
Abstract
Background
Pivotal phase III studies demonstrated that abrocitinib, an oral, once-daily, JAK1-selective inhibitor, is effective treatment for moderate-to-severe atopic dermatitis (AD) as monotherapy and in combination with topical therapy.
Objective
The aim of this study was to evaluate the long-term safety of abrocitinib 200 mg and 100 mg in an integrated analysis of a phase IIb study, four phase III studies, and one long-term extension study.
Methods
Two cohorts were analyzed: a placebo-controlled cohort from 12- to 16-week studies and an all-abrocitinib cohort including patients who received one or more abrocitinib doses. Adverse events (AEs) of interest and laboratory data are reported.
Results
Total exposure in the all-abrocitinib cohort (n = 2856) was 1614 patient-years (PY); exposure was ≥ 24 weeks in 1248 patients and ≥ 48 weeks in 606 (maximum 108 weeks). In the placebo-controlled cohort (n = 1540), dose-related AEs (200 mg, 100 mg, placebo) were nausea (14.6%, 6.1%, 2.0%), headache (7.8%, 5.9%, 3.5%), and acne (4.7%, 1.6%, 0%). Platelet count was reduced transiently in a dose-dependent manner; 2/2718 patients (200-mg group) had confirmed platelet counts of < 50 × 103/mm3 at week 4. Incidence rates (IRs) were 2.33/100PY and 2.65/100 PY for serious infection, 4.34/100PY and 2.04/100PY for herpes zoster, and 11.83/100PY and 8.73/100PY for herpes simplex in the 200-mg and 100-mg groups, respectively. IRs for nonmelanoma skin cancer, other malignancies, and major adverse cardiovascular events were < 0.5/100PY for both doses. Five venous thromboembolism events occurred (IR 0.30/100PY), all in the 200-mg group. There were three deaths due to gastric carcinoma (diagnosed at day 43), sudden death, and COVID-19.
Conclusion
Abrocitinib, with proper patient and dose selection, has a manageable tolerability and longer-term safety profile appropriate for long-term use in patients with moderate-to-severe AD.
Trial Registries
ClinicalTrials.gov: NCT02780167, NCT03349060, NCT03575871, NCT03720470, NCT03627767, NCT03422822.
Video abstract
Integrated safety analysis of abrocitinib for the treatment of moderate-to-severe atopic dermatitis from the Phase II and III clinical trial program (MP4 1,02,272 kb)
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Digital Features for this article can be found at https://doi.org/10.6084/m9.figshare.14794854. |
This is the first report of an integrated safety analysis for abrocitinib, using data pooled from one phase IIb study, four phase III studies, and one long-term extension study. |
The most common adverse events were nausea, headache, and acne, which were all non-serious; most patients had events that were mild or moderate in severity. |
Results suggest that abrocitinib, with proper patient and dose selection, has a manageable tolerability and long-term safety profile appropriate for long-term use in patients with moderate-to-severe atopic dermatitis. |
1 Introduction
Atopic dermatitis (AD) is a common, chronic inflammatory skin disease associated with intense pruritus, pain, sleep disturbance, and diminished quality of life [1,2,3,4]. Patients with moderate-to-severe disease who do not respond adequately to topical therapies have few effective approved treatment options [1, 5, 6].
Abrocitinib is an oral, once-daily, Janus kinase-1 (JAK1)-selective inhibitor under investigation for treatment of moderate-to-severe AD. Signaling through JAK1 upregulates production of proinflammatory cytokines proposed to underlie the pathophysiology of AD, including interleukin (IL)-4, IL-13, IL-22, IL-31, and thymic stromal lymphopoietin [7, 8].
Abrocitinib development was predicated on the hypothesis that JAK1 selectivity would minimize dose-limiting safety findings associated with JAK inhibitor selectivity profiles, thereby allowing dosing to support robust efficacy. Meaningful abrocitinib efficacy at doses of 100 and 200 mg once daily, as monotherapy and in combination with topical therapies, was demonstrated in adolescent and adults with moderate-to-severe AD [9,10,11,12]. Although the 100-mg dose has efficacy comparable to dupilumab, abrocitinib 200 mg showed more rapid onset and more profound effect on itch (observed as early as day 2 after treatment initiation) and skin clearance. Abrocitinib also improved measures related to quality of life, depression, and anxiety [10].
Although efficacy of JAK1 inhibition is clear, safety of long-term JAK1 inhibition in the AD population has not yet been established. To date, JAK inhibitor safety was primarily established in patients with rheumatoid arthritis (RA) [13, 14]. JAK inhibition in RA is associated with risk of infections (including viral reactivation and opportunistic infections), malignancy, nonmelanoma skin cancer (NMSC), venous thromboembolism (VTE), increased low-density lipoprotein cholesterol (LDL-C) and high-density lipoprotein cholesterol (HDL-C), and various changes in hematologic parameters [13, 14].
The safety profile of abrocitinib in AD is likely to be impacted, in part, by differences between RA and AD patient populations. JAK1 selectivity is also among the characteristics that may contribute to the overall benefit/risk profile [7]. For example, increased risk of viral reactivation is likely to occur across JAK inhibitors due to effects on lymphocytes and interferon signaling; however, protection from tuberculosis or other mycobacterial infections might be observed with JAK1 inhibitors because of preserved IL-12 and IL-23 signaling [15,16,17]. Additionally, effects of JAK inhibition on hematologic physiology is complex, and differences in hematologic profiles might be anticipated based on JAK selectivity. Assessment of these safety aspects in AD patients requires larger data sets than are available from individual randomized controlled studies.
Herein we report an integrated safety analysis in patients with moderate-to-severe AD treated with abrocitinib in the phase II and III clinical trial program, including a long-term extension study. These pooled data provide the opportunity to understand how JAK1 inhibition and patient population influence the safety profile. Objectives of this analysis were to identify dose-related differences for adverse events and laboratory values in abrocitinib-treated patients and, where possible, to identify potential risk factors for adverse events (AEs). Most importantly, these data help determine whether safety and tolerability associated with JAK-1 inhibition support the risk/benefit balance for use of selected doses in the clinic.
