BMS, Galderma, Kyowa Kyrin, Leo Pharma, Pierre Fabre, UCB, Lilly, Pfizer, Janssen, MSD, Novartis, Sanofi-Regeneron, Sunpharma. Silvia Ferrucci has been principal investigator in clinical trials for ABBVIE, Almirall, Galderma, Leo Pharma, Sanofi, Amgen, Novartis, Bayer and received honoraria for lectures for Novartis and Menarini. Giampiero Girolomoni has received personal fees from AbbVie, Abiogen, Almirall, Amgen, Biogen, Boehringer-Ingelheim, Bristol-Myers Squibb, Eli-Lilly, Leo Pharma, Merck Serono, Novartis, Pfizer, Samsung and Sanofi. Niccolò Gori served as advisory board member and received honoraria for lectures for AbbVie, Sanofi, and Leo-Pharma. Angelo Valerio Marzano reports consultancy/advisory boards disease-relevant honoraria from AbbVie, Boehringer-Ingelheim, Novartis, Pfizer, Sanofi and UCB. Michela Ortoncelli has served as advisory board member and/or consultant and has received fees and speaker's honoraria or has participated for clinical studies for AbbVie, Leo Pharma, and Sanofi Genzyme. Ketty Peris has served on advisory board, received honoraria for lectures and/or research grants for Abbvie, Almirall, Lilly, Galderma, Leo Pharma, Pierre Fabre, Novartis, Sanofi, Sun Pharma, Janssen. Simone Ribero has served as advisory board member and/or consultant and has received fees and speaker's honoraria or has participated for clinical studies for AbbVie, Almirall, Leo Pharma, Elli Lilly, Novartis, Pfizer and Sanofi Genzyme. Marco Romanelli has served as advisory board member and consultant and has received fees and speaker's honoraria or has participated in clinical trials for Abbvie Almirall, Bristol Myers Squibb, Leo Pharma, Lilly, Janssen Novartis, Sanofi Genzyme. The other authors have no competing interests to declare.
Data availability statement
Enquiries related to the data generated or analyzed during this study can be directed to the corresponding author.
Ethics statement
Approval of this study was obtained by the Local Ethics Committee—Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Università Cattolica del Sacro Cuore, Prot N.: 4434.
Patient consent to participate
Consent was required and obtained according to the ethical approval.
Patient consent to publish
The consent to publish was included in the consent form signed by all patients prior to participating in the study, according to the ethical approval.
Code availability
Not applicable.
Author contributions
All authors made substantial contribution to this manuscript and in detail: Andrea Chiricozzi conceptualized, designed the study, analyzed and interpretated the data, and drafted the article; Michela Ortoncelli, Donatella Schena, Silvia Mariel Ferrucci, Flaminia Antonelli, Riccardo Balestri, Michele Pellegrino, Aurora Parodi, Graziella Babino, Maddalena Napolitano, Mariateresa Rossi, Marco Romanelli, Alberto Maria Bertoldi, and Giovanni Palazzo acquired data and clinically managed patients; Annalisa Pitino and Giovanni Tripepi performed statistical analysis of data with graphical and tabular conceptualization; Gabriella Fabbrocini, Anna Balato, Angelo Valerio Marzano, Giampiero Girolomoni, Simone Ribero, Niccolò Gori, Maria Concetta Fargnoli, Luca Stingeni, and Ketty Peris reviewed the article critically for important intellectual content. All authors read and approved the final manuscript.
Additional information
The original online version of this article was revised due to correction in acknowledgment section.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Chiricozzi, A., Ortoncelli, M., Schena, D. et al. Long-term Effectiveness and Safety of Upadacitinib for Atopic Dermatitis in a Real-world Setting: An Interim Analysis Through 48 Weeks of Observation. Am J Clin Dermatol 24, 953–961 (2023). https://doi.org/10.1007/s40257-023-00798-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-023-00798-0