2 Methods
2.1 Study Design and Patients
Safety data were pooled from five short-term studies (phase IIb, MONO-1, MONO-2, COMPARE, REGIMEN [open-label period]) and a long-term extension study (EXTEND]) [9,10,11,12]. The data cutoff date for this analysis was April 22, 2020. Patients had moderate-to-severe chronic AD for ≥ 1 year prior to the study, were aged ≥12 years (MONO-1, MONO-2, REGIMEN, and EXTEND) or ≥ 18 years (phase IIb and COMPARE) and had a recent history of nonresponse/intolerance to topical medication or required systemic therapies to control AD (Table S1, see electronic supplementary material [ESM]). In the phase III studies, there were no exclusion criteria related to upper age limit, history or risk of VTE, or history of dupilumab use (except for COMPARE). Patients with history of systemic infection requiring parenteral therapy in the past 6 months, chronic or acute skin infection requiring treatment within the last week, any history of disseminated herpes zoster or herpes simplex, or with platelets < 140 × 103/mm3, lymphocytes < 0.5×103/mm3, neutrophils < 1.2 × 103/mm3, or hemoglobin < 10.0 g/dL or hematocrit < 30% were excluded.
2.2 Analysis Datasets
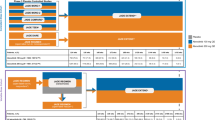
Safety data were assessed in the placebo-controlled cohort (Fig. 1a) and the all-abrocitinib cohort (Fig. 1b). The placebo-controlled cohort included participants in the 12- to 16-week studies who received abrocitinib 200 mg, 100 mg, or placebo. This dataset was used to assess abrocitinib safety relative to placebo, dose–response relationships for frequent adverse drug reactions, and laboratory changes. Additionally, this cohort allowed for evaluation of key subgroups: patients who received monotherapy (phase IIb, MONO-1, and MONO-2), patients who received abrocitinib with concomitant use of background topical therapy (COMPARE), and adolescent and adult patients (Fig. 1a).

Schematic of a placebo-controlled cohort and b all-abrocitinib cohort
The all-abrocitinib cohort included all patients who received one or more doses of abrocitinib 200 mg or 100 mg in placebo-controlled studies (if randomized to abrocitinib), 200 mg in the 12-week open-label portion of the REGIMEN trial and the EXTEND long-term extension trial (Table S1, see ESM; Fig. 1b). Day 1 of exposure in this pool was the first day of abrocitinib exposure; patients randomized to abrocitinib in parent studies continued to receive the same dose in the long-term extension study as received in the qualifying phase III study. This cohort enabled assessment of incidence rates (IRs; rate of occurrence of an AE during a given period), changes in laboratory parameters, and, where possible, risk factors for AEs.
2.3 Adverse Event Collection and Coding
AEs for the placebo-controlled cohort were coded using Medical Dictionary for Regulatory Activities (MedDRA) version 22.1 and AEs for the all-abrocitinib cohort were coded using MedDRA version 23.0 and classified as mild, moderate, or severe by the investigator (defined in Table S2, see ESM). AEs other than serious AEs (SAEs) were recorded from the day the patient entered the study through the last visit. SAEs were defined according to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Harmonised Tripartite Guideline Clinical Safety Data Management: Definitions and Standards for Expedited Reporting E2A [18]. SAEs were reported between the time a patient provided informed consent and 28 days after the last study dose or at any time after the last dose if the SAE was suspected to be related to drug treatment.
2.4 Adjudication of Events
Adjudication committees (external experts, blinded to treatment and independent from the study sponsor) were established to review opportunistic infections, malignancies, hepatic events, and cardiovascular events (including VTE).
2.5 Statistical Analyses
Statistical analyses were conducted in the safety analysis set, which included all patients who received one dose or more of study treatment. Continuous and categorical data were analyzed using descriptive statistics.
IRs were defined as the number of patients with events during the risk period divided by the sum of the durations of exposures of the patients during the risk period (time to last dose plus 28 days, time to death, time to last dose in the parent study for patients enrolled in the long-term extension study for the placebo-controlled cohort, or data cutoff date for ongoing studies for the all-abrocitinib cohort, whichever was smaller). For IR calculations, patient exposure was censored at the time of first event. IRs are presented as numbers of patients with events per 100 patient-years (PYs; i.e., incidence of events per 100 patients followed for 1 year) along with a 95% confidence interval (CI) computed using an exact method that assumed that the number of events followed a Poisson distribution.
Hazard ratios (HRs) were calculated from a Cox regression model with treatment group and parent study as covariates. Cox regression models were also used to identify risk factors in subgroups classified by intrinsic (e.g., age, sex, race) and extrinsic factors (i.e., background therapy, prior systemic therapy). Kaplan-Meier estimates were used to assess time to the first occurrence of an AE. Time to event was censored at the end of the risk period for patients who did not have an event.
3 Results
3.1 Patient Exposure and Demographics
The placebo-controlled cohort included 1540 patients: 590, 608, and 342 patients in the 200-mg, 100-mg, and placebo groups, respectively. The all-abrocitinib cohort included a total of 2856 patients: 1971 in the abrocitinib 200-mg and 885 in the 100-mg groups.
Baseline characteristics in the placebo-controlled cohort and all-abrocitinib cohort (200 or 100 mg) were comparable (Table 1). Median age was 33.0 and 31.0 years in the placebo-controlled and all-abrocitinib cohorts, respectively; proportions of adolescents were 8.1% and 12.7%, respectively; proportions of adults age ≥ 65 years were 6.1% and 5.1%, respectively. The majority of patients were male and White, although the number of Asian and Black or African American patients was substantial. Patients were from the United States, Canada, Australia, Asia (including Japan, China, Korea, and Taiwan), Europe, and South America. Approximately two-thirds of patients had moderate disease reflected in an Investigator’s Global Assessment (IGA) score of 3; almost half had previously used systemic therapy, including dupilumab. Baseline characteristics within each cohort were well balanced among the groups (Tables S3a and b, see ESM).
In the placebo-controlled cohort, mean (standard deviation [SD]) days of exposure was higher in abrocitinib groups (90.9 [21.9] days with 200 mg and 89.2 [25.2] days with 100 mg) than in the placebo group (83.1 [31.6] days) due to more discontinuations in placebo-treated patients. Among 2856 patients in the all-abrocitinib cohort, 1248 had ≥ 24 weeks and 606 had ≥48 weeks of cumulative abrocitinib exposure. Total exposure was 1614 PY. Among the 364 adolescent patients, 109 had ≥48 weeks of cumulative abrocitinib exposure (extent of exposure by dose is found in Table S4, see ESM). Maximum duration of exposure was 108 weeks.
3.2 Short-Term Safety and Tolerability
In the placebo-controlled cohort, AEs occurred in 68.3% of patients receiving abrocitinib 200 mg, 61.0% receiving 100 mg, and 55.0% receiving placebo; proportions of SAEs and severe AEs were similar across groups (Table 2). The most common AEs with a dose response and excess over placebo, which drove the difference in AEs overall, were nausea, headache, and acne. Most patients (94.2%) had events that were mild or moderate in severity. Most AEs were generally self-limited and seldom required interruption or permanent discontinuation of treatment.
3.2.1 Nausea
Nausea was the most frequently reported AE in the abrocitinib 200-mg group (Table 2). No nausea events were serious; one event (0.2%) in the 200-mg group was severe. Most resolved with no change or interruption to treatment: four events of nausea (two from each abrocitinib treatment group) led to discontinuation in the placebo-controlled cohort. Across all abrocitinib-treated patients, most nausea events occurred in the first week of treatment (63.5% of events with 200 mg and 72.3% of events with 100 mg). Median time to resolution of nausea was 15 days (17 days for 200 mg and 8 days for 100 mg). Female patients had higher frequency of nausea compared with male patients (18.1% vs 7.6%).
3.2.2 Headache
A dose-related increase in the proportion of patients with headache was observed in abrocitinib-treated patients (Table 2). No headache events were categorized as SAEs or severe AEs. Three abrocitinib-treated patients overall (0.3%) had headache events leading to study discontinuation (two in the 200-mg and one in the 100-mg group); no placebo-treated patients discontinued due to headache. Among patients who experienced headache, the initial event, in > 40%, occurred within the first week of treatment (47.6% with 200 mg; 39.6% with 100 mg); median time to resolution was 4 days (5 days in the 200-mg and 3.5 days in the 100-mg group).
3.2.3 Acne
There was a dose-related increase in acne events (4.7%, 1.6%, and 0% for 200-mg, 100-mg, and placebo groups, respectively; Table 2). There were no SAEs, severe AEs, or AEs that led to discontinuation. Median time to resolution was 247 days, with approximately one-third (33.0%) of the first events resolved by day 84 as calculated by Kaplan-Meier estimate. There was no clustering of acne events early or late in treatment, with 35.6% occurring after 8 weeks. Other details for acne events were not captured.
3.3 Longer-Term Safety and Adverse Events of Special Interest
The two dose groups in the all-abrocitinib cohort had similar proportions of AEs, SAEs, and severe AEs (Table 3). SAEs occurred most frequently in infections and infestations system organ class (SOC). Death occurred in three patients.
3.3.1 Serious Infections
The proportion of patients with serious infections (SAEs in the infections and infestation SOC) was similar across placebo and abrocitinib groups in the placebo-controlled cohort and in the 200-mg and 100-mg groups in the all-abrocitinib cohort (Table 4). The most frequent serious infections (0.2% or less for each type) in abrocitinib-treated patients included pneumonia (six events of pneumonia, pneumonia bacterial, or pneumonia pneumococcal), herpes simplex (four events of herpes simplex, ophthalmic herpes simplex, or oral herpes simplex), and herpes zoster (four events of herpes zoster or ophthalmic herpes zoster). Per protocol, serious infections that met SAE criteria required withdrawal of study drug and study termination. A 69-year-old Black or African American woman (200-mg group) tested positive for coronavirus disease 2019 (COVID-19) on day 84, was hospitalized, and died of COVID-19 on day 107. There were no events of tuberculosis or fungal or bacterial opportunistic infections.
3.3.2 Herpes Zoster
Incidence rates of herpes zoster in the placebo-controlled cohort were dose-related (Table 4). Most events overall were mild or moderate. Four events of herpes zoster (0.2%; all in the abrocitinib 200-mg group) resulted in permanent discontinuation of study treatment. Patients with herpes zoster ranged from 12 to ≥ 75 years of age. A multivariate analysis found that abrocitinib 200 mg, age ≥65 years, and severe disease at baseline were associated with higher risk of herpes zoster (Table S5, see ESM). All events of adjudicated opportunistic infections were cutaneous multidermatomal herpes zoster (0.60/100 PY [95% CI 0.29–1.10]). Similar to herpes zoster events overall, these events were mostly mild or moderate in severity and resolved with oral antiviral therapy.
3.3.3 Herpes Simplex and Eczema Herpeticum
Events of herpes simplex were dose-related, with higher incidence with abrocitinib than placebo; however, no dose–response relationship was observed for eczema herpeticum events (there were none in the abrocitinib 200-mg group; Table 4). When eczema herpeticum and herpes simplex events were considered together, there was no dose response in the abrocitinib 200-mg and 100-mg groups (16.21 and 16.05/100 PY, respectively), but these IRs were higher than in the placebo group (9.88/100 PY). Similar dose–response relationships were observed in the all-abrocitinib cohort.
There were 23 total cases of eczema herpeticum observed in abrocitinib-treated patients. Four events were serious (all in the 100-mg group); 91.7% resolved and five led to study discontinuation (in one patient [0.1%] in the 200-mg group and four [0.5%] in the 100-mg group). Using multivariate analysis, the most significant risk factors for herpes simplex or eczema herpeticum included having medical history of herpes simplex or eczema herpeticum and region other than the United States, Canada, or Australia (Table S6, see ESM). Events of eczema herpeticum in the all-abrocitinib cohort were less frequent in patients with IGA of 0 or 1 (Table S7, see ESM) when events were stratified by IGA score before or immediately after the event start day. Among the 23 events of eczema herpeticum, 21 (91.2%) occurred in patients with uncontrolled AD (IGA >1).
3.3.4 Malignancy
There were seven cases of NMSC: four patients (0.2%) in the 200-mg and three patients (0.3%) in the 100-mg group. The four cases in the 200-mg group were squamous cell carcinoma; cases in the 100-mg group included two cases of basal cell carcinoma and one case of T-cell cutaneous lymphoma. Most cases (5/7; 71.4%) were in patients ≥ 60 years old and occurred in the first 3 months of abrocitinib treatment.
There were three events of adjudicated malignancies, including two of prostate cancer. One event occurred in a 73-year-old White patient (100-mg group) with a history of an enlarged prostate and another occurred in a 68-year-old White patient (200-mg group). The remaining case, in a 78-year-old White woman (200-mg group), was a gastric adenocarcinoma diagnosed on study day 43 by computed tomography showing carcinomatosis with multifocal hepatic metastases. This patient died of the gastric adenocarcinoma approximately 9 months after study discontinuation.
3.3.5 Cardiovascular Safety
In the all-abrocitinib cohort, three SAEs were adjudicated as major adverse cardiovascular events (MACE; two events of myocardial infarction and one event of sudden death). A 73-year-old White woman (100-mg group) with a history of aortic sclerosis and calcification and untreated hypertension experienced sudden death on day 107, which occurred 22 days after discontinuation of abrocitinib; a 60-year-old White man (200-mg group) with a body mass index (BMI) of 32 kg/m2 and a history of impaired glucose tolerance had myocardial infarction on day 104; and a 63-year-old White man (200-mg group) with a history of hypertension, chronic obstructive pulmonary disease, and hypercholesterolemia had myocardial infarction on day 159. The IR for MACE was 0.18/100 PY (95% CI 0.04–0.52).
Two patients were adjudicated as having had a transient ischemic attack, although neither event was reported as such. A 55-year-old White woman (100-mg group) with a medical history of seizure disorder, meningitis, and pulmonary hypertension had an event reported as seizure, and a 75-year-old White man (100-mg group) had an event reported as vertigo.
There were five events of VTE (IR 0.30/100 PY), all in the 200-mg group. Three SAEs were adjudicated as pulmonary embolism (PE): a 55-year-old White male (200-mg group) with a medical history of back pain, vertigo, asthma, and environmental allergies had a PE on day 80; a 68-year-old White female (200-mg group) with estrogen use and a history of uterine prolapse, menopause, hypertension, increased blood cholesterol, and first-degree atrioventricular block had a PE on day 98; and a 16-year-old Black or African American male (200-mg group) with morbid obesity and an extensive family history of PE (including an 18-year-old brother with PE) had a PE on day 565 (Table S8, see ESM). No consistent pattern of change in platelet counts was observed in these patients, suggesting such changes were unrelated to VTE occurrence (Table S8, see ESM). The IR for PE was 0.18/100 PY (95% CI 0.04–0.52). Two AEs were adjudicated as deep venous thrombosis (DVT). A 44-year-old White female (200-mg group) experienced calf thrombosis after arthroscopic surgery on day 232 and a 50-year-old Asian female (200-mg group) with a BMI of 33.7 kg/m2 and a history of hypertension experienced superficial thrombophlebitis with Doppler ultrasound showing small thrombus formation in the left great saphenous vein and left superficial femoral vein on day 48. The IR for DVT was 0.12/100 PY (95% CI 0.01–0.43).
3.3.6 Pregnancy
Pregnancy occurred in seven patients: one pregnancy is ongoing without complication; there were three healthy births, two spontaneous miscarriages, and one unknown outcome.
3.3.7 Laboratory Abnormalities
There was a dose-dependent decrease in platelets in the placebo-controlled cohort, with median values reaching nadir at week 4 (Fig. 2a). Median platelet counts subsequently increased and plateaued at week 12, with final 12-week values remaining below baseline. Most patients (> 95%) maintained platelet value > 100 × 103/mm3. Nine patients in the 200-mg group had confirmed platelet values < 75 × 103/mm3 between week 2 and month 1 of exposure; all patients but one were ≥ 65 years of age. Two of 2718 (0.1%) patients (among the nine in the 200-mg group with confirmed platelet values < 75 × 103/mm3) met discontinuation criteria for platelet count (confirmed < 50 × 103/mm3). No clinically meaningful AEs of hemorrhage were associated with low platelet counts.


Clinical safety data from baseline to week 16 for a platelets, b absolute lymphocyte count, c absolute neutrophil count, and d hemoglobin. IQR interquartile range, Q quarter. Boxes span the IQR; stars represent the median value; whiskers delineate the maximum and minimum values (maximum = Q3 + 1.5 × IQR; minimum = Q1 − 1.5 × IQR); circles represent outliers
In the all-abrocitinib cohort, there were no changes over time in median absolute lymphocyte count (ALC; Fig. 2b), absolute neutrophil count (ANC; Fig. 2c), or hemoglobin values (Fig. 2d). Four of 2832 (0.1%) patients met discontinuation criteria for lymphocytes (confirmed ALC < 0.5 × 103/mm3), all in the 200-mg group. These four patients were ≥ 65 years of age and events in three patients occurred in the first four treatment weeks. No patients had confirmed ANC < 1 × 103/mm3. One patient in the 200-mg group had confirmed hemoglobin < 8 g/dL: a 66-year-old Black or African American female with no known cause for decreased hemoglobin.
Abrocitinib had a dose-dependent effect on LDL-C (Fig. 3a) and HDL-C from baseline to week 16; there was no notable change in LDL-C/HDL-C ratio over 16 weeks (Fig. 3b). There were more shifts to above patients’ baseline in the abrocitinib groups relative to the placebo group (Table S9, see ESM) at both week 4 and the final visit (week 12/week 16). Most patients had baseline values < 130 mg/dL, and most values remained < 130 mg/dL throughout the study period. There was a dose-related increase in creatine kinase that began at week 4 and plateaued at week 8 (Fig. 4). There was no meaningful difference in the proportions of patients with creatine kinase 5× the upper limit of normal (ULN) (2.7%, 2.3%, 3.8%) or 10× ULN (1.8%, 0.3%, 1.5%) in the placebo, abrocitinib 100-mg, or abrocitinib-200 mg groups, respectively. Dose-related increase in blood creatine phosphokinase AEs was observed with abrocitinib (Table 2); no cases of rhabdomyolysis were reported.

Mean percent change from baseline in a LDL-C and b LDL/HDL ratio by visit in the placebo-controlled cohort. CI confidence interval, HDL high-density lipoprotein, LDL low-density lipoprotein, LDL-C low-density lipoprotein cholesterol. Error bars represent standard error

Line plot of median (Q1, Q3) creatinine kinase (U/L) data by visit in the placebo-controlled cohort. Q quarter. Error bars represent interquartile range
In the placebo-controlled cohort, there was no meaningful change over time in measures of central tendency for alanine aminotransferase (ALT) or aspartate aminotransferase. There was no dose-related increase in patients with ALT >3 × ULN in the abrocitinib 100-mg or 200 mg-groups (0.5% in each) relative to placebo (1.5%). No AEs were adjudicated as Hy’s law, highly likely drug-induced liver injury, or definite drug-induced liver injury.
4 Discussion
This is the first report of an integrated safety analysis for abrocitinib, a selective JAK1 inhibitor with robust efficacy in adults and adolescents with moderate-to-severe AD. This initial analysis demonstrates that abrocitinib, with appropriate dose and patient selection, has a manageable longer-term safety and tolerability profile that supports its use in patients with moderate-to-severe AD.
Most AEs were mild, self-limited, and infrequently required interruption or permanent discontinuation of abrocitinib therapy. The most common dose-related, drug-related AEs included nausea, vomiting, upper abdominal pain, herpes simplex, headache, dizziness, acne, and creatine phosphokinase increase (without rhabdomyolysis). The most common adverse reactions (nausea and headache) first occurred within 2 weeks of treatment initiation and seldom led to discontinuation. Two phase I studies that examined abrocitinib in fed and fasted states (NCT02163161 and NCT04065633) found that gastrointestinal symptoms, including nausea, occurred only when patients were in the fasted state, suggesting that nausea may be related to local gastric concentration of abrocitinib (Pfizer, data on file), and the effects may be mitigated with food. Acne events are seen across the JAK class; acne occurred in 14% of patients with moderate-to-severe AD treated with upadacitinib 30 mg once daily in a 16-week study [19]. No acne events in abrocitinib-treated patients were severe, and none led to treatment discontinuation. Further study will be required to understand the nature of the acne and mechanisms involved.
These pooled data allowed for an assessment of the potential impact of JAK1 selectivity on infections and hematologic changes. Serious and opportunistic infections and viral reactivation are associated with JAK inhibition [20, 21]. This analysis of abrocitinib in patients with AD found no dose-response relationship for serious infections or an excess of events compared with placebo. The most frequent serious infections were herpetic in nature, and a dose-related increase in herpes zoster infections was observed, which is consistent with the viral reactivation seen with other JAK inhibitors [14]. This was anticipated, as JAK1 inhibitors block signaling of type 1 and type 2 interferons and cytokines via the gamma chain of the IL-2 receptor and are important in lymphocyte development [22,23,24]. Unlike with tofacitinib and baricitinib [21, 25], an increased rate of herpes zoster in Asian patients was not observed. There were no opportunistic events of tuberculosis, fungal infections (including invasive fungal infections), mycobacterial infections, or infections with other intracellular bacteria. Differing results may be due to age of enrolled patients, as older patients are more at risk for infections [7, 26, 27]. Additionally, JAK inhibitor selectivities are different. JAK1-selective inhibitors are expected to preserve signaling of IL-12 and IL-23, cytokines thought to be important in protecting against opportunistic infections [15,16,17]. Although this suggests preserved protection with JAK1 inhibition, monitoring for tuberculosis prior to the initiation of abrocitinib will be important.
One aspect that may be unique to the AD population is the relationship between herpes simplex and eczema herpeticum. Patients with AD have a propensity to develop herpes simplex, and in a minority of patients, these events may develop into eczema herpeticum [28]. In this analysis, there was a dose-related increase in herpes simplex events; however, eczema herpeticum events were more frequent in the placebo and abrocitinib 100-mg groups. This suggests that, although herpes simplex is more frequent with abrocitinib 200 mg, these herpes simplex infections do not tend to become disseminated eczema herpeticum. Patients who develop eczema herpeticum have more severe AD, as defined by the Eczema Area and Severity Index, and a more T helper 2-polarized disease, with higher concentrations of immunoglobulin E and eosinophils [29]. Effects of JAK inhibition on eczema herpeticum may be related to improvement in skin barrier function and immunologic abnormalities [30, 31].
JAK selectivity may affect the pattern of changes in laboratory values associated with JAK inhibition. Treatment with the JAK1/2 inhibitor baricitinib was shown to increase platelet counts [32]. In contrast, abrocitinib was associated with an early, dose-related, transient decrease in platelets that recovered toward baseline over the course of treatment. Changes in platelets associated with JAK inhibitors are proposed to occur via mechanisms that affect synthesis and degradation of thrombopoietin (TPO), the primary regulator of platelet homeostasis [33, 34]. One such mechanism is driven by blockade of JAK2-mediated inhibition of TPO uptake and degradation and is associated with increased platelet production, as observed with baricitinib [35]. Preclinical studies showed that deletion of JAK2 in the megakaryocyte lineage results in increased platelet production [36]. Given the in vitro selectivity of abrocitinib targeting JAK1 compared with JAK2 (28-fold) at therapeutic doses [37], a TPO-driven mechanism is less likely. The other mechanism, which occurs via JAK1-dependent IL-6–mediated hepatic TPO production, is associated with transient decreased platelet production, consistent with effects observed with abrocitinib [34, 35]. Emerging evidence also points to a direct effect of JAK1-mediated IL-6 signaling on hematopoietic stem and progenitor cell pools [38,39,40], which could lead to transient reduction of IL-6–dependent hematopoietic progenitors, resulting in low platelet and lymphocyte production [35]. Although no change in ALC was noted with abrocitinib over time, four patients (0.2%) had confirmed ALC < 0.5 × 103/mm3 in the first 4 weeks of therapy.
Interactions between JAK1- and JAK2-mediated activity are complex. Our data suggest that abrocitinib preserves JAK1 selectivity and that decreased platelet levels associated with abrocitinib may occur via IL-6–mediated decreases in TPO synthesis. This is supported by evidence that IL-6 inhibition with monoclonal antibody tocilizumab is also associated with a decrease in platelet concentration [41].
Both lymphocyte and platelet effects are more pronounced in older patients. These may be due to age-related decreases in immune function in bone marrow and peripheral blood [42]. Because effects on platelets and lymphocytes are predictable regarding timing and patient age, they are amenable to monitoring in the clinic. No clinically meaningful changes in neutrophil or hemoglobin levels were noted with abrocitinib treatment; any such changes are manageable with appropriate monitoring early in treatment.
Other events related to JAK inhibition, such as malignancy and VTE, are likely to be managed similarly across this class of agents, either because they occur regardless of selectivity or occur infrequently enough that it is difficult to determine differences based on JAK selectivity. The number of malignancies in the abrocitinib development program was small, limiting IR precision. Because JAK inhibitors may theoretically increase risk of malignancies [43], including lymphoma and NMSC, this potential risk remains for abrocitinib. Most cases of NMSCs occurred within the first 3 months of abrocitinib treatment, with none accruing thereafter. Identification of cases within 3 months may be due to more frequent assessment by dermatologists in clinical trials or related to skin clearance, and not necessarily to JAK inhibition. Periodic examination of patients for NMSC should be considered, particularly those with risk factors (e.g., older age, history of UV phototherapy).
Dose-related events of VTE occurred in patients treated with abrocitinib and other JAK inhibitors [44, 45]. A population-based cohort study in a Danish health care registry found VTE IR of 0.14 (95% CI 0.12–0.16) per 100 PY in individuals with AD compared with 0.12 (95% CI 0.11–0.12) per 100 PY for an age- and sex-matched general population control group (Pfizer, data on file). In our analysis, adjudicated IR for VTE in the all-abrocitinib cohort was 0.3/100 PY (95% CI 0.1–0.7), with all five events occurring in patients receiving abrocitinib 200 mg. This suggests that appropriate patient selection (e.g., excluding patients with high risk of VTE such as previous PE or recent hospitalization) to minimize this risk may need to be considered. Note that exclusion criteria for VTE risk were added later in the abrocitinib development program as the class risk for VTE became clearer.
There were three adjudicated MACE with abrocitinib (IR 0.18/100 PY [95% CI 0.04–0.52]). In a population-based cohort study of patients in a Danish health care registry, IRs for patients with AD and matched controls were 0.26 (95% CI 0.24–0.29) per 100 PY and 0.22 (95% CI 0.21–0.23) per 100 PY, respectively (Pfizer, data on file).
Appropriate dose selection can minimize risk related to abrocitinib. The more robust efficacy of abrocitinib 200 mg, both its rapidity and depth of effect, suggests that this dose may be preferred for many patients. The safety and tolerability profile described here supports this dose for most patients, including adolescents. However, the 100-mg dose may be a more appropriate starting dose for patients with higher risk for or lower tolerance for adverse reactions. Although most patients aged ≥ 65 years tolerated abrocitinib 200 mg well, many dose-related effects, such as hematologic changes and herpes zoster, occurred at higher rates in this age group. Vaccination for herpes zoster should be given to eligible patients prior to initiating abrocitinib [21]. Most abrocitinib-treated patients did not have characteristics traditionally associated with cardiovascular risk (i.e., older age, smoking, history of cardiovascular disease, and diabetes). Although most patients with moderate-to-severe AD are unlikely to need lipid monitoring and management, patients with those risk factors may need closer attention.
5 Conclusions
This is the first report of longer-term safety of abrocitinib in patients with moderate-to-severe AD. Abrocitinib, with proper patient and dose selection, has a manageable tolerability and longer-term safety profile appropriate for long-term use. The long-term extension study is ongoing, and future analyses and additional data will provide further information on the long-term safety profile of abrocitinib.
Change history
02 May 2022
A peer-reviewed video abstract was retrospectively added to this publication.
13 October 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40257-021-00638-z
References
Boguniewicz M, Fonacier L, Guttman-Yassky E, Ong PY, Silverberg J, Farrar JR. Atopic dermatitis yardstick: practical recommendations for an evolving therapeutic landscape. Ann Allergy Asthma Immunol. 2018;120(1):10-22 e2.
Newton L, DeLozier AM, Griffiths PC, Hill JN, Hudgens S, Symonds T, et al. Exploring content and psychometric validity of newly developed assessment tools for itch and skin pain in atopic dermatitis. J Patient Rep Outcomes. 2019;3(1):42.
Silverberg JI, Gelfand JM, Margolis DJ, Boguniewicz M, Fonacier L, Grayson MH, et al. Pain is a common and burdensome symptom of atopic dermatitis in United States Adults. J Allergy Clin Immunol Pract. 2019;7(8):2699-706.e7.
Vakharia PP, Chopra R, Sacotte R, Patel KR, Singam V, Patel N, et al. Burden of skin pain in atopic dermatitis. Ann Allergy Asthma Immunol. 2017;119(6):548-52 e3.
Katoh N, Ohya Y, Ikeda M, Ebihara T, Katayama I, Saeki H, et al. Japanese guidelines for atopic dermatitis 2020. Allergol Int. 2020;69(3):356–69.
Wollenberg A, Barbarot S, Bieber T, Christen-Zaech S, Deleuran M, Fink-Wagner A, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part II. J Eur Acad Dermatol Venereol. 2018;32(6):850–78.
He H, Guttman-Yassky E. JAK inhibitors for atopic dermatitis: an update. Am J Clin Dermatol. 2019;20(2):181–92.
Howell MD, Kuo FI, Smith PA. Targeting the Janus kinase family in autoimmune skin diseases. Front Immunol. 2019;10:2342.
Gooderham MJ, Forman SB, Bissonnette R, Beebe JS, Zhang W, Banfield C, et al. Efficacy and safety of oral Janus kinase 1 inhibitor abrocitinib for patients with atopic dermatitis: a phase 2 randomized clinical trial. JAMA Dermatol. 2019;155(12):1371–9.
Silverberg JI, Simpson EL, Thyssen JP, Gooderham M, Chan G, Feeney C, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156(8):863–73.
Simpson EL, Sinclair R, Forman S, Wollenberg A, Aschoff R, Cork M, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396(10246):255–66.
Bieber T, Simpson EL, Silverberg JI, Thaçi D, Paul C, Pink AE, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;864(12):1101–12.
Harigai M, Honda S. Selectivity of Janus kinase inhibitors in rheumatoid arthritis and other immune-mediated inflammatory diseases: is expectation the root of all headache? Drugs. 2020;80(12):1183–201.
Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13(4):234–43.
Boisson-Dupuis S. The monogenic basis of human tuberculosis. Hum Genet. 2020;139(6–7):1001–9.
Jindal AK, Suri D, Guleria S, Rawat A, Garg S, Bal A, et al. Recurrent salmonella typhi infection and autoimmunity in a young boy with complete IL-12 receptor β1 deficiency. J Clin Immunol. 2019;39(4):358–62.
Smith NL, Denning DW. Clinical implications of interferon-gamma genetic and epigenetic variants. Immunology. 2014;143(4):499–511.
ICH Harmonised Tripartite Guideline: Clinical Safety Data Management: Definitions and Standards for Expedited Reporting E2A [Internet]. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use [Updated October 24 1994]. https://database.ich.org/sites/default/files/E2A_Guideline.pdf. Accessed 1 April 2021.
Guttman-Yassky E, Thaci D, Pangan AL, Hong HC, Papp KA, Reich K, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol. 2020;145(3):877–84.
Harigai M. Growing evidence of the safety of JAK inhibitors in patients with rheumatoid arthritis. Rheumatology (Oxford). 2019;58(suppl 1):i34–42.
Sunzini F, McInnes I, Siebert S. JAK inhibitors and infections risk: focus on herpes zoster. Ther Adv Musculoskelet Dis. 2020;12:1759720X20936059.
Lee AJ, Ashkar AA. The dual nature of type I and type II interferons. Front Immunol. 2018;9:2061.
Waickman AT, Park JY, Park JH. The common gamma-chain cytokine receptor: tricks-and-treats for T cells. Cell Mol Life Sci. 2016;73(2):253–69.
Gadina M, Hilton D, Johnston JA, Morinobu A, Lighvani A, Zhou YJ, et al. Signaling by type I and II cytokine receptors: ten years after. Curr Opin Immunol. 2001;13(3):363–73.
Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, Del Carmen ML, Reyes Gonzaga J, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med. 2017;376(7):652–62.
Marra F, Parhar K, Huang B, Vadlamudi N. Risk factors for herpes zoster infection: a meta-analysis. Open Forum Infect Dis. 2020;7(1):ofaa005-ofaa.
Toruner M, Loftus EV Jr, Harmsen WS, Zinsmeister AR, Orenstein R, Sandborn WJ, et al. Risk factors for opportunistic infections in patients with inflammatory bowel disease. Gastroenterology. 2008;134(4):929–36.
Wollenberg A, Zoch C, Wetzel S, Plewig G, Przybilla B. Predisposing factors and clinical features of eczema herpeticum: a retrospective analysis of 100 cases. J Am Acad Dermatol. 2003;49(2):198–205.
Beck LA, Boguniewicz M, Hata T, Schneider LC, Hanifin J, Gallo R, et al. Phenotype of atopic dermatitis subjects with a history of eczema herpeticum. J Allergy Clin Immunol. 2009;124(2):260–9.
Khattri S, Shemer A, Rozenblit M, Dhingra N, Czarnowicki T, Finney R, et al. Cyclosporine in patients with atopic dermatitis modulates activated inflammatory pathways and reverses epidermal pathology. J Allergy Clin Immunol. 2014;133(6):1626–34.
Tintle S, Shemer A, Suarez-Farinas M, Fujita H, Gilleaudeau P, Sullivan-Whalen M, et al. Reversal of atopic dermatitis with narrow-band UVB phototherapy and biomarkers for therapeutic response. J Allergy Clin Immunol. 2011;128(3):583-93 e1-4.
Olumiant. Prescribing information. Eli Lilly and Company; 2020.
Hoffmeister KM. TPO-logy accepted. Blood. 2018;132(6):555–7.
Kaser A, Brandacher G, Steurer W, Kaser S, Offner FA, Zoller H, et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: role in inflammatory thrombocytosis. Blood. 2001;98(9):2720–5.
Koride S, Nayak S, Banfield C, Peterson MC. Evaluating the role of Janus kinase pathways in platelet homeostasis using a systems modeling approach. CPT Pharmacometr Syst Pharmacol. 2019;8(7):478–88.
Meyer SC, Keller MD, Woods BA, LaFave LM, Bastian L, Kleppe M, et al. Genetic studies reveal an unexpected negative regulatory role for Jak2 in thrombopoiesis. Blood. 2014;124(14):2280–4.
Vazquez ML, Kaila N, Strohbach JW, et al. Identification of N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfonamide (PF-04965842): a selective JAK1 clinical candidate for the treatment of autoimmune diseases. J Med Chem. 2018;61(3):1130–52.
Bernad A, Kopf M, Kulbacki R, Weich N, Koehler G, Gutierrez-Ramos JC. Interleukin-6 is required in vivo for the regulation of stem cells and committed progenitors of the hematopoietic system. Immunity. 1994;1(9):725–31.
Ikebuchi K, Wong GG, Clark SC, Ihle JN, Hirai Y, Ogawa M. Interleukin 6 enhancement of interleukin 3-dependent proliferation of multipotential hemopoietic progenitors. Proc Natl Acad Sci USA. 1987;84(24):9035–9.
Tie R, Li H, Cai S, Liang Z, Shan W, Wang B, et al. Interleukin-6 signaling regulates hematopoietic stem cell emergence. Exp Mol Med. 2019;51(10):1–12.
Actemra. Prescribing information. Genentech Inc; 2020.
Pritz T, Weinberger B, Grubeck-Loebenstein B. The aging bone marrow and its impact on immune responses in old age. Immunol Lett. 2014;162(1 pt B):310–5.
Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;17(1):78.
Taylor PC, Weinblatt ME, Burmester GR, Rooney TP, Witt S, Walls CD, et al. Cardiovascular safety during treatment with baricitinib in rheumatoid arthritis. Arthritis Rheumatol. 2019;71(7):1042–55.
Bieber T, Thyssen JP, Reich K, Simpson EL, Katoh N, Torrelo A, et al. Pooled safety analysis of baricitinib in adult patients with atopic dermatitis from 8 randomized clinical trials. J Eur Acad Dermatol Venereol. 2021;35(2):476–85.
Acknowledgments
Editorial/medical writing support under the guidance of the authors was provided by Irene Park, PhD, and Mariana Ovnic, PhD, at ApotheCom, San Francisco, CA, USA, and was funded by Pfizer Inc., New York, NY, USA, in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med. 2015;163:461–4).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Pfizer, Inc.
Conflicts of interest and financial disclosures
E. L. Simpson reports grants from Pfizer Inc., Eli Lilly, Kyowa Kirin, LEO Pharma, Merck, and Regeneron and personal fees from Pfizer Inc., Bausch Health (Valeant), Dermira, Eli Lilly, Galderma, LEO Pharma, Menlo Therapeutics, Novartis, Regeneron, and Sanofi Genzyme. J. I. Silverberg is an investigator for AbbVie, Celgene, Eli Lilly, GSK, Kiniksa, LEO Pharma, Menlo Therapeutics, Realm Therapeutics, Regeneron, Roche, and Sanofi; a consultant for Pfizer Inc., AbbVie, Anacor, AnaptysBio, Arena Pharmaceuticals, Asana Biosciences, Dermira, Dermavant, Eli Lilly, Galderma, GSK, Glenmark, Incyte, Kiniksa, LEO Pharma, MedImmune, Menlo Therapeutics, Novartis, Realm Therapeutics, Regeneron, and Sanofi; a speaker for Regeneron and Sanofi; and is on advisory boards for Pfizer Inc., Dermira, LEO Pharma, and Menlo Therapeutics. A. Nosbaum is an investigator for AbbVie, Eli Lilly, Incyte, LEO Pharma, Novartis, and Sanofi; a consultant for Pfizer Inc., AbbVie, Eli Lilly, Galderma, LEO Pharma, Novartis, and Sanofi; a speaker for AbbVie, Regeneron, and Sanofi; and is on advisory boards for Pfizer Inc., AbbVie, LEO Pharma, and Sanofi. K. L. Winthrop reports research grants from Bristol Myers Squibb and consultant honorarium from Pfizer, Eli Lilly, Bristol Myers Squibb, UCB, AbbVie, Gilead, Roche, Novartis, Regeneron, and Sanofi. E. Guttman-Yassky is an advisory board member for Celgene, Dermira, Galderma, Glenmark, Medimmune, Novartis, Pfizer, Regeneron, Sanofi, Stiefel/GlaxoSmithKline, Vitae, and Asana Biosciences (honorarium); consultant for AbbVie, Anacor, Celgene, Dermira, Galderma, Glenmark, LEO Pharma, Medimmune, Novartis, Pfizer, Regeneron, Sanofi, Stiefel/GlaxoSmithKline, Vitae, Mitsubishi Tanabe, Eli Lilly, Asana Biosciences, Kiowa Kirin, and Almirall (honorarium); and investigator for Celgene, LEO Pharma, Medimmune, Regeneron, and Eli Lilly (grants to institution). K. M. Hoffmeister has received honoraria as consultant from Pfizer. A. Egeberg has received research funding from Pfizer, Eli Lilly, Novartis, AbbVie, Janssen Pharmaceuticals, the Danish National Psoriasis Foundation, the Simon Spies Foundation, and the Kgl Hofbundtmager Aage Bang Foundation, and honoraria as consultant and/or speaker from AbbVie, Almirall, LEO Pharma, Galápagos NV, Samsung Bioepis Co., Ltd., Pfizer, Eli Lilly and Company, Novartis, Galderma, Dermavant, UCB, Mylan, Bristol Myers Squibb, and Janssen Pharmaceuticals. H. Valdez, M. Zhang, S. A. Farooqui, W. Romero, A. J. Thorpe, R. Rojo, and S. Johnson are employees of and shareholders in Pfizer.
Ethics approval
Ethics approval was obtained for individual studies included in this integrated safety analysis.
Informed consent
Informed consent was obtained from all participants of the individual studies included in this integrated safety analysis.
Data availability
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Author contributions
EGY, AE, HV, MZ, WR, AJT, RR, and SJ contributed to the conceptualization and design of the analysis. ELS, AN, KLW, KMH, AE, MZ, WR, AJT, and SJ performed or supported the data analysis. All authors contributed to the data interpretation, critically reviewed the manuscript and approved the submitted version.
Additional information
The original online version of this article was revised due to revised supplement file.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Simpson, E.L., Silverberg, J.I., Nosbaum, A. et al. Integrated Safety Analysis of Abrocitinib for the Treatment of Moderate-to-Severe Atopic Dermatitis From the Phase II and Phase III Clinical Trial Program. Am J Clin Dermatol 22, 693–707 (2021). https://doi.org/10.1007/s40257-021-00618-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-021-00618-3